Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(5):500-510. doi:10.7150/ijbs.8065 This issue Cite
Research Paper
Profiling of MicroRNAs under Wound Treatment in Aquilaria sinensis to Identify Possible MicroRNAs Involved in Agarwood Formation
Zhi-Hui Gao1, Yun Yang2, Zheng Zhang1, Wen-Ting Zhao1, Hui Meng2, Yue Jin1, Jun-Qing Huang1, Yan-Hong Xu1, Li-Zi Zhao1, Juan Liu1, Jian-He Wei1,2 ![]()
1. Institute of Medicinal Plant Development, Chinese Academy of Medical Science & Peking Union Medicinal College (National Engineering Laboratory for Breeding of Endangered Medicinal Materials), Beijing 100193, China
2. Hainan Branch, Institute of Medicinal Plant Development (Hainan Provincial Key Laboratory of Resources Conservation and Development of Southern Medicine), Chinese Academy of Medical Science & Peking Union Medicinal College, Wanning 571533, China
Received 2013-11-6; Accepted 2014-4-3; Published 2014-4-25
Abstract
Agarwood, a kind of highly valued non-timber product across Asia, is formed only when its resource trees -- the endangered genus Aquilaria are wounded or infected by some microbes. To promote the efficiency of agarwood production and protect the wild resource of Aquilaria species, we urgently need to reveal the regulation mechanism of agarwood formation. MicroRNAs (miRNAs) are a group of gene expression regulators with overwhelming effects on a large spectrum of biological processes. However, their roles in agarwood formation remain unknown. This work aimed at identifying possible miRNAs involved in the wound induced agarwood formation. In this study, the high-throughput sequencing was adopted to identify miRNAs and monitor their expression under wound treatment in the stems of A. sinensis. The miR171, miR390, miR394, miR2111, and miR3954 families remained at the reduced level two days after the treatment. 131 homologous miRNAs in the 0.5 h library showed over three-fold variation of read number compared with the control library, of which 12 exhibiting strong expression alterations were further confirmed by real-time quantitative PCR. Target prediction and annotation of the miRNAs demonstrated that the binding, metabolic process, catalytic activity, and cellular process are the most common functions of the predicted targets of these newly identified miRNAs in A.sinensis. The cleaveage sites of three newly predicted targets were verified by 5'RACE.
Keywords: microRNA, Agarwood, Aquilaria sinensis, wound, small RNA
Introduction
Agarwood is a highly valued fragrant non-timber products used in medicine, perfumes, and incense across Asia, Middle East, and Europe [1, 2]. It is produced by species of the tropical tree genus Aquilaria, of which the population is dramatically declining due to overexploitation. Nine species of this genus have been listed in the IUCN Red List of Threatened Plants published by The World Conservation Union (IUCN) since 1998 [3]. Furthermore, all of the Aquilaria species are regulated under the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES). Because agarwood is formed only after the trunk or branch is wounded or infected with microbes [4, 5], even though Aquilaria trees have been planted in South China and many Southeast Asian countries on a large scale, the global demand for agarwood still considerably exceeds the supply. The stimuli could induce defensive response and changes of secondary metabolism network which lead to the accumulation of sesquiterpenes and phenylethyl chromone derivatives and the deposition of resin [6]. Our previous researches [7-9] showed that wound induced expression of many genes in the pathway of sesquiterpenes biosynthesis and could induce the biosynthesis of sesquiterpenes in A. sinensis. This kind of wound-induced production of useful secondary metabolites also exists in many economic plants, including coniferous trees (for resin production), Dracaena angustifolia, Santalum album L., and Dalbergia odorifera (for medicinal compounds production), etc. Further understanding of the regulation mechanism of agarwood formation will help to reveal the relationship between plant defensive response and secondary metabolism, and supply information for efficient agarwood production without damage to the wild resource of Aquilaria. However, the molecular mechanism of the agarwood induction remains largely unknown.
MiRNAs, a group of endogenous non-coding small RNAs (sRNAs) have been proved to be crucial post-transcriptional and translational as well as transcriptional gene expression regulators [4, 10-12]. The functional mature miRNAs are generally 20-24 nucleotides and generated from their precursors (pre-miRNAs) which are usually 70-300 nt in length in plants and can form imperfect stem-loop structures. The mature miRNAs regulate gene expression mainly by targeting mRNA for mRNA cleavage or repressing the protein translation [4]. In plants, the miRNA and the target mRNA interact in a near-perfect sequence complementary way [13].
MiRNAs have been shown to be involved in a large variety of biological processes in plants, including developmental processes and stress responses. Some plant miRNAs were observed to respond to stress conditions and some miRNA targets were found to be stress-associated, indicating that miRNAs may participate in plant stress responses. Employing miRNA macroarrays, miRNAs responded to nutrient stresses in three organs of common bean were identified [14]. Yakovlev et al. [15] identified 24 novel and 16 conserved miRNAs in Norway spruce and analyzed their expression in cold and warm conditions with real-time RT-PCR. By deep sequencing, ten conserved and a series of novel miRNAs that responded to heat stress were identified in Brassica rapa [16]. With the same approach, eight and 13 miRNA families were found to be significantly regulated by toxic heavy metal cadmium in roots or shoots of Brassica napus, respectively [17]. MiRNAs respond to stresses including drought [18, 19], salt [20-22], cold [23, 24], oxidation [18, 25], nutrient deprivation [26-28], and aluminum oxide nanoparticles [29] were also identified.
