Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(6):643-651. doi:10.7150/ijbs.8974 This issue Cite
Research Paper
Androgen Receptor Mutations and Polymorphisms in African American Prostate Cancer
Shahriar Koochekpour1, 2 ![]() , Erick Buckles3, 4, Mojgan Shourideh1, SiYi Hu4, Dhyan Chandra5, Jovanny Zabaleta4, 6, Kristopher Attwood7
, Erick Buckles3, 4, Mojgan Shourideh1, SiYi Hu4, Dhyan Chandra5, Jovanny Zabaleta4, 6, Kristopher Attwood7
1. Department of Cancer Genetics, Center for Pharmacology and Genetics, Elm and Carlton Streets, Roswell Park Cancer Institute, Buffalo, New York.
2. Department of Urology, Center for Pharmacology and Genetics, Elm and Carlton streets, Roswell Park Cancer Institute, Buffalo, New York.
3. Department of Biology, Dillard University, New Orleans, Louisiana.
4. Stanley S. Scott Cancer Center, Louisiana State University Health Sciences Center, New Orleans, Louisiana.
5. Department of Pharmacology and Therapeutics, Roswell Park Cancer Institute, Buffalo, New York.
6. Department of Pediatrics, Louisiana State University Health Sciences Center, New Orleans, Louisiana.
7. Department of Biostatistics and Bioinformatics, Roswell Park Cancer Institute, Buffalo, New York.
Received 2014-3-1; Accepted 2014-3-25; Published 2014-6-5
Abstract
The Androgen receptor (AR) plays a central role in the normal development of the prostate gland, in prostate carcinogenesis, and in the progression of prostate cancer (PCa) to advanced metastatic disease. African American (AA) men with PCa present with higher tumor volume, more advanced tumor stage, and higher Gleason score. This could be in part related to the AR expression or activity in the prostate tissue of AA men, or to unique mutations or polymorphisms of the AR. In Caucasian Americans (CAs), AR mutations are rare or infrequent in organ-confined tumors, but occur at a higher rate in advanced, metastatic, or castrate-recurrent disease. In AAs, the prevalence, clinical, and biological significance of AR mutations in PCa are unknown. In this study, we investigated the occurrence of somatic and germline AR mutations in patients with primary PCa in AAs compared with CAs. Due to very limited data available on allelic distribution of E213 (G/A) single nucleotide polymorphism (SNP), we also assessed this in patients with sporadic PCa and in unrelated healthy individuals from both ethnic populations. Somatic missense AR mutations were detected at a higher rate in AAs (17 out of 200 cases) than in CAs (2 out of 100 cases). In AAs, the majority of these mutations (41.1%) were from Gleason 7 tumors, a small portion (23.5%) from Gleason 8 tumors, and the rest (35.2%) from Gleason 6 tumors. Analysis of genomic DNAs extracted from white blood cells of patients with sporadic PCa revealed that the rate of germline AR mutations were also higher (~4 times) in AAs than in CAs. With respect to E213 (G/A) SNP, the E213 A-allele expression was 5.85 times higher in healthy unrelated AA men than in CA men. However, in AAs with somatic AR mutation, the E213 G-allele distribution was almost equal to the A-allele. Silencing of one of the somatic AR mutations (i.e., 597 Ser>Gly) in a primary AA-PCa cell line (e.g., E006AA) revealed that similar AR mutation can be associated simultaneously with both “gain-of-function” phenotype (cell migration and invasion) and a “loss-of-function” phenotype (proliferation). Our data demonstrated a higher susceptibility for genetic alterations in the AR in the form of somatic mutations in sporadic PCa or in the form of germline mutations in AAs as compared with CAs. These data may support the idea that AR-specific hypermutator phenotype in combination with other genes, might serve as a contributing factor to ethnic differences in PCa and potentially different clinical outcome in AAs as a high-risk population.
Keywords: Androgen receptor, prostate cancer, African American, mutations
Introduction
Prostate cancer (PCa) is the most prevalent cancer among men living in the United States and is a significant contributor to morbidity and mortality in older men [1]. It is well established that PCa does not affect racial/ethnic populations similarly. PCa has been accounted for 37% of all cancers in African-American (AA) men in 2013 [2]. AA men have 1.6-1.9 times higher incidence rate and 2-3 times greater mortality rate than Caucasian Americans (CAs). AA men with PCa present with higher tumor volume, more advanced tumor stage, higher Gleason grade, and higher prostate specific antigen (PSA). Overall, AA men have a worse prognosis than their Caucasian counterparts. The underlying reasons for such disproportionate ethnic differences in PCa prognosis and mortality may reflect genuine racial differences in cancer biology, socio-cultural differences, or access to health care systems. Also it is possible that AA men at any stage of disease present with a biologically more aggressive PCa and hence, a worse prognosis.
