Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(7):702-714. doi:10.7150/ijbs.9326 This issue Cite
Research Paper
Targeting the Sheddase Activity of ADAM17 by an Anti-ADAM17 Antibody D1(A12) Inhibits Head and Neck Squamous Cell Carcinoma Cell Proliferation and Motility via Blockage of Bradykinin Induced HERs Transactivation
Yanchao Huang1,2 ![]() , Nathan Benaich1,4, Christopher Tape1,5, Hang Fai Kwok1,3*
, Nathan Benaich1,4, Christopher Tape1,5, Hang Fai Kwok1,3* ![]() , Gillian Murphy1*
, Gillian Murphy1*
1. Cancer Research UK Cambridge Institute, University of Cambridge, Department of Oncology, UK
2. IONTAS Ltd, Hopkins Building, Tennis Court Rd., Cambridge CB2 1QW, UK (current address)
3. Faculty of Health Sciences, University of Macau, Macau (current address)
4. Centre for Stem Cells and Regenerative Medicine, King's College London School of Medicine, 28th Floor, Tower Wing, Guy's Hospital, Great Maze Pond, London SE1 9RT, UK (current address)
5. The Institute of Cancer Research, 237 Fulham Road, London, SW3 6JB, UK (current address).
* Joint senior authors.
Received 2014-4-8; Accepted 2014-5-18; Published 2014-6-21
Abstract
A disintegrin and metalloproteinase 17 (ADAM17) regulates key cellular processes including proliferation and migration through the shedding of a diverse array of substrates such as epidermal growth factor receptor (EGFR) ligands. ADAM17 is implicated in the pathogenesis of many diseases including rheumatoid arthritis and cancers such as head and neck squamous cell carcinoma (HNSCC). As a central mediator of cellular events, overexpressed EGFR is a validated molecular target in HNSCC. However, EGFR inhibition constantly leads to tumour resistance. One possible mechanism of resistance is the activation of alternative EGFR family receptors and downstream pathways via the release of their ligands. Here, we report that treating human HNSCC cells in vitro with a human anti-ADAM17 inhibitory antibody, D1(A12), suppresses proliferation and motility in the absence or presence of the EGFR tyrosine kinase inhibitor (TKI) gefitinib. Treatment with D1(A12) decreases both the endogenous and the bradykinin (BK)-stimulated shedding of HER ligands, accompanied by a reduction in the phosphorylation of HER receptors and downstream signalling pathways including STAT3, AKT and ERK. Knockdown of ADAM17, but not ADAM10, also suppresses HNSCC cell proliferation and migration. Furthermore, we show that heregulin (HRG) and heparin-binding epidermal growth factor like growth factor (HB-EGF) predominantly participate in proliferation and migration, respectively. Taken together, these results demonstrate that D1(A12)-mediated inhibition of cell proliferation, motility, phosphorylation of HER receptors and downstream signalling is achieved via reduced shedding of ADAM17 ligands. These findings underscore the importance of ADAM17 and suggest that D1(A12) might be an effective targeted agent for treating EGFR TKI-resistant HNSCC.
Keywords: ADAM17, inhibitory antibody, HER receptor and ligands, cell proliferation and motility.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide [1]. Up to 40% of HNSCC patients present with metastatic disease and the 5-year overall survival rate has remained around 50% for decades [2]. The epidermal growth factor receptor (EGFR) [also known as HER1], one of four members of the HER family of transmembrane tyrosine kinase growth factor receptors, is the most validated therapeutic target in HNSCC [2]. Upon activation by its growth factor ligands [e.g. amphiregulin (AREG), epiregulin (EPG), and heparin-binding epidermal growth factor like growth factor (HB-EGF)], EGFR forms homodimers and may pair with another member of the EGFR family receptor to create an activated heterodimer. EGFR dimerization stimulates tyrosine autophosphorylation of the EGFR intracellular domain. This autophosphorylation elicits downstream activation and signalling transduction cascades, leading to cell proliferation, migration and invasion to drive malignant progression [3, 4].