In the last few years, several studies have investigated the wound response or the defensive roles of miRNAs in plants. The mechanical stress responsive miRNAs were identified in Populus [30]. Some wounding/topping responsive small RNAs and viral defensive miRNAs were identified by sequencing or bioinformatics approach [31-33]. The highly abundant 22nt miRNAs were shown to be master regulators of NB-LRR defense gene family in legume by small RNA sequencing and bioinformatics analysis [34]. Functional study proved that the miR319 regulated TCP (TEOSINTE BRANCHED/CYCLOIDEA/PCF) transcription factors were responsible for the synthesis of defensive hormone jasmonic acid in Arabidopsis [35]. Recently, miR393 was shown to repress the auxin signalling and prevent it from antagonizing SA signalling, thus can fine-tune plant defences and prioritize resources in Arabidopsis [36].
Such a class of critical regulator might function in the stress response and agarwood formation of A. sinensis. However, the responsive activities and responsive patterns of miRNAs after wound treatment are almost totally unknown in plant stems and in Aquilaria species. In the previous study, we developed a method for the miRNA identification of the non-conserved plant species like A. sinensis using the transcriptome data and examined the expression of several stress-responsive miRNAs after wound treatment by real-time PCR [37]. To further investigate the responsive pattern of all miRNAs under wound treatment and identify potential critical miRNAs in agarwood biosynthesis, we predicted Aquilaria miRNA and monitored the expression changes of the small RNAs after wound treatment in the stems of A. sinensis by small RNA sequencing.
Materials and Methods
Plant materials and wound treatment
Stem samples were collected from three-year-old A. sinensis cultivated in the Hainan Branch, Institute of Medicinal Plant Development, Chinese Academy of Medical Science & Peking Union Medicinal College. For wound treatment, stems were incised by single side razor blades at every 1-2 cm. The wounded stems were collected at different time points including 0.5 h, 4 h, and 48 h, according to previous studies [6, 7, 38]. Samples of all time points were collected from different seedlings. For each time point, over five seedlings were used and the all treated stems were collected together. For sequencing, the samples were mixed. For RT-PCR, The samples from different seedlings were used as biological repeats. These samples and untreated stems were ground in liquid nitrogen for RNA isolation or stored at -80℃ until use.
RNA isolation and sRNA sequencing
RNAs were extracted from all samples using the Norgen Total RNA Extraction Kit (Cat#17200). The quantity and quality of each RNA sample were examined by NanoDrop ND2000 (Thermo Fisher, USA). The RNA integrity was measured by agarose gel electrophoresis. For sRNA sequencing, 10 μg of total RNA isolated from each sample was pooled. Illumina-solexa sequencing of sRNA was performed at the MininGen Biotechnology Co. Ltd, China. RNA was purified by polyacrylamide gel electrophoresis (PAGE) to enrich the molecules in the range of 18-30 nt, and ligated with proprietary adaptors to both 5' and 3' termini of the RNA. Ligated samples were used as templates for cDNA synthesis and then amplified with 18 PCR cycles to produce the sequencing libraries that were subjected to Illumina's proprietary sequencing-by-synthesis method.
Prediction of conserved miRNAs and novel miRNAs
Individual sequence reads with the base quality scores were produced by Illumina. Adapter sequences were removed from both ends of Illumina reads. All identical sequences were counted and redundant sequences were eliminated from the initial data set. The resulting set of unique sequences with associated read counts is referred to as sequence tags. These reads were mapped onto the transcriptome data of A. sinensis, which contained 89,137 unigenes generated from 454 sequencing. The reads were then classed into two sets, the perfectly matched reads and not perfectly matched reads. For the perfectly matched data, the lowly expressed reads (read counts <2) were filtered first. Then, the 150 nt upstream and 150 nt downstream sequences of the candidate unique reads on the transcriptome sequences were extracted for secondary structure prediction. The Einverted of Emboss [39] was used to find the inverted repeats (step loops or hairpin structure), with parameters threshold =30, match score = 3, gap penalty = 6, and maximum repeat length = 240 as described by Jones-Rhoades and Bartel [40]. The secondary structure of the inverted repeat was predicted by RNAfold. Unique reads in the inverted repeats were evaluated by MirCheck [40]. All mature sequences of candidate miRNAs predicted by previous secondary structure analysis were searched against the plant mature miRNAs of Sanger miRBase (Release 16) using the program Patscan. Two mismatches were allowed while identifying homologs of known plant miRNAs. These homologs were then identified as conserved miRNAs and the sequences that could not match to known plant miRNAs were considered as candidate novel miRNAs. The reads in the not perfectly matched set were also searched against the plant mature miRNAs of Sanger miRBase by the same approach. If one read could match to known plant miRNAs and was sequenced more than 100 times in the four libraries, it was deemed as “homolog miRNAs”.
Target prediction
A search for miRNA target genes was then performed using an approach described previously [41]. All identified miRNA sequences were used to query the transcriptome library for potential target sequences using Patscan with default parameters. Three mismatches, 0 insertions, and 0 deletions could be permitted. Only hits that with less than two mismatches in positions 1-9, no mismatches in positions 10 and 11, and less than three mismatches after position 11 in the mature miRNAs were considered good target sequences. Target genes were annotated with Swissprot Database (downloaded from European Bioinformatics Institute by Feb 8th, 2010) [42], with e value≤ 1e-10, as well as by Interproscan [43]. WEGO [44] was used to do GO classification.
Determination of miRNA expression profile
We employed IDEG6 [45], which is similar to credibility interval approaches reported for the analysis of SAGE data [46], to identify miRNAs showing statistically significant difference in relative abundance (as reflected by total count of individual sequence reads) between each two small RNA libraries. We used the general Chi2' method, because it resulted in the most efficient tests [45]. Finally, miRNA with a P-value≤ 0.01 was deemed to be significantly different between the two samples.