The androgen receptor (AR) is a nuclear transcription factor and a member of the steroid hormone receptor superfamily of genes [3]. The AR gene is located on the X chromosome at position Xq11-12 and spans ~90 kb containing eight exons that code for a ~2,757 bp open reading frame within a 10.6-kb mRNA. Like many other steroid receptors, AR consists of separate domains responsible for ligand-binding (LBD; 295 amino acid coded by exons 4-8), well-conserved DNA-binding domain (DBD; 68-amino acid coded by exon 2 and 3), nuclear localization (amino acid 628-657), and transactivation units such as AF-1, AF-5, and AF-2 encoded by exon 1 and 8. The most variable region is the N-terminal domain (NTD) of exon 1 with 555 amino acid residues. This domain has several regions of highly repetitive DNA sequences, such as CAG and GGC repeats. The most recognized repeat is a CAG triplet that begins at codon 58 with an average of 22 repeats. Recent studies do not support the association between CAG-repeat length and PCa risk or aggressiveness [4,5].
The normal growth, development, and maintenance of the prostate are dependent on androgen acting through the AR. In the early and intermediate stages, the vast majority of PCa are androgen and AR-dependent. These facts serve as the basis for androgen-ablation therapy which has been an important treatment modality [6]. Since PCa is highly heterogeneous, several mechanisms simultaneously may contribute to castrate-recurrent or resistant (CR) PCa within the same patient. In addition to growth factors, oncogenes and tumor suppressor genes, and cellular and acellular stromal factors, the AR plays a central role in PCa development. Several AR-related mechanisms have been proposed for metastatic or CR-progression of PCa including but not limted to: 1) AR mutations or amplifications leading to hypersensitive receptor or being responsive to anti-androgens or non-androgenic ligands, 2) AR-splice variants, and 3) AR co-regulators [7,8]. When activated by the endogenous androgen ligands, testosterone (T) and dihydrotestosterone (DHT), the ligand-receptor complex translocates into the nucleus and binds to specific genomic DNA sequences in the regulatory regions of AR-regulated genes. Binding of the androgen-AR complex to these androgen response elements regulates expression of androgen target genes such as prostate-specific antigen (PSA).
In addition to the classically known oncogenes, the AR has been considered as an oncogene that is involved specifically in prostate carcinogenesis and PCa recurrence. Functional AR is expressed at any stage of PCa including prostate intraepithelial neoplasia (PIN), primary PCa, metastatic PCa, and castration-recurrent PCa (CRPCa). The AR is among the most mutated type of the steroid receptors. So far, more than 700 mutations of the AR have been reported, most of which led to different, non-malignant clinical categories of androgen-insensitivity syndrome [9]. Overall, AR mutations in Caucasian patients are rarely found in untreated primary organ-confined (localized) PCa (<2%), but are detected at a high frequency in androgen-ablated, metastatic, or CR-tumors [9,10]. The frequency of AR mutation varies greatly among different studies, with up to 25% in androgen-stimulated tumors and up to 50% in metastatic or CR-tumors [11]. The gain of function mutations of the AR in PCa are detected in different functional domains and rarely in 5'- and 3'-untranslated regions (UTRs) of the gene. Most of these mutations are single base substitutions that directly or indirectly affect the AR function. About 51% of the substitution mutations are located in the LBD, 41% at the NTD, and 7% at DBD [12].
Unlike somatic mutations, germline AR mutations are rarely identified [3]. The R726L mutation was reported in Finnish patients with sporadic or familial PCa [13,14]. Additional reports include two unrelated PCa patients with G2T and C214A mutations within the 5'-UTR (non-coding) region of the AR [15], and one final report showed the AR-Q798E mutation in the PCa tissue and genomic (g) DNA of a patient [16]. AR somatic mutation was recently discovered in an AA-PCa cell line (i.e., E006AA) [17]. In addition, during screening for genomic alterations in AAs with familial PCa, we identified a novel germline AR missense mutation (A1675T: T559S) in three siblings with early-onset PCa, referring to the X-linked transmission pattern [18].
In the present study, the occurrence of somatic AR mutation in primary PCa tissues and germline AR mutations in patients with sporadic PCa was investigated in AAs compared with CAs. In addition, we determined the distribution of E213(G/A) single nucleotide polymorphism (SNP) in unrelated healthy individuals of both ethnic populations. Functional consequences of silencing a specific somatic AR-mutation (599 Ser > Gly) was also tested in in vitro assays.
Materials and Methods
Source of tissue specimens, ethical considerations, and genomic DNA Extraction
Sample collection was performed after obtaining written informed consent from patients for the use of their samples for diagnostic and scientific purposes. All clinical samples were provided based on a unique and random alpha-numerical coded system without referring to the patients by name or using any other personal identifier term. During data processing and analyses, samples were assigned a different random number. Ethical approval was obtained from the Institutional Review Boards (IRBs) at Louisiana State University Health Sciences Center (LSUHSC), School of Medicine, New Orleans, LA and Roswell Park Cancer Institute (RPCI), Buffalo, NY. The study conformed to the principles outlined in the Declaration of Helsinki. All tissue biospecimens were obtained from the biospecimen core facilities at the Louisiana Cancer Research Consortium (LCRC) affiliated with Tulane Medical School and School of Medicine, LSUHSC, New Orleans (http://www.louisianacancercenter.org/collaborative-initiatives/sharedresources/biospecimen-core) (Additional File 1: supplementary Table 1).