Two classes of EGFR inhibitors are in clinical use for treating HNSCC: monoclonal antibodies like cetuximab and small molecule inhibitors [5, 6, 7]. Gefitinib, one of the small molecule inhibitors, directly inhibits kinase activity of EGFR to activate downstream signalling pathways. Although the activity of EGFR inhibitors in HNSCC patients is clear, modest clinical results have been reported, suggesting either the presence of acquired or intrinsic resistance pathways [8, 9, 10]. One potential mechanism of resistance to anti-EGFR agents is the activation of alternative HER receptors via the release of their ligands [11, 12]. It has been previously shown that HNSCC cells resistant to the monoclonal antibody cetuximab exhibited increased EGFR levels and upregulation and activation of HER2, HER3 and compared with the sensitive parental cell line [13]. Moreover, overexpression of HER4 is associated with a higher proliferative rate in HNSCC [14]. Thus, there is a pressing need to develop novel agents which can block multiple HER receptors and downstream signalling pathways to overcome resistance to EGFR inhibition.
A disintegrin and metalloproteinase 17 (ADAM17), also called tumour necrosis factor-α-converting enzyme(TACE), is a member of the ADAM protein family of disintegrins and metalloproteases that releases membrane-bound tumour necrosis factor (TNF)-α precursor into a soluble form [15]. ADAM17 is involved in regulating HER family activation via shedding of HER ligand precursors such as transforming growth factor (TGF)-α, EGF, AREG, HB-EGF, heregulin (HRG) and EPG, resulting in enhanced downstream signalling pathways and regulating tumour growth and metastasis as well as tumour angiogenesis [16, 17]. Increased mRNA or protein level of ADAM17 was shown in several different tumour tissues such as breast [18], colon [19], pancreas [20], prostate [21] and renal cancer [22]. In breast cancer, ADAM17 was indicated as a prognostic marker [23]. Overexpression of its HER ligand substrates TGF-α, HB-EGF, AREG and EPG has been correlated with poor outcomes for patients receiving therapy [24, 25, 26, 27]. To date, there is evidence that ADAM17 might be an attractive therapeutic target for the treatment of diverse cancers including HNSCC [28, 29]. Similarly, therapies that decrease ADAM17 activity in conjunction with current treatments may enhance response rates in HNSCC. These questions have however remained unexplored in HNSCC.
We previously developed a specific human ADAM17 inhibitory antibody, D1(A12), which suppresses EGFR ligand and TGF-α shedding in vitro and in vivo [30, 31]. Here, we postulate that therapeutic response in HNSCC could be ameliorated by dual treatment with EGFR and ADAM17 inhibitors. In the present study, we exposed a panel of HNSCC cell lines to D1(A12) either alone or in combination with gefitinib to evaluate its anti-tumour effects using a variety of cell-based assays. We also studied the mechanism of the inhibitory effects of D1(A12) in order to generate further understanding of ADAM17 function in HNSCC.
Materials and methods
Cell culture and reagents
The following cell lines were obtained commercially: SCC9, SCC12, SCC13, and SCC25 from the American Type Culture Collection (Manassas, VA, USA) (Rheinwald and Beckett, 1981). SJG15 cells were established from freshly resected HNSCC specimens of patients treated at Addenbrooke's Hospital, Cambridge. Normal primary human keratinocyte cells (KN) were obtained from operations to remove excess skin at the same hospital. Work with human material was carried out in compliance with the UK Human Tissue Act (2004), approved by the National Research Ethics Service, and appropriate informed consent was obtained from patients prior to their operation.
All cells were maintained in DMEM-F12 medium supplemented with 10% fetal bovine serum (FBS) (Life technologies) in a humidified atmosphere of 5% CO2 at 37°C. Dialysed FBS was obtained from Sigma-Aldrich. Recombinant human AREG, HB-EGF, HRG, EPG, TGF-α and control human IgG were purchased from R&D Systems (Minneapolis, MN, USA). Polyclonal antibodies against (Phospho)-EGFR, (Phospho)-HER2, (Phospho)-HER3, (Phospho)-extracellular signal-regulated kinase (ERK), (Phospho)-AKT, (Phospho)- Signal Transduction And transcription (STAT)3 were from Cell Signalling Technology (Danvers, Massachusetts, USA). Anti-ADAM17 antibody (ab39162) was purchased from Abcam (Cambridge, UK). Gefitinib was obtained from Selleckchem (USA). AG 1478, tissue inhibitor of matrix metalloproteinase 3 (TIMP-3) and TNF protease inhibitor 2 (TAPI-2) were from Calbiochem (San Diego, CA, USA). ADAM17 and β-actin antibodies were from Abcam (Cambridge, UK). 24 well BioCoat™ Growth Factor Reduced BD Matrigel ™ Invasion Chamber was from Science laboratory supplier (Nottingham, UK). RayBio human EGFR phosphorylation antibody 1 was from RayBiotech (Norcross, USA).