Real-time reverse transcription-PCR
The mature miRNA reverse transcription was performed with MIR-specific stem-loop primers. These MIR-specific primers (Additional File 1: Supplementary table 4) were designed according to the mature miRNA sequence [47]. 0.5 μg total RNA for each sample was used in the 25 μl reaction system containing 200 U M-MLV transcriptase (Promega), 20 U RNase inhibitor (Promega), 0.2 mM dNTP, and 0.1 μM primer. The PCR process was performed as previously described by Pant et al. The real-time PCR reaction was performed using SYBR Green as fluorescence dye and run on 96-well plates with an iCycler iQ thermocycler (BioRad). PCR was performed with 40 cycles of 95℃ for 15 s and 60℃ for 1 min. Each PCR was repeated at least three times and each miRNA was tested in triplicated samples. Expression data were collected by BioRad iQ5 Optical System Software version 2.0, 2-⊿⊿Ct method was adopted to assess the relative expression level of stress-responsive miRNAs and tubulin (TUA) was used as a reference according to our previous work [37].
5'RACE-PCR
The SMARTER RACE cDNA amplification kit (Clontech) was used for the 5'RACE-PCR to detect the cleavage sites of the miRNA-target genes. A gene-specific primer was used for each amplification of cDNA ends. The PCR products from a positive 5'RACE reaction were gel purified and cloned into the pMD19-T simple vector for sequencing.
Results
Deep sequencing of sRNAs in wound and control stems of A. sinensis
After Solexa sequencing and removing the low-quality sequences, adaptor sequences and the sequences shorter than 18 nt, the four sRNA libraries (CK, 0.5 h, 4 h, 48 h), which represented the healthy stems, stems after short-term, middle-term, and long-term wound treatment produced 7,683,107, 7,196,203, 7,493,219, and 6,791,432 high-quality reads, respectively (Table 1). Among them, the 0.5 h data group showed the highest sequence redundancy with 57% redundancy rate and the CK group showed the lowest redundancy with 37% redundancy rate.
Data profile of sequenced reads in four libraries
| Class | Total Reads | High-quality Reads | Total Unique Clean Reads | Redundancy(%)a | Perfect Matchedb Unigene Datac | % | Total of Perfect Matched | % |
|---|---|---|---|---|---|---|---|---|
| CK | 9015257 | 7684107 | 5071927 | 34.0 | 626697 | 12.36 | 2227879 | 28.99 |
| 0.5 h | 8656112 | 7196203 | 3097915 | 57.0 | 422494 | 13.64 | 2655741 | 36.90 |
| 4 h | 8625731 | 7493219 | 4589050 | 38.8 | 590850 | 12.88 | 2368534 | 31.61 |
| 48 h | 8097027 | 6791432 | 3578524 | 47.3 | 516943 | 14.45 | 2733023 | 40.24 |
| Total | 34394127 | 29164961 | 14625419 | 49.9 | 1623045 | 11.10 | 9985177 | 34.24 |
a Redundancy=100-(Total Unique Clean Reads/Total Clean Reads x 100)
b using soap1.11 aligner: soap.huge -s 6 -v 0 -r 2
c There are 89137 unigenes, generated from 454 sequencing
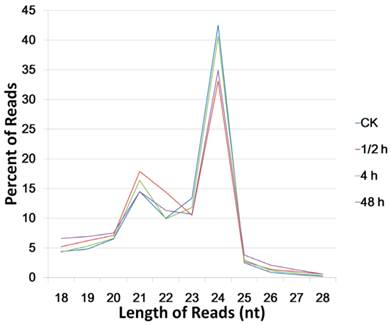
For the length distribution of total sequences, 20-24 nt reads amounted to over 80% in each library, while the 24 nt reads were the most abundant, and the 21 nt reads took the second place. In the CK sample, the 24 nt and 21 nt reads amounted to 42.5% and 14.5%, respectively. After 0.5 h of wound treatment, the 21 nt sequences grew to 17.9%, whereas the 24 nt sequences rapidly decreased to 33.1% (Figure 1). The unique sequences in these two libraries did not exhibit significant difference, demonstrating that the wound treatment may strongly and rapidly induce the expression of 21 nt sRNA which represents the miRNAs group.
Length distribution of total small RNAs (high quality reads) in the four small RNA libraries.
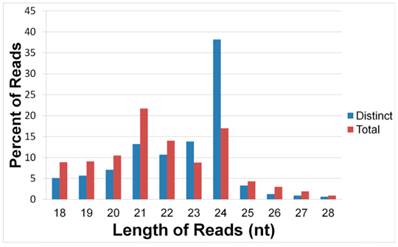
For stem-loop structure prediction of potential miRNAs, all unique sequences were mapped to the 454 transcriptome libraries of healthy and wounded stems of A.sinensis. The perfectly matched distinct reads respectively made up 12.36%, 13.64%, 12.88%, and 14.45% of the four libraries (Figure 2). The length distribution analysis showed that, in the perfectly matched sRNAs, the 21 nt class had the highest level of abundance, although the 24 nt sRNAs had the most distinct sequences.
Length distribution of the perfect matched sRNAs. The distinct sRNA is obtained by counting each sRNA sequence for once, the total sRNA include all reads of each sRNA sequence.