Somatic AR mutations were examined using gDNA extracted from fresh frozen PCa tissue with 80% or higher tumor content. Examination of germline variations of AR gene sequence was carried out using white blood cells (WBCs) of patients with primary PCa. Distribution of E213 (G/A) SNP was determined using gDNAs extracted from WBCs of unrelated healthy individuals with no clinical history or evidence of cancer. DNAs were extracted using the QIAGEN DNeasy Blood & Tissue Kit, as recommended by the manufacturer. DNA samples were quantitated using a NanoDrop 8000 Spectrophotometer (Thermo Scientific).
PCR, gel-purification, and bi-directional pyrosequencing
Polymerase chain reaction (PCR) amplification was performed by using the previously well-characterized exon-specific primer sequences covering AR gene intron/exon boundaries [19] (Additional File 1: Supplementary Table 2). Fifty nanograms of gDNA was amplified by 35 cycles of PCR in 50 μL containing 0.2 μM of each primer, 0.2 mM of dNTPs , 1.5 mM of MgCl2 and 2.5 units of GoTaq DNA polymerase (Promega, Madison, WI, USA). PCR conditions were 95°C for 5 min, followed by 95 °C for 45 s, 56 to 61 °C for 45 s and 72 °C for 1 min, with a 10-min extension at 72 °C after the last cycle. The correct band size was verified by running a 1.2% agarose gel. PCR products were gel-purified by using a Qiagen PCR-cleaning kit (Qiagen, Inc., Valencia, CA, USA). Sequencing was performed in both directions and repeated independently to ensure the accuracy of the data. The reported sequence was examined by Chromas LITE software (version 2.0) and compared with the AR gene in the NCBI gene database (Accession No. NM_000023).
Cell culture, reagents, and antibodies
E006AA was established from an AA patient with an organ-confined Gleason 6 tumor as described previously [20]. Cos-7 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). E006AA and Cos-7 cell lines were cultured in DMEM-10% Fetal Bovine Serum (FBS) and RPMI-1640 supplemented with 10% FBS, 1 mM sodium pyruvate, and 10 mM HEPES, respectively. All tissue culture media were from Invitrogen (Carlsbad, CA). Anti-human AR (H-441), GAPDH, and secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
AR-silencing in E006AA cell line
E006AA cells were cultured up to 70% confluency, trypsinized, seeded at 2 x 105 cells/well in 6-well plates, and incubated overnight. Cells were transfected with 10 ul (50 pmol) of AR-siRNA oligo (sc-29204) or control siRNA oligo (sc-37007) from SantaCruz and 10 µl lipofectamine 2000 for 8 h. After removal of transfection medium, cells were incubated in complete medium for 36 h before being harvested for western blotting or cell proliferation, migration, or invasion assays.
Western analysis
After transient transfections and incubating cells for 36 h in their maintenance medium, the culture plates were washed three times with cold PBS. Cells were lysed in 0.5 ml of lysis buffer (20 mM PIPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 0.1% SDS, pH 7.4) containing protease inhibitors (10 μg/ml aprotinin, 10 μg/ml leupeptin, 1mM PMSF and 1 mM sodium orthovandate). The collected lysate in eppendorf tubes was placed on ice for 30 min. After centrifugation (20 min; 16,000 g at 4ºC), supernatants were removed and the protein concentration was determined using the BCA protein assay (Pierce, Rockford, IL). Twenty micrograms of protein from each sample were resolved on 4-15% Tris-Glycine gels under reducing condition, and transferred to PVDF membranes (Millipore, Bedford, MA). Membranes were blocked with 5% BSA in TBS-T (150 mM NaCl, 200 mM Tris, 0.1% Tween-20) for 1 h and then incubated with 2 μg/ml of anti-AR or -GAPDH. After 1h at room temperature, membranes were washed with TBS-T and incubated with a 1:1000 dilution of HRP-conjugated secondary antibody (1:1000 dilution; Santa Cruz Biotechnology) for 1 h at room temperature. Membranes were then washed for 30 min, treated with the ECL detection system (Amersham, Arlington Heights, IL), and exposed to Biomax MR film (Kodak).
Cell proliferation assay
Cells were seeded in 96-well tissue culture plates in their respective complete culture media for 3 days. After changing the medium to serum-free, cell were treated with purified recombinant human saposin C (characterized before) [6]. After 2 days of incubation, the cell number was determined by MTS assay (based on a novel tetrazolium compound) using CellTiter 96 AQueous One Solution Cell Proliferation / Cytotoxicity Assay kit (Promega, Madison, WI). For each cell line, the total cell number was determined using a standard curve based on absorption (O.D) versus cell number. We used twelve replicates for each treatment concentration. The assay was repeated at least three times.
Cell migration and invasion assays
The effect of AR down-modulation on E006AA cell migration and invasion was performed using 8-µm transwell filters (Corning) with minor modification as described previously [21]. For the invasion assay, the upper compartment was coated with 50 µg Matrigel (BD Biosciences) to form a matrix barrier. A suspension of cells (4 × 104 in 200 µl) in basal medium containing 0.1% BSA was added to the upper compartment. The lower compartment was filled with 400 µl basal medium containing 1% FBS as a chemoattractant. After 24 h, the non-migratory cells on the upper surface were removed by a cotton swab and the cells on the lower surface were fixed and stained with the Diff-Quick solution (Dade Behring, Deerfield, Illinois). Migrated or invaded cells in each transwell filter were counted in ten randomly selected fields (at 40X magnification). The experiment was performed in quadruplicates and repeated at least three times independently.