Enzyme-linked immunosorbent assay (ELISA)
For detection of endogenous soluble AREG, TGF-α and HB-EGF in cell culture media, cells were grown in tissue culture treated 96 well plates (Corning, UK) and then serum deprived for 24 h. The culture medium was replaced with 100 µl of medium supplemented with 1% dialysed FBS (Sigma-Aldrich). After treatment for the indicated time period, the supernatants were harvested for determination using a commercially available sandwich ELISA (DuoSet kit; R&D Systems) according to the manufacturer's instructions. All the samples were assayed in quadruplicate.
Cell proliferation assay
To assess cell viability, cells were treated in a similar way as in ELISA. For multiple dosing treatments, the IncuCyte (Essen Bioscience) live cell imaging system was used to measure the cell growth based on confluence over time. For single dose treatment and ligands induced cell growth, the CellTiter-Glo (CTG) Luminescent Cell Viability Assay (Promega) was used to measure the number of viable cells following the manufacturer's instruction. After 15 min of incubation, the luminescence signal was read on the Clarity™ Luminescence Microplate Reader (BioTek).
In vitro motility assays
To assess the migratory and invasive capacity of SCC9 and SCC13 cells using the transwell chamber assay, cells were grown in complete medium, serum starved for 24 h before dissociation and resuspended in serum free medium. For migration, 24 well non-coated transwell inserts (8 µm pore size, BD Biosciences) were used. 500 µl of 1×105 cells were seeded per transwell insert. For invasion, 500 µl of 2×105 cells were seeded per insert in 24 well BD BioCoat™ Growth Factor Reduced Matrigel ™ invasion chamber. 750 µl of 10% fetal calf serum (FCS) and serum free medium were used as chemoattractant and negative control, respectively. After 48 h incubation time, media was aspirated and cells on the inner side of the membrane were removed using cotton swabs. For migration, each insert was fixed for 10 min in 100% ice cold MeOH, washed with PBS and then stained with 0.5% crystal violet for 20 min. The membranes were then washed with PBS and 10% HAC was used to elute the dye. The absorbance was measured at 600 nm. For invasion, the invaded cells were counted manually. Each condition was performed in triplicate.
The wound closure assay was performed using a 96 well Essen Imagelock plate (EssenBioscience, UK). Cells were grown to confluence followed by 48 h serum starvation. Wounds were made using a 96-well WoundMaker, and cells were washed 2×PBS before adding drugs. Following assay initiation, images of all 96-wells were obtained every three hours until assay completion using the IncuCyte imaging system. Each image was automatically analysed using phase contrast image based algorithms. The Relative Wound Density (%) metric that analyses both the inside of the wound and the outside cell region is used to express kinetic wound closure. Each condition was performed in 8 replicates. At all conditions, the assay was performed at least three times independently.
Immunoblotting analysis
Five HNSCC cell lines and KN were grown in 10% FCS for 72 h, collected and lysed in ice-cold RIPA lysis buffer (1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 150 mM NaCl and 10 mM Tris-HCl) containing a protease inhibitor cocktail and phosphatase inhibitor tablet (Roche). Immunoblotting was also used to evaluate the protein levels of EGFR, HER2, HER3, AKT, STAT3, ERK and their phosphorylated forms with or without BK stimulation. SCC9 and SCC13 cells were serum starved 48 h, treated with 0.5 µM of D1(A12) and human IgG for 2 h in serum free medium and then exposed to 10 nM BK for 10 min. Cells were harvested and lysed as above.
The total protein concentration was determined using Direct Detect (Millipore). Equivalent amounts of proteins (20 μg) were then separated by 10% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). After blocking in Phosphate buffered saline (PBS) containing 4% non-fat milk for 1 h, the membranes were incubated with primary antibodies at room temperature for 2 h and then with horseradish peroxidase (HRP) conjugated anti-rabbit antibody (GE Healthcare) at a dilution of 1:2000 at room temperature for 1 h. Signals were detected on X-ray film using the ECL detection system (GE Healthcare). Equal protein loading was assessed by the expression of β-actin.
RNA interference studies
Cells were seeded in 6 well plates and 24 h later 10 nM of ADAM17, ADAM10 or non-target (NT) siRNAs (Life technologies) were added in DMEM-F12 plus 10% FBS. 4 h later the medium was refreshed and another 10 nM of siRNAs were added. Transfection reagent of INTERFERin (Polyplus) was used for siRNA transfection. For signalling studies, 72 h after siRNA transfection the cells were serum starved for 24 h then challenged with BK. Cells were washed with PBS and lysed in RIPA buffer. For transwell migration assay, after 48 h siRNA transfection cells were serum starved for 24 h then following the procedures described in migration assay.