Conserved and candidate novel miRNAs identified in A.sinensis
The conserved miRNAs in A. sinensis were identified by the homology-based method using the plant miRNA sequences in the miRBase. For precursor prediction, because only a few nucleotide sequences of A. sinensis were available in GenBank, we employed the transcriptome sequence data of A. sinensis obtained by 454 sequencing to search for the inverted repeats and stem-loop structures as previously described [48]. Finally, 11 sequences were identified as conserved miRNAs (Additional File 1: Supplementary table 1), and their hairpin structures are shown in Additional File 2: Supplementary figure 1. As conserved miRNAs with precursors are quite limited, another strategy previously used [37, 49, 50] was also adopted to identify miRNAs in A. sinensis. Based on homology, all reads without precursors were blasted against the mature miRNA sequences in miRBase. If an identified homology read was sequenced more than 100 times in the four libraries, it would be deemed as “Homologues microRNA” (homo-miRNA). A total of 283 sRNAs in A. sinensis were identified as homo-miRNAs (Additional File 1: Supplementary table 2).
To uncover novel miRNAs candidate from A. sinensis, all unannotated sRNAs were searched against our transcriptome sequence data of A. sinensis to identify perfectly matched sequences. After searching for potential miRNA precursors and predicting their secondary structures, 174 sequences were identified as candidate novel miRNAs with stem-loop precursors in A. sinensis (Additional File 2: Supplementary figure 2). The detailed miRNA sequences and their precursors are described in Additional File 1: Supplementary table 3.
Expression pattern of miRNA families after wound treatment
To compare the expression patterns of the miRNA families in A. sinensis after wound treatment, the read numbers of all members of each family in the four libraries were summed up. Two groups were divided according to the read numbers. In the highly expressed group were miR156, miR159, miR162, miR164, miR168, miR172, miR319, and miR396 while in the lowly expressed group were miR160, miR166, miR167, miR169, miR171, miR390, miR393, miR394, miR2111, and miR3954. The results showed in Figure 3 demonstrated that the miR159 family was the most abundant in all libraries, with over 20,000 reads for each library. The miR396 was another highly accumulated miRNA family. Except for the 0.5 h after wounding, miR396 family had over 20,000 reads per million reads at all time points, but all less than the numbers of miR159 family in the same library. Relatively speaking, miR160, miR390, and miR3954 were the three least accumulated families which had less than 200 reads in each library.
Expression levels of conserved miRNA families in the four small RNA libraries of A. sinensis. A: expression pattern of the highly expressed miRNA families; B: expression patterns of the lowly expressed miRNA families.
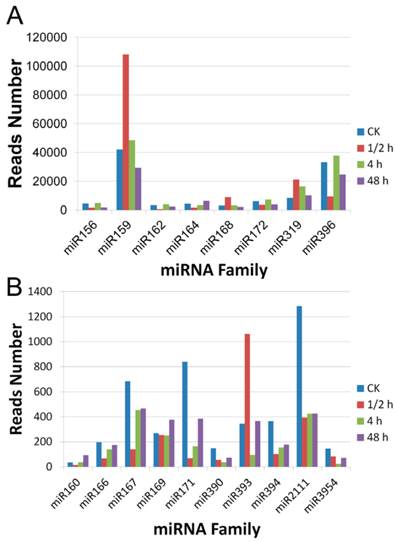
All miRNA families except for the miR169 and the miR172 showed significant variation in the four libraries. Most families showed a great change at the 0.5 h after wound treatment. For example, miR159, miR168, and miR393 were increased by over twofold compared with the CK, whereas miR156, miR162, miR164, miR396, miR160, miR166, miR167, miR171, miR390, miR394, and miR2111 were all drastically decreased. After these dramatic changes, most miRNA families including miR159, miR162, miR164, miR168, miR319, miR396, miR166, and miR167 returned to the level close to that of the untreated sample. Noteworthy, miR171, miR390, miR394, miR2111, and miR3954 remained at the reduced level until 2 days after the treatment. We also observed that the miR160 family had a decreased level at 0.5 h, but gradually accumulated to a level higher than that of the untreated sample thereafter.
Differently expressed miRNAs under wound treatment
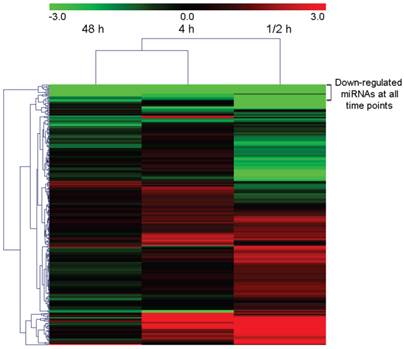
The expression levels of all identified homologous/conserved miRNAs in the four samples were compared using the Cluster 3.0 algorithm [51]. Expression clustering with different samples showed that the expression characteristic of the 0.5 h sample which represented the short-term response to the wound treatment was different from that of other samples (Figure 4). The 4 h response and 48 h response were the closest among all libraries. In general, 131 miRNAs in the 0.5 h library showed an over three-fold variation in read number compared with those in the CK library. 70 and 29 in the 4 h library and 48 h library respectively showed over three-fold variance. 13 miRNAs including the conserved miRNA miR473 remained at a constant level in all the three time points after treatment.
Heat map showing differently expressed conserved and homo-miRNAs in wound tissues compared with the 0h sample.