Statistical analysis
All analyses were conducted in SAS v9.3 (Cary, NC) at a nominal significance level of 0.05. The association between scores and group was evaluated using an ANOVA model, using Dunnet's method for comparisons between non-transfected and both AR-siRNA and control-siRNA. For both migration and invasion assays, the ANOVA test indicated a significant association (p < 0.001) and the post-hoc tests demonstrated a significant difference between non-transfected and AR-siRNA (p < 0.001). For cell proliferation, the ANOVA test was non-significant (p = 0.371) and as a result the post-hoc tests are also non-significant.
Results
Somatic AR mutations in primary prostate cancers occurs at a higher rate in African Americans than in Caucasian Americans
To determine the relative occurrence of somatic AR mutations, we used gDNAs extracted from untreated primary PCa of AAs (n=200) and CAs (n=100) (Additional File 1: Supplementary Table 1). Using the well-established and optimized primers covering the AR gene intron/exon boundaries (Additional File 1: Supplementary Table 2), PCR, gel-purification, and bi-directional pyrosequencing, we screened the entire coding region for mutations. Somatic AR mutations were confirmed by comparing the sequence with those from matched normal tissues. In total, 17 somatic AR mutations were identified in AAs (Table 1). Of these, 9 mutations (52.9%) were located in LBD, 3 in DBD (17.6%), and 5 in NTD (29.4%). All of these mutations were missense mutation. No insertions or deletions were observed. The majority of the mutations (7 out of 17; 41.1%) were from Gleason 7 tumors. A small portion of the mutations were from Gleason 8 tumors (4 out of 17; 23.5%) and the rest were from low grade tumors (Gleason ≤ 6, 6 out of 17; 35.2%). We detected only two missense mutations in CAs with Gleason 7 tumors. Overall our data indicated that somatic AR mutation was more frequent in AAs (4.25 times) than in CAs. We observed that 1 of 3 mutations (597 Ser > Gly) in DBD from a Gleason 6 tumor was identical to a somatic AR mutation that was previously reported in the E006AA cell line, an AA-PCa cell line established from a patient with an organ-confined primary untreated tumor [20]. The 597 Ser>Gly mutation in the reference AR protein sequence (NM_000044.3) corresponds with the 599 Ser>Gly in E006AA, due to the two additional amino acid increase in the CAG (glutamine) and GGC (Glycine) repeat tracts [17].
Somatic AR missense mutations and E213 (G/A) polymorphism in primary prostate cancers in African Americans versus Caucasian Americans.a
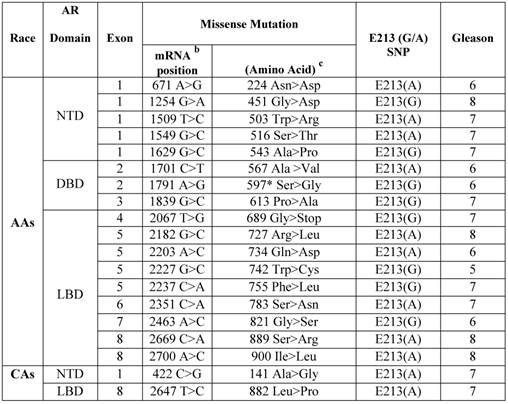
a Archival genomic DNAs (200 African American and 100 Caucasian Americans with PCa).
b AR mRNA sequence: NCBI gene bank/ Accession # NM_000044.3.
c AR protein sequence / NP_000035.2
In our analysis, the observed allele frequency for PCa patients and normal individuals of AA origin for CAG and GGC repeat length polymorphism was similar. The number of repeats varied from 9 to 30 in CAG (PCa patients: Mean=18, SD=3.31; in normal cases: Mean=19, SD=3.41), and from 8 to 20 in GGC (PCa patients: Mean= 14.45, SD=2.0; in normal cases: Mean=15, SD=2.21). In Caucasians, the number of repeats varied from 9 to 28 in CAG (PCa patients: Mean=22, SD=2.32; in normal cases: Mean=21, SD=3.13), and from 8 to 18 in GGC (PCa patients: Mean= 16, SD=3.2; in normal cases: Mean=16, SD=2.49).
Germline mutations of AR in African American men versus Caucasian American men with sporadic prostate cancer
To examine germline mutation in AR, we screened the entire exonic sequence of AR and its 5'-UTR and 3'-UTR regions in gDNAs extracted from WBCs of 162 patients (88 AAs and 74 CAs) with sporadic PCa (Supplementary Table 1). As shown in Table 2, the germline mutations were detected in 10 out of 88 (11.3%) AAs. Of these, 2 mutations were detected at 5'-UTR and 8 others in exon 1 (NTD region). Four of exon I mutations were in the form of substitution and 4 others as synonymous type mutations. Two of the substitution type mutations in AAs were from patients with Gleason 7 tumor, one mutation from a patient with Gleason 6, and 1 from Gleason 8 tumor. Three of the synonymous type mutations were from patients with Gleason 7 and 1 from a patient with Gleason 8 tumor. However, in CAs, we detected only 2 synonymous mutations (2.7%) in the NTD region. One of these mutations was from a patient with Gleason 7, and the other one was from a Gleason 6 tumor. Overall, 10 out of 12 (83.3%) germline mutations in AAs and CAs were detected in patients who had Gleason 6 or 7 tumors and only 2 out of 12 (16.6%) were from patients with high grade tumors (Gleason 8). These data also indicated that germline mutations occurred at a higher rate (4.18 times) in AAs as compared with CAs.