Phosphorylation protein array
To analyse the alterations in phosphorylation of EGFR, HER2, HER3 and HER4 with the treatment of D1(A12), cells were treated as described in immunoblotting. Cells were washed once with ice-cold PBS and then extracted with cell lysis buffer provided in the RayBio Human EGFR Phosphorylation Antibody Array kit (RayBiotech). To use the RayBio Phosphorylation antibody Array 1, equal amount of cell lysates (100 µg) were added into antibody array membrane. After 2 h incubation, the antibody array membranes were washed and cocktail of biotinylated anti-EGFR was used to detect phosphorylated proteins on activated receptors. After incubation with HRP-conjugated streptavidin, the signals were visualized by chemiluminescence. 4 positive controls were used to normalize the signals.
Statistical analysis
To evaluate significant differences between groups, a Student's t test was performed, with P< 0.05 considered significant.
Results
Analysis of EGFR, HER2, HER3 and downstream signalling proteins in human HNSCC cell lines and normal human keratinocytes
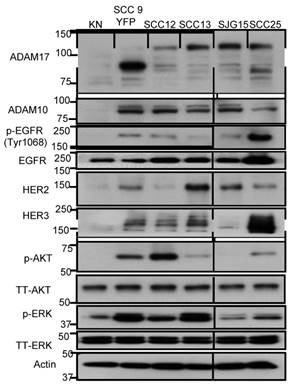
We first set out to evaluate the protein expression levels of EGFR, HER2, HER3, HER4, AKT, ERK, ADAM17 and ADAM10 in five HNSCC cell lines and normal human keratinocytes using immunoblotting. Compared to normal keratinocytes, HNSCC cells overexpressed EGFR, HER2, HER3, AKT, ERK, ADAM17 and ADAM10 (Fig. 1). HER4 was undetectable. HNSCC cells overexpressed both the full-length (120 kD with prodomain) and the mature form (85 kD without prodomain) of ADAM17 compared to normal keratinocytes. Interestingly, HER2, HER3, p-AKT and p-ERK were also highly expressed in SCC9 and SCC13 cells. Total AKT and ERK were equally expressed in all five HNSCC cell lines and normal human keratinocytes. Based on these results, we selected SCC9 and SCC13 cells as cell line models to study the effects of inhibition of ADAM17 activity and cellular function of HER family in HNSCC.
Characterization of (p-) EGFR, HER2, HER3, (p-) AKT, (p-) ERK1/2 and ADAM10/17 expression in 5 human HNSCC cell lines and normal human keratinocyte cells (KN). Cell lysates (20 µg) were prepared, resolved by SDS-PAGE, and subjected to immunoblotting analysis with the indicated antibodies. SCC9 cells are yellow fluorescent protein (YFP) labelled. The prefix “p-” and “TT” refer to phosphorylation form and total form of the parentheses. β-actin was used as a loading control.
Effect of inhibition of ADAM17 and HER ligands on cell proliferation
Previous studies indicated that HER ligands shed by ADAM17 can activate HER receptors and downstream signalling pathways, leading to the induction of cell proliferation in prostate cancer cells [32]. We therefore investigated whether inhibition of ADAM17 sheddase activity by D1 (A12) could decrease HNSCC cell proliferation in vitro. Treating SCC13 cells with 0.05 µM and 0.5 µM D1 (A12) inhibited proliferation by 7% and 19% (P<0.001) compared to PBS treated cells, respectively (Fig. 2A). A similar result was obtained in SCC9 cells (data not shown). We confirmed the growth inhibition effect elicited by multiple doses of D1 (A12) in SCC13 cells using automated live-cell imaging in vitro. Fig. 2B shows that 72 h treatment with three doses of 0.25 and 0.5 µM D1 (A12) significantly inhibited cell growth compared to human IgG treated cells. The broad-spectrum metalloprotease inhibitor CT1746 was used as a positive control [33]. These results suggest that the sheddase activity of ADAM17 substantially contributed to cell proliferation in HNSCC cells.