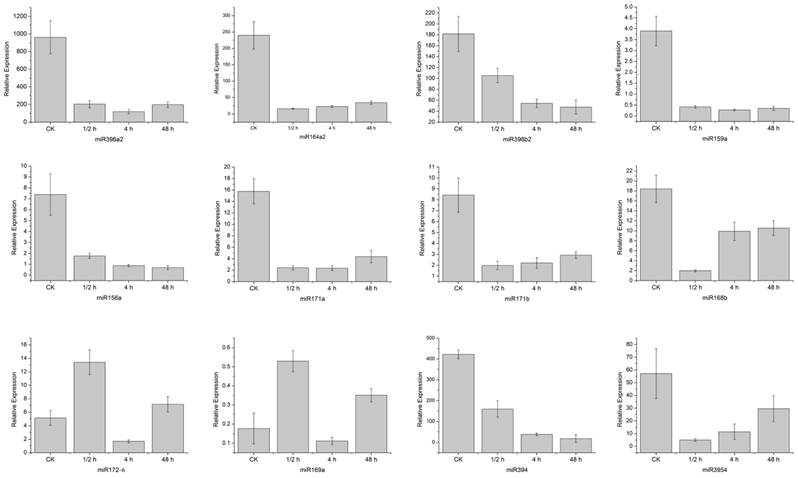
Twelve homo-miRNAs showing different expression levels in the control and wounded samples were selected for verification by the real-time quantitative PCR (Figure 5). Except for miR168 which showed no dramatic increase at the 4 h time point as the sequencing data, all these miRNAs exhibited the same expression pattern with their sequencing results. That is, miR396a2, miR164a2, miR396b2 and miR159a, miR156a, miR171a, miR171b, miR168b, miR394, and miR3954 were down-regulated by the wound treatment, while miR172 and miR169 were up-regulated.
Confirmation of 12 differently expressed homo-miRNAs by quantitative real-time PCR. Stem samples were collected during a time course after being hurt by single side razor blades. Error bars indicate one standard deviation of three biological replicates.
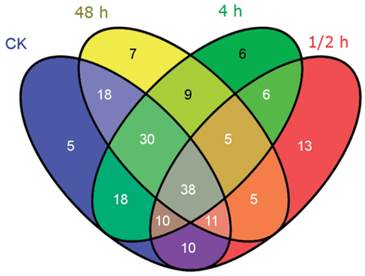
The distribution of all novel miRNA candidates identified from the four libraries was displayed by Venn diagram (Figure 6). 140, 98, 122, and 123 miRNAs were respectively indentified in the CK, 0.5 h, 4 h, 48 h libraries. Among them, 38 novel miRNA candidates were common in all libraries. Similar to the homo-miRNAs, the 0.5 h time point shared the least common novel miRNA candidates with the control sample. A total of 13 novel miRNA candidates were only identified at the 0.5 h time point, making up 13.3% of the novel miRNA candidates in this library. 6 and 7 novel miRNA candidates were specific for the 4 h time point and 48 h time point, respectively, making up 4.9% and 5.7% in the corresponding library.
Venn diagram indicating the common and unique novel miRNAs in the four small RNA libraries.
Biological processes regulated by miRNA changes during the wound treatment
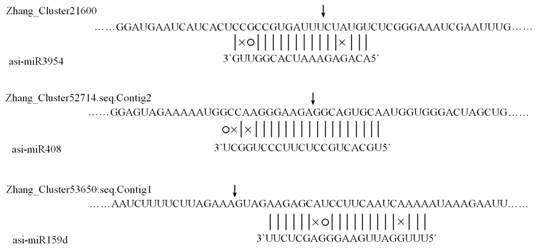
Since miRNAs function via regulating the expression of their target genes, to further dig the associations between agarwood formation and miRNAs as well as to predict the underlying regulation mechanisms, targets of all identified miRNAs in A. sinensis were predicted using the 454 transcriptome sequences. 3,125 sequences were identified as the targets of the conserved and novel miRNAs in A. sinensis by the computational algorithm. 285 and 3,125 sequences were respectively identified as the putative targets of homo-miRNAs and conserved/novel miRNAs in A.sinensis. Compared to the conserved/novel miRNAs, the targets of homo-miRNAs identified were limited. This might be caused by the fact that the homo-miRNAs were universally highly expressed, leading to the low expression level of their targets in the transcriptome data. To further confirm the newly predicted targets of the Aquilaria miRNAs, 5' RACE PCR were conducted to define the cleavage sites of three newly identified targets. As shown in Figure 7, the transcript of zhang_cluster21600, zhang_cluster52714.seq.Contig2, and zhang_cluster53650.seq.Contig1 were cleaved by asi-miR3954, asi-miR408, and asi-miR159d respectively.
Cleavage sites of three newly predicted targets identified by the 5' RACE PCR. The position of dominant 5'RACE products of three transcripts are indicated by a vertical arrow in the expanded region.
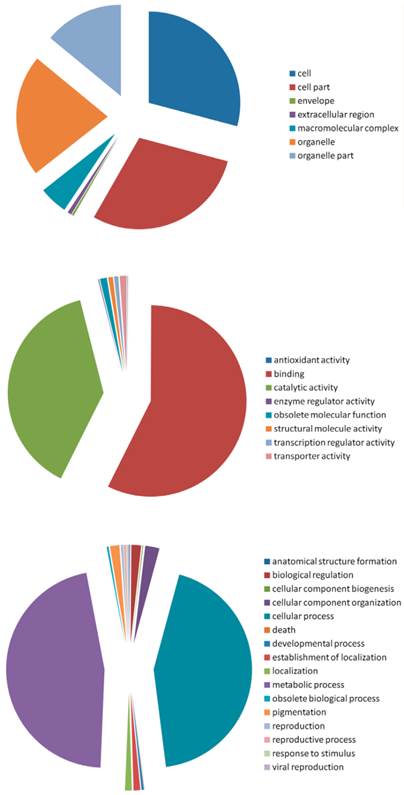
The gene ontology categories were assigned to these predicted targets according to their cellular component, molecular function and the biological processes in which they were involved (Figure 8). In the cellular component group, the targets fell into seven categories, with cell, cell part and organelle as the three most represented. In the molecular function group, the targets were classified into eight categories, with the most falling into the binding and the catalytic activity categories. Among the 16 categories of the biological process group, most of the targets were classified into the metabolic process and cellular process. Taken all these targets together, binding, metabolic process, catalytic activity, and cellular process were the most enriched.