Germline mutation of AR in African American and Caucasian American Men with sporadic Prostate Cancer. a
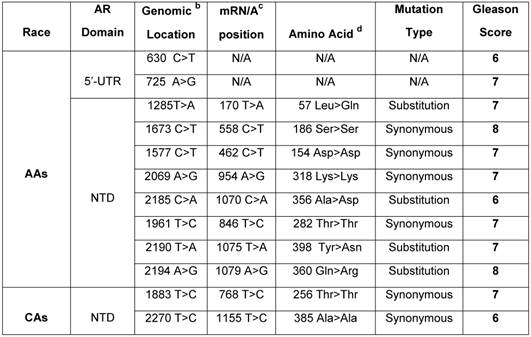
a Archival genomic DNAs (88 African American and 74 Caucasians with PCa).
b Genomic AR sequence: NCBI gene bank/Accession # NM_000023. c AR mRNA sequence: NCBI gene bank/ Accession # NM_000044.3. d AR protein sequence / NP_000035.2
Comparative analysis of AR-E213 G/A SNP in patients with primary prostate cancer and in unrelated normal individuals
The AR-Stu I restriction site, commonly known as E213 G/A SNP (rs6152) is a synonymous change (GAG > GAA) for glutamic acid (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=6152). Very limited data is available on allelic distribution of E213 SNP in the general population or in PCa patients in either AAs or CAs. Therefore, we analyzed the occurrence of the E213(G/A) polymorphism in gDNAs extracted from PCa tissues. As shown in Table 1, 9 out of 17 (52.9%) AA-patients with somatic AR mutations presented with E213-A alleles compared with 8 out of 17 (47%) who had G-allele. The proportion of A-alleles was the same among AA patients with Gleason 6, 7, or 8 tumors. However, 4 out of 7 patients with the G-allele had Gleason 7 tumor, 2 with Gleason 6 tumor, and only 1 with Gleason 8 tumor.
To determine the E213 (G/A) polymorphism in the general population, we used gDNAs extracted from a population of unrelated healthy AA and CA males (Table 3) (Additional File 1: Supplementary Table 1). Analysis of bi-directional sequencing showed that in AA males, 34 out of 61 (55.7%) had the A-allele and 26 out 61 (42.62%) had the G-allele. Among CA males, 6 out of 63 had (9.52%) had the A-allele and 58 out of 63 (92%) had the G-allele. Overall, E213 A-allele expression was 5.85 times higher in AA men than in CA-men. However, the E213 G-allele presented at a higher rate (~2.6 times) in CA men compared with AA men.
Relative allelic distribution of E213 (G/A) SNP in unrelated healthy population of African American and Caucasian American men.a
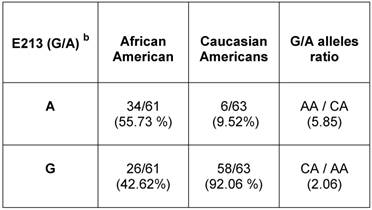
a AR mRNA sequence: NCBI gene bank/ Accession # NM_000044.3
b E213 (G/A) SNP is located at nucleotide 1754 according to NM_000044.3
Silencing AR (599 Ser>Gly) in the E006AA cell line decreased migration and invasion but had no effect on proliferation
AR (599 Ser>Gly) somatic mutation in our current and previous study were both discovered in PCa tissues and cells obtained from AA patients. We used commercial siRNA-oligos to transiently silence AR in a racially-compatible PCa cell line, E006AA (Fig. 1A). Transient silencing of AR did not affect cell proliferation in the E006AA cell line (Fig. 1B).
The effect of transient silencing of mutant AR (599 Ser>Gly) on proliferation, migration, and invasion in the E006AA cell line. A. Western blotting of AR in the E006AA Cell line transiently transfected with control- or AR-siRNA oligos. B. Transfected cells were seeded in 96-well tissue culture plates in complete culture medium. After 3 days of incubation, the cell number was determined by MTS assay using CellTiter 96 AQueous One Solution Cell Proliferation / Cytotoxicity Assay. Total cell number was determined using a standard curve based on absorption (O.D) versus cell number. Experiments were performed in triplicate and the assay was repeated at least three times independently. C. Transfected cells were subjected to migration and invasion assays as described in “Materials and Methods”. A representative photomicrograph from control and AR-siRNA transfected cells is presented at 400× magnification using a phase-contrast microscope. D. Cells migrated or invaded were counted from ten random fields. Each bar represented the mean ± SEM of three independent experiments, each performed in triplicate. For cell proliferation assay, the ANOVA test was non-significant (p = 0.371) and as a result the post-hoc tests are also non-significant. For migration and invasion assays, the ANOVA test indicated a significant association (p < 0.001) and the post-hoc tests demonstrated a significant difference between non-transfected or control-siRNA and AR-siRNA (p < 0.001).