(A) Effect of a single dose of D1(A12) on the growth of SCC13 cells. Serum starved cells were treated with indicated concentrations of D1(A12) in medium supplemented with 1% dialyzed fetal calf serum (FCS) for 72 h. (B) Effect of ADAM17 inhibition with multiple doses of D1(A12) on the growth of SCC13 cells. Cells were treated in the same way as single dose treatment and the growth was monitored by IncuCyte. After 72 h incubation the cell confluence was calculated. CT1746 and human IgG were used as positive and negative controls respectively. Each point is the mean ± SD of 8 replicates. (C) D1(A12) sensitizes SCC9 cells and SCC13 cells (D) to gefitinib. Serum starved cells were treated with various concentrations of gefitinib and 0.5 µM D1(A12) for 72 h. The viability was measured by CTG and plotted as a percentage of viable cells relative to DMSO treatment. Each point is the mean ± SD of 8 replicates. (E) Effect of ADAM17 siRNA on the growth of SCC13 cells. Transfection of ADAM17 siRNA #3 in SCC13 cells significantly reduced cell proliferation, compared to non-targeting siRNA control. Each point is the mean ± SD of 4 replicates. (F) Effect of HER ligands on the growth of SCC13 cells. Serum starved cells were treated indicated concentrations of HRG, HB-EGF with or without 10 µg/ml neutralizing HER3 and HB-EGF antibodies in serum free medium for 72 h. Each point is the mean ± SD of 4 replicates. (G) Effect of HER ligands on the growth of SCC13 cells. Serum starved cells were treated indicated concentrations of AREG and EPG in serum free medium for 72 h. Each point is the mean ± SD of 4 replicates.
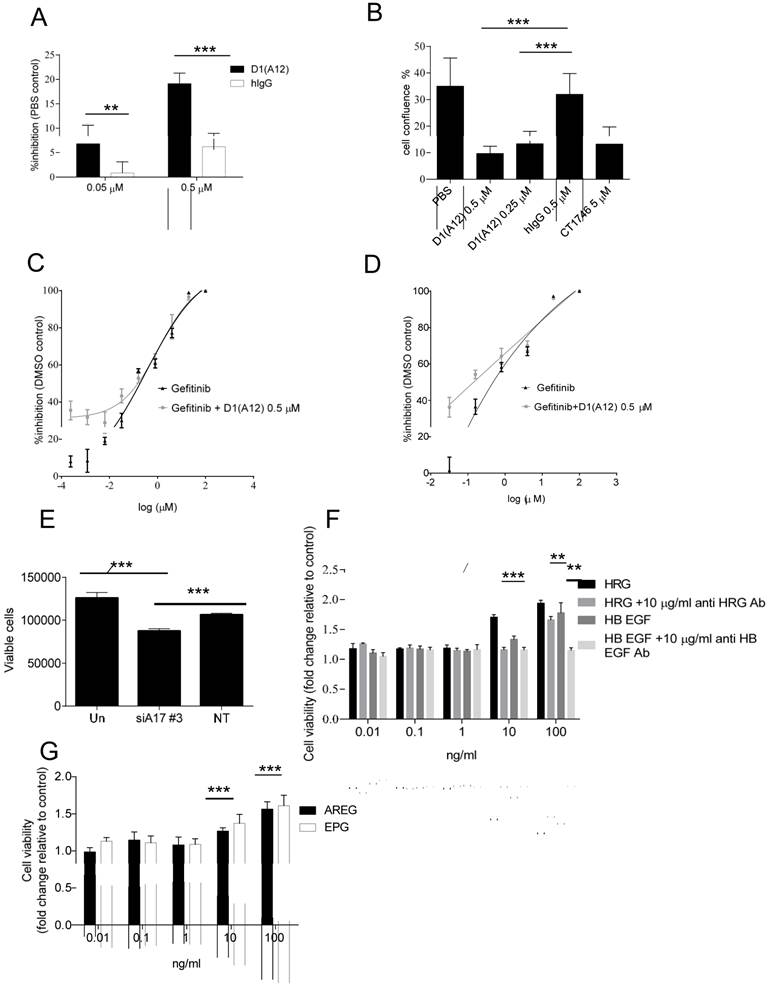
Gefitinib is a widely-used inhibitor of the EGFR tyrosine kinase domain that reduces tumour cell proliferation [34]. Due to the high level of EGFR in SCC9 and SCC13 cells, we next examined the combined effect of treating cells with gefitinib and D1 (A12). As expected, exposure to gefitinib (0.0064-100 µM) resulted in a dose dependent growth inhibition. Addition of 0.5 µM D1 (A12) sensitized the SCC9 cells (Fig. 2C) and SCC13 cells (Fig. 2D) to gefitinib at lower concentrations (0.8-0.0064 µM), indicating potential cooperative effects on EGFR.