Gene Ontology of the predicted targets of the conserved and novel miRNAs in A.sinensis. Categorization of miRNA-target genes was performed according to the cellular component (A), molecular function (B) and biological process (C).
Some predicted targets of these miRNAs have functional relevance to wound. For example, the targets of miR396-4 and miR396-44 were annotated with oxidoreductase activity; the target of miR157-2 was related to cell redox homeostasis; because oxidative burst is a major response triggered by wound, these enzymes might all be involved in the defence response. The targets of miR396-4 and miR394-1 were predicted having aspartic-type endopeptidase activity which might also as a part of defence response. For the candidate novel miRNAs, a possible target of novel-100b-5p was predicted to have glutathione peroxidase activity and response to oxidative stress which always happened after wound; the target of novel-5-3p and novel-10-3p was predicted to have function in viral reproduction; furthermore, more and different types of peptidases were predicted to be the targets of these novel miRNAs. The possible actions of miRNAs after wound were described in the Additional File 2: Supplementary Figure 3.
For most miRNAs, multiple targets with diverse functions were identified, reflecting the high efficiency of this group of short RNAs adopted by biological organisms. The targets of several differently expressed miRNAs were identified. For example, the targets of miR169-1 which accumulated significantly in the 0.5 h library have protein kinase activities. The target of miR396b2 which was down-regulated in all wound libraries has glutamyl-tRNA reductase activity and was predicted to bind NADP, a function involved in the production of NADPH. The target of miR159b which was up-regulated in the 0.5 h and 4 h libraries has transcription factor activities. The target of miR3954 which was down-regulated in all wound libraries has DNA binding and aminoacyl-tRNA ligase activity to regulate the protein translation. In the novel miRNAs, miR-novel-094-3p which was expressed only in the three wound libraries had the target with transcription factor activity.
Discussion
Novel miRNA candidates in A. sinensis and their possible function
As the agarwood resource, Aquilaria is a significant tree genus for the economic development of many countries in Asia. However, the wound-induced nature of the agarwood formation restricts the agarwood yield and threatens the populations of Aquilaria trees. Since miRNAs are a class of critical regulator in various biological processes, this work aimed to identify miRNAs involved in the wound stress in A. sinensis and to find the possible association between miRNAs and wound-induced agarwood formation. Thus, we investigated the specific miRNAs and their time course expression patterns in the healthy and especially the wounded stems of A. sinensis.
Through the high-throughput small RNA sequencing, 34,394,127 small RNA sequences were obtained. Among them 11 conserved and 136 novel miRNA candidates were identified by precursor prediction using the transcriptome sequence data. Target prediction of these miRNAs indicated their possible functions in A. sinensis. It was intriguing that most functions of these targets were concentrated in four GO categories: binding, metabolic process, cellular process and catalytic activity. Because the agarwood formation is a progress of defensive response accompanied with the accumulation of secondary metabolites mainly in sesquiterpenes and chromones. The results indicated that the candidate novel miRNAs involved in the metabolic process and catalytic activity might have direct function in the secondary metabolites accumulation under the wound treatment. And the miRNAs involved in the binding and cellular processes might participate in the transcription regulation, signal communication, and energy supply for secondary metabolism.
Wound induced miRNAs and their possible roles in agarwood formation
For the most conserved miRNAs such as miR159 and miR396 families, the most significant different expression appeared at the 0.5 h time point and then the difference quickly disappeared, indicating that they might function at the upstream of wound response. MiR473 was an opposite example, of which the expression not only quickly reduced at the early response phase but also kept at a lower level in the consequent response phase. In Populus trichocarpa, the expression of ptr-miR473 was reduced in mechanically stressed woody tissues and it is predicted to target an UV-B resistant gene (UVR8), which is believed to positively regulate phenylpropanoid metabolism associated with cinnamate 4-hydroxylase for the production of cell wall polyphenolics, such as lignin and flavonoids [30]. Further identification of the target of miR473 in A. sinensis would help to reveal the exact role of miR473 in the wound-induced agarwood formation. In the homo-miRNAs, miR171 members showed a consistently low expression level in the wounded stems compared to the healthy stems. In previous studies, the function of miR171 family has been designated to be associated with the plant development [52, 53]. However, this family was found to be involved in the drought stress in Solanum tuberosum by sequencing [54] and also down-regulated in the Cd-stressed rice by microarray [55]. Our result gave another evidence for participation of the miR171 family in plant stress response. This also implied the relationship of plant stress and development. Several other miRNAs which remained at a constant expression level in all three wound libraries were also identified. As agarwood formation is a long-term process, these miRNAs were predicted to have more direct functions other than the early wound responses such as signal transduction, defensive hormone synthesis, and the expression of regulatory genes. The target of miR396b2 which were down-regulated in all wound libraries has glutamyl-tRNA reductase activity and was predicted to bind NADP, these functions were involved in the production of NADPH, which is the cofactors of HMGR and DXR, the two key enzymes in terpene biosynthesis [56, 57]. This demonstrates that this miRNA is a direct effector for the accumulation of agarwood constituents. Since plant wound response may include various aspects of biological activities, whether miR396b2 and other wound-responsive miRNAs are critical in the agarwood formation remains to be revealed to further uncover and confirm their targets as well as the corresponding target functions.
In previous work, we selected ten stress-related miRNAs according to some published papers[37], and examined their expressions after wound treatment by RT-PCR. These miRNAs include members from the same family of the miRNAs we examined by RT-PCR here, such as miR159, miR168, miR171, miR172 and miR396 families, these miRNAs from a same family showed different expression patterns. Furthermore, our results of the expression pattern of the miRNA families and individual miRNAs also showed that miRNAs in a same family always have diverse expression pattern. This was consistent with previous works [58-60], demonstrating distinct functions performed in a miRNA family.