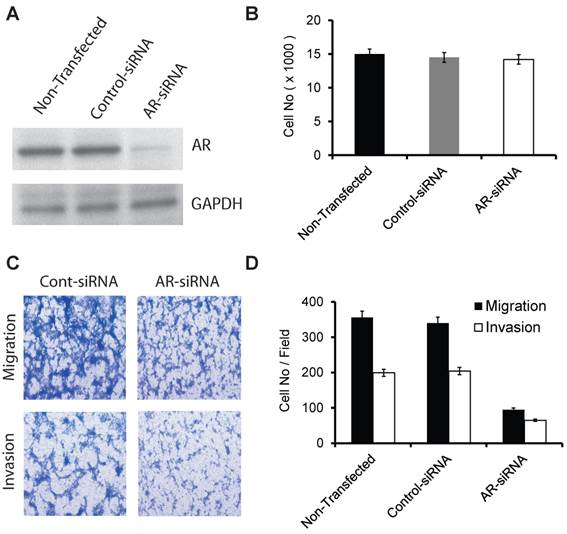
However, we found that AR-silencing significantly decreased E006AA cell migration and invasion by 73.3% and 67.3%, respectively (Fig. 1C and D).
Discussion
AR is expressed in a significant portion of CRPCa patients and it seems that even in cases under maximal androgen ablation therapy, the presence of AR is often required for castrate-resistance or proliferating metastatic PCa cells [22-25]. Collectively, it indicates the importance of AR, its expression and activity, and/or the function of AR-target genes and their regulators in PCa. Limited information is available on race and AR expression or mutation in AA-PCa. In a single report, AR protein expression was analyzed in benign and malignant prostate tissues obtained from radical prostatectomy specimens of 25 AAs and 25 CAs [26]. Using a visual scoring method, AR immunostaining was detected more intensely in both benign and malignant prostate epithelial nuclei in AAs than in CAs. In AAs compared with CAs, PCa cells were 27% more likely stained for AR, and among immunopositive benign prostate cells, AR protein expression was 81% greater [26]. In contrast to limited investigation in AA-PCa patients, AR mutation has been intensively investigated in primary, metastatic, and CRPCa patients in CAs [3,11].
While investigating the AR sequence in the E006AA cell line derived from a Gleason 6 organ-confined tumor, we observed X chromosome duplication, AR gene mutation (at the DNA-binding domain), and its genomic amplification. Somatic AR mutation (599 S>G) in this cell line was proved to be a dominant-negative or loss-of-function type [17]. This study also showed that after androgenic stimulation, the mutated-AR (Ser599Gly) in E006AA cell line is able to translocate to the nucleus, binds to putative androgen-response elements (DNA-binding sites), but fails to stimulate AR-target genes expression. The occurrence of AR 599 Ser>Gly mutation in one of the PCa tissues and in the E006AA cell line led us to suspect that this may be a germline mutation that exists more frequently in familial PCa. Our analysis of the matched normal and PCa tissues obtained from PCa patients excluded the possibility of a germline mutation. In addition, we did not find AR599 (Ser>Gly) mutation in the sequencing data obtained from a previous study that included PCa-affected members of 30 high-risk AA-families [18]. Furthermore, analyses of gDNAs extracted from WBCs of unrelated healthy AA excluded the possibility of the observed mutation as a polymorphic variant or SNP.
Transient silencing of AR did not affect cell proliferation in the E006AA cell line. However, it significantly decreased cell migration and invasion. This data was in agreement to a previous study that demonstrated lentiviral-shRNA silencing did not affect E006AA cell growth [17]. Overall, our data highlights the potential significance of AR mutations that might contribute or intensify migratory and invasive abilities independent of growth characteristics in PCa. In addition, site-specific genetic alterations of AR in PCa may contribute at the same time to “loss-of-function” phenotype (cell proliferation) and “gain-of-function” phenotypes such as migration and invasion. The relationship between AR genotype and phenotype in PCa is complicated and remains largely unknown. It has been reported that similar AR mutations can be associated with both a “gain-of-function” in one individual and a “loss-of-function” phenotype in another individual. For example, Gottlieb et al have reported that 13 AR mutations in different individuals were associated with variable phenotypes such as partial androgen-insensitivity syndrome (PAIS), complete androgen insensitivity syndrome (CAIS), mild androgen insensitivity syndrome (MAIS) and PCa [12]. However, 32 AR mutations were associated with CAIS, PAIS, and/or MAIS, without PCa. Overall, these data strongly support the contribution of non-genomic, post-transcriptional, or post-translational events which might alter the functional consequences of AR mutations. In addition, the presence of multiple AR mutations in multi-focal PCa tissues will further complicate the net-effect or outcome of “gain-of-function” or “loss-of-function” mutations in PCa biology and clinical outcome. Taking into consideration the inter-focal heterogeneity, it will not be surprising to detect different AR mutations in the localized tumor, or in different metastatic locations in the same patient.