We next examined whether siRNA-mediated knockdown of ADAM17 could impair cell proliferation in SCC13 cells. After 72 h incubation, we observed that transfection with ADAM17 siRNA significantly reduced cell proliferation compared to non-transfected cells and cells transfected with non-targeting siRNA control, respectively (both P<0.001) (Fig. 2E).
To investigate which HER ligands are responsible for driving cell proliferation in SCC13 cells, we stimulated serum-starved cells with a serial dilution of recombinant human HB-EGF, AREG, EPG and HRG for 72 h followed by CTG assay. We observed that 10 ng/ml and 100 ng/ml HRG significantly promoted cell growth to the highest extent compared to other ligands at the same concentration (both P<0.001) (Fig. 2F). HB-EGF was less potent than HRG in promoting cell proliferation. 10 ng/ml and 100 ng/ml AREG and EPG could also stimulate cell proliferation, but to a lesser extent than HRG and HB-EGF (Fig. 2G). To confirm the specificity of the HRG effect, we neutralized the HRG ligand with human HER3 antibody and found that 10 µg/ml HER3 antibody significantly inhibited the induced proliferation by 10 ng/ml and 100 ng/ml HRG (P<0.001 and P<0.01, respectively). We observed a similar effect with anti-HB-EGF antibody as with HER3 antibody (Fig. 2F). In addition, we also tested the effect of human HER2 antibody on cell proliferation in SCC13 cells and we found that 10 µg/ml HER2 antibody did not significantly prevent cell proliferation (data not shown). Taken together, these studies demonstrated that HRG plays a predominant role of inducing proliferation in SCC13 cells.
Effect of inhibition of ADAM17 and HER ligands on cell motility
Previously it was demonstrated that ADAM17 contributes to tumour cell migration and invasion in vitro [34] and EGFR inhibition with gefitinib can decrease cancer cell metastasis in vivo [36]. We therefore examined whether treatment with inhibitory antibody D1 (A12) could affect cell motility in the presence or absence of gefitinib in an in vitro wound closure assay. Treating SCC9 cells (Fig. 3A) and SCC13 cells (Fig. 3B) with D1 (A12) alone showed about 15% and 40% (P<0.05) inhibition compared to PBS and human IgG, respectively. In both cell lines, gefitinib (0.05 µM~5 µM) showed a concentration dependent inhibition of cell migration. Moreover, treatment with 0.05 µM gefitinib caused a significant decrease in cell migration in conjunction with 0.5 µM D1 (A12). The effect of inhibition of proliferation by D1 (A12) and gefitinib (Fig. 2C and 2D) might also contribute to the reduction of cell motility because cell growth occurs during the process of cell migration. These data indicate that inhibition of ADAM17 proteolytic activity resulted in a substantial reduction of cell migration in HNSCC cells treated with gefitinib or not.
(A-B) Effect of D1(A12) and gefitinib on cell migration and invasion. Cells were treated with indicated concentrations of gefitinib in the presence or absence of D1(A12). As a single agent, D1(A12) reduced SCC9 (A) and SCC13 (B) cell migration, compared to human IgG treated cells. Combined 0.05 µM gefitinib with 0.5 µM D1(A12) showed a significant reduction of cell migration in SCC9 (A) and SCC13 (B) by a wound closure assay. Each point is the mean ± SD of 8 replicates. (C) Effect of D1(A12) on SCC13 cell migration by a transwell migration assay. 0.2 µM D1(A12) significantly decreased cell migration compared 0.2 µM human IgG treated cells. Each point is the mean ± SD of 3 replicates. (D-a) Effect of ADAM17 siRNA on SCC9 cell migration. Transfection of ADAM17 siRNA #3 in SCC9 cells significantly reduced cell migration by a transwell migration assay, compared to non-targeting siRNA control (*P<0.05). Each point is the mean ± SD of 3 replicates. (D-b) immunoblotting analysis of ADAM17 in cells transfected with ADAM17 siRNA, non-targeting siRNA and non-transfected cells. (E-a) Effect of ADAM17 siRNA on SCC13 cell migration. Transfection of ADAM17 siRNA #3 in SCC13 cells significantly reduced cell migration by a transwell migration assay, compared to non-targeting siRNA control (*P<0.05). Each point is the mean ± SD of 3 replicates. (E-b) immunoblotting analysis of ADAM17 in cells transfected with ADAM17 siRNA, non-targeting siRNA and non-transfected cells. (F) Effect of HB-EGF and AREG on SCC13 cell migration by a wound closure assay. Cells were treated with indicated agents with or without inhibitors. Data are shown as percentage of control. The values above the x-axis mean the inhibition of cell migration and the values below the x-axis mean the promotion of cell migration. TNF protease inhibitor 2 (TAPI-2), a broad-spectrum inhibitor of ADAMs and other matrix metalloproteinases, was used as a positive control. (G) Effect of D1(A12) on SCC13 cell invasion by a transwell invasion assay. D1(A12) treated cells significantly decreased invasion ability compared to human IgG treated or untreated cells. Each point is the mean ± SD of 3 replicates. Each point is the mean ± SD of 8 replicates.