Wound-associated biological processes in A. sinensis
A large number of wound-responsive conserved/homo miRNAs have been reported to participate in many developmental processes in other plants. For example, miR159, which mainly targets the MYB transcription factors in Arabidopsis, affects the leaf development and embryogenesis [61]. miR396 regulates GROWTH-REGULATING FACTORs (GRFs) which are required for coordination of cell division and differentiation during leaf development in Arabidopsis [62]. Even though these two families are all related to the leaf development in Arabidopsis, they were the two most abundant miRNA families in the stem of A. sinensis. Thus, their novel functions maybe expected in different plants and tissues or under different conditions. With a large group of members in these two families, divergent expression pattern also appeared while most members shared similar expression patterns. This phenomenon demonstrates that the function diversity may exist among different family members.
As wound-responsive miRNAs related to plant development, miR156 control the floral organ identity and flowering time, and miR164 controls the boundary in meristem, organ formation and separation. Since these miRNAs have great potential to participate in the development of A. sinensis too, their responses to the wound treatment imply that wound treatment may influence many aspects of the development of A. sinensis.
Supplementary Material
Additional File 1Supplementary Table 1-Supplementary Table 4.
Supplementary Figure 1-Supplementary Figure 3.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81001607 and No. 31100220), the Program for New Century Excellent Talents in University Funded by the Ministry of Education of China (No. 2008), National Key Technology R&D Program (No. 2011BAI01B07), and the Key Project in the Science & Technology Program of Hainan Province (No. ZDXM20120033, ZDZX20100006).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yagura T, Shibayama N, Ito M. et al. Three novel diepoxy tetrahydrochromones from agarwood artificially produced by intentional wounding. Tetrahedron Lett. 2005;46(25):4395-4398
2. Persoon GA. Growing 'the wood of the gods': agarwood production in southeast Asia. Smallholder Tree Growing for Rural Development and Environmental Services: Lessons from Asia. 2008;5:245-262
3. Oldfield S, Lusty C, MacKinven A. The world list of threatened trees. Combridge,UK: World conservation press. 1998
4. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215-233
5. Pojanagaroon S, Kaewrak C. Mechanical methods to stimulate aloes wood formation in Aquilaria crassna Pierre ex H.Lec. (Kritsana) trees. WOCMAP III: Conservation, Cultivation and Sustainable Use of MAPs. 2005(676):161-166
6. Ito M, Kumeta Y. Characterization of delta-guaiene synthases from cultured cells of Aquilaria, responsible for the formation of the sesquiterpenes in agarwood. Plant Physiol. 2010;154(4):1998-2007
7. Zhang Z, Yang Y, Wei J. Response of endogenous jasmonates and sesquiterpenes to mechanical wound in Aquilaria sinensis stem. Acta Horticulturae Sinica. 2013;40(1):163-168
8. Gao Z, Zhao WT, Jin Y. Expression patterns of terpene synthases of endangered Aquilaria sinensis under different stresses. Chinese Trad Herb Drugs. 2013;44(6):749-754
9. Zhang Z, Zhang X, Yang Y. et al. Hydrogen peroxide induces vessel occlusions and stimulates sesquiterpenes accumulation in stems of Aquilaria sinensis. Plant Growth Regul. 2014;72(1):81-87
10. Khraiwesh B, Arif MA, Seumel GI. et al. Transcriptional control of gene expression by microRNAs. Cell. 2010;140(1):111-122
11. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281-297
12. Baek D, Ville´n J, Shin C. et al. The impact of microRNAs on protein output. Nature. 2008;455:64-71
13. Bartel B, Bartel DP. MicroRNAs: at the root of plant development? Plant Physiol. 2003;132(2):709-717
14. Valdes-Lopez O, Yang SS, Aparicio-Fabre R. et al. MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytol. 2010;187(3):805-818
15. Yakovlev IA, Fossdal CG, Johnsen Ø. MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010;187(4):1154-1169
16. Yu X, Wang H, Lu Y. et al. Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J Exp Bot. 2012;63(2):1025-1038
17. Zhou ZS, Song JB, Yang ZM. Genome-wide identification of Brassica napus microRNAs and their targets in response to cadmium. J Exp Bot. 2012;63(12):4597-4613
18. Jagadeeswaran G, Saini A, Sunkar R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta. 2009;229(4):1009-1014
19. Li W-X, Oono Y, Zhu J. et al. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell. 2008;20(8):2238-2251
20. Liu D, Yu D. MicroRNA (miR396) negatively regulates expression of ceramidase-like genes in Arabidopsis. Prog Nat Science. 2009;19(6):781-785
21. Lu S, Sun Y-H, Chiang VL. Stress-responsive microRNAs in Populus. Plant J. 2008;55(1):131-151
22. Sun G, Stewart CN Jr, Xiao P. et al. MicroRNA expression analysis in the cellulosic biofuel crop switchgrass (Panicum virgatum) under abiotic stress. PLoS One. 2012;7(3):e32017
23. Zhou X, Wang G, Sutoh K. et al. Identification of cold-inducible microRNAs in plants by transcriptome analysis. BBA - Gene Regul Mech. 2008;1779(11):780-788
24. Zhang J, Xu Y, Huan Q. et al. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics. 2009;10(1):449
25. Sunkar R, Kapoor A, Zhu J-K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006 tpc.106.041673