The amino-terminal transactivation domain of exon 1 in AR contains 3 polymorphic sequences that include two trinucleotide polymorphic repeats (CAGn and GGCn) and the stable dimorphic AR-E213(G/A) SNP located between the CAG/GGC repeat regions. The association between E213(G/A) SNP and PCa has been investigated in only one relatively large study in Australian patients [27]. Based on this population-based case-control study (n=719 controls and n=811 PCa patients), there was no association between the E213-G allele and risk of PCa. However, the frequency of A-allele careers (5%) in metastatic PCa (Stage IV, T4N+M+) was less than in organ-confined non-metastatic tumors (Stage I-II, 16%, P = 0.03). In an earlier study, the E213(G/A) polymorphism was reported to associate with androgenic alopecia [28].
Searching the NCBI-SNP data base revealed that E213 A- and G-alleles were reported at 52.1% and 47%, respectively in self-described African/African-American heritage PCa patients / (n=24). However, the E213 A-and G-alleles were presented at 18.3% and 81.7%, respectively in self-described Caucasian heritage (n=31). In our investigation, 52.9% of AA-patients with somatic AR mutations had the E213-A allele compared with 47% who had the G-allele (Table. 1). The proportion of A-alleles among low, moderate-, and high grade tumors were the same. However, 4 out of 7 patients with the G-allele had Gleason 7 tumor, 2 with Gleason 6 tumor, and only 1 with Gleason 8 tumor. To determine the E213 (G/A) allelic distribution in the normal US-male population, we analyzed the gDNAs extracted from WBCs of 124 unrelated healthy AA and CA males (Table. 3). In AAs, the E213 A-allele was presented in 55.7% and G-allele was detected in 42.62% of cases. However, 92% of CA males had the G-allele and only 9.52% of them had the A-allele. These data showed that E213 A-allele expression was detected at 5.85 times higher in AA men than in CA men. The E213 G-allele existed at a higher rate (~2.6 times) in CA-men compared with AA-men. Taking into consideration our data and NCBI-SNP data base, it appears that the relative distribution of the A- and G-allele in normal male populations and PCa patients is not necessarily associated with PCa. Using qRT-PCR, we tested the effect of a transcription inhibitor (i.e., actinomycin D) on AR-mRNA expression in Cos-7 cells (with no endogenous AR expression) transiently transfected with vectors expressing E213 G-allele or A-allele. This experiment showed that E213 A-allele only slightly (~10%) increased AR-mRNA expression levels in Cos-7 cells (data not shown). This data indicate that E213 A-allele may increase AR-mRNA stability. Additional studies are needed to confirm this statement.
It is a fact that tumors are genetically unstable. Formation of mutations is an inseparable and inherent characteristic of different types of cancers. On the other hand, progression of tumors appears to be a genetically evolutionary and multistep process. The final outcome will be dictated by the selection of the desirable mutations in tumor cells within their microenvironment. It is also conceivable that in certain type of cancers, some critical genes may be more susceptible to mutations than others. Oncogenic addiction of PCa to AR-dependent signaling and activity, high somatic mutation rate in AR, and formation of site-specific AR mutation in response to first or second generation antiandrogens in CRPCa cells and tissues, may collectively support the hypothesis that the AR gene has the characteristic hypermutable or hypermutator phenotype. It is naturally expected that such hypermutator phoenotypes should lead to gain-of-function type and adaptive mutations. It is likely that the AR hypermutator phenotype could be intensified or affected by other demographic (e.g., race) or environmental factors.
Our data indicated a higher rate for genomic alterations in the AR in the form of somatic mutations in primary PCa or in germline mutations in AAs as compared with CAs. We hypothesize that there is a potential for the AR-specific hypermutator phenotype that either alone, or in combination with other genes, might serve as a contributing risk factor to ethnic differences in PCa incidence and potentially different clinical outcome in AAs as a high-risk population. Future large-scale investigations are required to determine the contribution of gene-specific or site-specific AR somatic and/or germline mutations in patient populations in relation to demographic and other clinicohistopathological determinants of PCa aggressiveness or progression.
Supplementary Material
Additional File 1Supplementary Tables 1-2.
Acknowledgements
Part of this study was presented at the Annual Meetings of the American Urological Association (AUA). This work was supported in part by the Dillard-LSUHSC Minority Health and Health Disparities Research Center (P20MD004817 to J. Wilson, J. Estrada, E. Buckles, and S.K), R21CA143589 (to S.K.), R21CA183892 (to S.K.), and R21CA149137 (to S.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Minority Health and Health Disparities or the National Cancer Institute. The authors wish to thank Dr. Suzanne Hess for her editorial assistance.