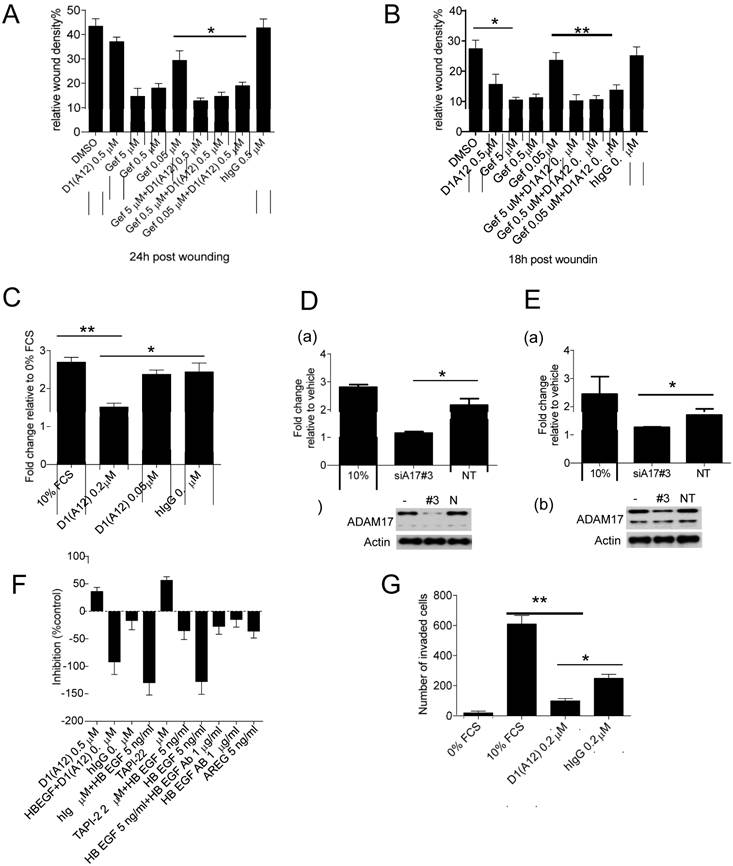
To further test the effect of D1 (A12)-mediated inhibition of cell migration, we coaxed SCC13 cells to migrate across uncoated transwell membranes towards the chemoattractant of 10% FCS in vitro for 48 h. Treatment with 0.2 µM D1 (A12) decreased the migration of SCC13 cells by 44% (P<0.01) and 38% (P<0.05) compared to 10% FCS and human IgG controls, respectively (Fig. 3C). Using growth factor reduced Matrigel-coated transwell membranes, we found that the invasive behaviour of SCC13 cells was impaired by 84% (P<0.01) and 61% (P<0.05) due to 0.2 µM D1(A12) compared to 10% FCS and human IgG controls, respectively after 48 h (Fig. 3G).
We next examined whether siRNA-mediated knockdown of ADAM17 could interfere with SCC9 and SCC13 cell migration. We confirmed potent knockdown of ADAM17 at the protein level in SCC9 (Fig. 3D (b)) and SCC13 cells (Fig. 3E (b)) by immunoblotting. After 24 h of transwell migration, we found that transfection with ADAM17 siRNAs significantly reduced the migration of SCC9 cells by 47% (P<0.05) (Fig. 3D (a)) and SCC13 cells by 26% (P<0.05) (Fig. 3E (a)), compared to cells transfected with non-targeting siRNA controls.
In order to identify which EGFR ligands play a role on cell motility, we performed a wound closure assay to examine the effects of HB-EGF and AREG on SCC13 cell motility. SCC13 cells were treated with 5 ng/ml HB-EGF and 5 ng/ml AREG alone or with inhibitors and 10 µg/ml anti HB-EGF antibody in serum free medium. After 15 h the motility of the HB-EGF and AREG treated cells was significantly increased by 120% and 70% over untreated cells, respectively. Anti HB-EGF antibody and 0.5 µM D1 (A12) inhibited HB-EGF induced migration by 75% and 30%, respectively (Fig. 3F). TGF α and HRG did not enhance SCC13 cell migration, while EPG and AREG similarly induced cell migration (data not shown). Taken together, these data suggest that HB-EGF plays a leading role in promoting the migratory activity of SCC13 cells compared to other growth factors, which is consistent with previous findings [37].