26. Chiou T-J, Aung K, Lin S-I. et al. Regulation of phosphate homeostasis by microRNA in Arabidopsis. Plant Cell. 2006;18(2):412-421
27. Abdel-Ghany SE, Pilon M. MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J Biol Chem. 2008;283(23):15932-15945
28. Kawashima CG, Yoshimoto N, Maruyama-Nakashita A. et al. Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. Plant J. 2009;57(2):313-321
29. Burklew CE, Ashlock J, Winfrey WB. et al. Effects of aluminum oxide nanoparticles on the growth, development, and microRNA expression of tobacco (Nicotiana tabacum). PLoS One. 2012;7(5):e34783
30. Lu S, Sun Y-H, Shi R. et al. Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell. 2005;17(8):2186-2203
31. Tang S, Wang Y, Li Z. et al. Identification of wounding and topping responsive small RNAs in tobacco (Nicotiana tabacum). BMC Plant Biol. 2012;12:28
32. Perez-Quintero AL, Neme R, Zapata A. et al. Plant microRNAs and their role in defense against viruses: a bioinformatics approach. BMC Plant Biol. 2010;10:138
33. Perez-Quintero AL, Quintero A, Urrego O. et al. Bioinformatic identification of cassava miRNAs differentially expressed in response to infection by Xanthomonas axonopodis pv. manihotis. BMC Plant Biol. 2012;12(1):29
34. Zhai J, Jeong DH, De Paoli E. et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011;25(23):2540-2553
35. Schommer C, Palatnik JF, Aggarwal P. et al. Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol. 2008;6(9):e230
36. Robert-Seilaniantz A, MacLean D, Jikumaru Y. et al. The microRNA miR393 re-directs secondary metabolite biosynthesis away from camalexin and towards glucosinolates. Plant J. 2011;67(2):218-231
37. Gao ZH, Wei JH, Yang Y. et al. Identification of conserved and novel microRNAs in Aquilaria sinensis based on small RNA sequencing and transcriptome sequence data. Gene. 2012;505(1):167-175
38. Reymond P, Weber H, Damond M. et al. Differential gene expression in response to mechanical wounding and insect feeding in Arabidopsis. Plant Cell. 2000;12(5):707-719
39. Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics. 2000;16(6):276-277
40. Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14(6):787-799
41. Schwab R, Palatnik JF, Riester M. et al. Specific effects of microRNAs on the plant transcriptome. Dev cell. 2005;8(4):517-527
42. Altschul SF, Gish W. [27] Local alignment statistics. In: (ed.) Russell FD. Methods in Enzymology. vol. 266. Academic Press. 1996:460-480
43. Consortium GO. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(suppl 1):D258-D261
44. Ye J, Fang L, Zheng H. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(suppl 2):W293-W297
45. Romualdi C, Bortoluzzi S, d'Alessi F. et al. IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics. 2003;12(2):159-162
46. Vêncio RZN, Brentani H, Pereira CAB. Using credibility intervals instead of hypothesis tests in SAGE analysis. Bioinformatics. 2003;19(18):2461-2464
47. Chen C, Ridzon DA, Broomer AJ. et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33(20):e179
48. Gao ZH, Wei JH, Yang Y. et al. Selection and validation of reference genes for studying stress-related agarwood formation of Aquilaria sinensis. Plant Cell Rep. 2012;31(9):1759-1768
49. Subramanian S, Fu Y, Sunkar R. et al. Novel and nodulation-regulated microRNAs in soybean roots. BMC Genomics. 2008;9(1):160
50. Yang Y, Chen X, Chen J. et al. Differential miRNA expression in Rehmannia glutinosa plants subjected to continuous cropping. BMC Plant Biol. 2011;11:53
51. de Hoon MJL, Imoto S, Nolan J. et al. Open source clustering software. Bioinformatics. 2004;20(9):1453-1454
52. Kasschau KD, Xie Z, Allen E. et al. P1/HC-Pro, a viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA function. Dev Cell. 2003;4(2):205-217
53. Llave C, Xie Z, Kasschau KD. et al. Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297(5589):2053-2056
54. Hwang E-W, Shin S-J, Yu B-K. et al. miR171 family members are involved in drought response in Solanum tuberosum. J Plant Biol. 2011;54(1):43-48
55. Ding Y, Chen Z, Zhu C. Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa). J Exp Bot. 2011;62(10):3563-3573
56. Ma Y, Yuan L, Wu B. et al. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J Exp Bot. 2012;63(7):2809-2823
57. Nagegowda DA. Plant volatile terpenoid metabolism: Biosynthetic genes, transcriptional regulation and subcellular compartmentation. FEBS lett. 2010;584(14):2965-2973
58. Maher C, Stein L, Ware D. Evolution of Arabidopsis microRNA families through duplication events. Genome Res. 2006;16(4):510-519
59. May P, Liao W, Wu Y. et al. The effects of carbon dioxide and temperature on microRNA expression in Arabidopsis development. Nat Commun. 2013 4
60. Yumul RE, Kim YJ, Liu X. et al. POWERDRESS and diversified expression of the MIR172 gene family bolster the floral stem cell network. PLoS Genet. 2013;9(1):e1003218
61. Palatnik JF, Wollmann H, Schommer C. et al. Sequence and expression differences underlie functional specialization of Arabidopsis microRNAs miR159 and miR319. Dev Cell. 2007;13(1):115-125
62. Wang L, Gu X, Xu D. et al. miR396-targeted AtGRF transcription factors are required for coordination of cell division and differentiation during leaf development in Arabidopsis. J Exp Bot. 2010;62(2):761-773
Author contact
![]() Corresponding author: Phone: 86-10-57833363; Fax: 86-10-57833363; Email addresses: JW: wjianhnet
Corresponding author: Phone: 86-10-57833363; Fax: 86-10-57833363; Email addresses: JW: wjianhnet