Competing interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer J Clin. 2014;64:9-29
2. DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA Cancer J Clin. 2013;63:151-66
3. Koochekpour S. Androgen receptor signaling and mutations in prostate cancer. Asian Journal of Andrology. 2010;12:639-57
4. Price DK, Chau CH, Till C, Goodman PJ, Baum CE, Ockers SB. et al. Androgen receptor CAG repeat length and association with prostate cancer risk: results from the prostate cancer prevention trial. Journal of Urology. 2010;184:2297-302
5. Lindström S, Ma J, Altshuler D, Giovannucci E, Riboli E, Albanes D. et al. A large study of androgen receptor germline variants and their relation to sex hormone levels and prostate cancer risk. Results from the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. J Clin Endocrinol Metab. 2010Sep;95(9):E121-7
6. Huggins C, Hodges CV. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Research. 1941;1:293-7
7. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nature Review Cancer. 2001;1:34-45 Review
8. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocrinology Reveiw. 2007;28:778-808
9. Gottlieb B, Beitel LK, Wu JH, Trifiro M. The androgen receptor gene mutations database (ARDB). Hum Mutation. 2004;23:527-33
10. Bubendorf M, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, Willi N, Mihatsch MJ, Sauter G, Kallioniemi OP. Survey of gene amplifications during prostate cancer progression by high-throughput fluorescence in situ hybridization on tissue microarrays. Cancer Research. 1999;59:803-6
11. Linja MJ, Visakorpi T. Alterations in AR and prostate cancer. Journal of Steroid Biochemistry Mollecular Biology. 2004;92:255-264
12. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Human Mutation. 2012;33:887-94
13. Mononen N, Syrjäkoski K, Matikainen M, Tammela TL, Schleutker J, Kallioniemi OP, Trapman J, Koivisto PA. Two percent of Finnish prostate cancer patients have a germ-line mutation in the hormone-binding domain of the androgen receptor gene. Cancer Research. 2000;60:6479-81
14. Gruber SB, Chen H, Tomsho LP, Lee N, Perrone EE, Cooney KA. R726L androgen receptor mutation is uncommon in prostate cancer families in the United States. Prostate. 2003;54:306-9
15. Crocitto LE, Henderson BE, Coetzee GA. Identification of two germline point mutations in the 5'UTR of the androgen receptor gene in men with prostate cancer. Journal of Urology. 1997;158:1599-601
16. Evans BA, Harper ME, Daniells CE, Watts CE, Matenhelia S, Green J & Griffiths K. Low incidence of androgen receptor gene mutations in human prostatic tumors using single strand conformation polymorphism analysis. Prostate. 1996;28:162-71
17. D'Antonio JM, Vander Griend DJ, Antony L, Ndikuyeze G, Dalrymple SL, Koochekpour S, Isaacs JT. Loss of androgen receptor-dependent growth suppression by prostate cancer cells can occur independently from acquiring oncogenic addiction to androgen receptor signaling. PLoS One. 2010;8:e11475
18. Hu SY, Liu T, Liu ZZ, Ledet E, Velasco-Gonzalez C, Mandal DM, Koochekpour S. Identification of a novel germline missense mutation of the androgen receptor in African American men with familial prostate cancer. Asian J Androl. 2010;12:336-43
19. Lubahn DB, Brown TR, Simental JA, Higgs HN, Migeon CJ. et al. Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity. Proceeding of National Academy of Science, USA. 1989;86:9534-8
20. Koochekpour S, Maresh GA, Katner A, Parker-Johnson K, Lee TJ, Hebert FE. et al. Establishment and characterization of a primary androgen-responsive African-American prostate cancer cell line, E006AA. Prostate. 2004;60:141-52
21. Hu S, Delorme N, Liu Z, Liu T, Velasco-Gonzalez C, Garai J, Pullikuth A, Koochekpour S. Prosaposin down-modulation decreases metastatic prostate cancer cell adhesion, migration, and invasion. Molecular Cancer. 2010;9:30
22. Tamburrino L, Salvianti F, Marchiani S, Pinzani P, Nesi G, Serni S. et al. Androgen receptor (AR) expression in prostate cancer and progression of the tumor: Lessons from cell lines, animal models and human specimens. Steroids. 2012;77:996-1001
23. Donovan MJ, Osman I, Khan FM, Vengrenyuk Y, Capodieci P, Koscuiszka M. et al. Androgen receptor expression is associated with prostate cancer-specific survival in castrate patients with metastatic disease. British Journal of Urology International. 2010;105:462-7
24. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S. et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discovery. 2013;3:1030-43
25. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D. et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discovery. 2013;3:1020-9
26. Gaston KE, Kim D, Singh S, Ford OH 3rd, Mohler JL. Racial differences in androgen receptor protein expression in men with clinically localized prostate cancer. Journal of Urology. 2003;170:990-3
27. Hayes VM, Severi G, Eggleton SA, Padilla EJ, Southey MC, Sutherland RL. et al. The E211 G>A androgen receptor polymorphism is associated with a decreased risk of metastatic prostate cancer and androgenetic alopecia. Cancer, Epidemiology, Biomarkers, and Prevention. 2005;14:993-6
28. Ellis JA, Scurrah KJ, Cobb JE, Zaloumis SG, Duncan AE, Harrap SB. Baldness and the androgen receptor: the AR polyglycine repeat polymorphism does not confer susceptibility to androgenetic alopecia. Human Genetics. 2007;121:451-7
Author contact
![]() Corresponding author: Shahriar Koochekpour, MD. PhD. Departments of Cancer Genetics and Urology, Center for Pharmacology and Genetics, Elm and Carlton Streets, Roswell Park Cancer Institute, Buffalo, New York, NY 14263. E-mail: Shahriar.koochekpourorg.
Corresponding author: Shahriar Koochekpour, MD. PhD. Departments of Cancer Genetics and Urology, Center for Pharmacology and Genetics, Elm and Carlton Streets, Roswell Park Cancer Institute, Buffalo, New York, NY 14263. E-mail: Shahriar.koochekpourorg.