Effect of inhibition of ADAM17 on shedding of HER ligands
Given that inhibiting the sheddase activity of ADAM17 using D1 (A12) suppresses HNSCC cell proliferation and motility, we next investigated whether treatment with D1 (A12) reduces the levels of ligands known to be shed by ADAM17 into cell culture medium. AREG was abundantly present in HNSCC cell culture media compared to other EGFR ligands such as HB-EGF, TGF-α and TNF-α (data not shown). Analysis of supernatants after a 72 h incubation by ELISA showed that the endogenous shedding of AREG was significantly decreased by the addition of D1 (A12) at a serial of concentrations (0.05 µM~ 0.5 µM) in SCC9 cells (Fig. 4A) and SCC13 cells (Fig. 4B). This was also true for the shedding of HRG in SCC13 cells (at 0.1 µM, P<0.05; at 0.2 µM and 0.5 µM, P<0.01) (Fig. 4C). Human IgG was used as negative control. A similar result was also seen in SJG15 cells (data not shown). These results indicate that ADAM17 is a major sheddase for AREG in SCC9 and SCC13 cells. We only measured soluble HRG (a specific ligand of HER3) in SCC13 cell culture media because we observed that D1 (A12) treatment resulted in the reduction of phosphorylation of HER3 only in SCC13 cells (see Fig. 6A).
(A-C) D1(A12) inhibits endogenous AREG and HRG shedding in SCC9 and SCC13 cells. Cells were seeded in complete medium, followed by 24 h serum starvation and then treated with a serial of dilutions of D1(A12) and human IgG for 72 h in medium supplemented with 1% dialyzed fetal calf serum (FCS). AREG and HRG were measured by ELISA in the supernatants collected after treatments. Each point is the mean ± SD of 4 replicates. (D) Effect of ADAM10/ 17 siRNA on AREG shedding in SCC13 cells. Transfection of ADAM17 siRNA #3 significantly decreased the production of soluble form of AREG, compared to non-targeting siRNA control (P<0.001) and to cells transfected with ADAM10 siRNA (P<0.01). Each point is the mean ± SD of 4 replicates.
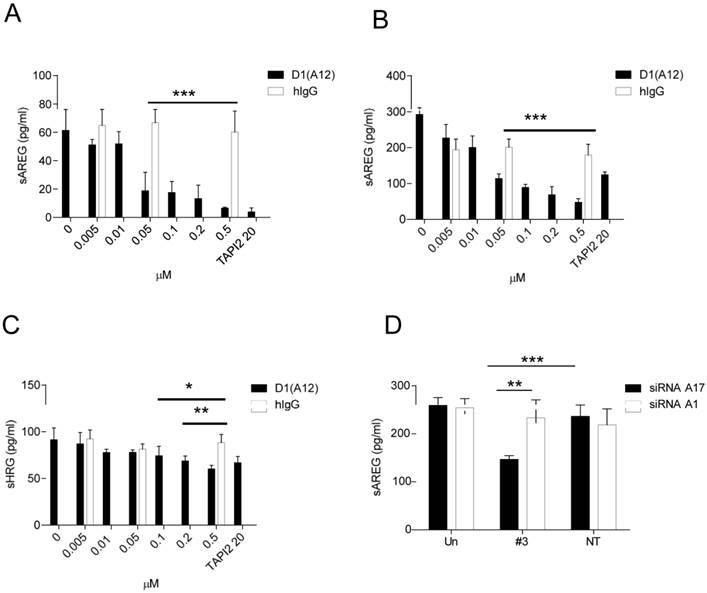
ADAM10, which is closely related to ADAM17, was found to be the major sheddase for betacellulin and EGF [38]. We next sought to determine which of these ADAMs is responsible for shedding of AREG. ELISA analysis of cell culture media after a 72 h siRNA treatment revealed that the endogenous shedding of AREG was significantly inhibited by ADAM17 siRNA compared to cells transfected with ADAM10 siRNA (P<0.01) and with non-targeting siRNA controls (P<0.001), respectively (Fig. 4D). As expected, betacellulin was significantly suppressed by ADAM10 siRNA not by ADAM17 siRNA (data not shown).
Effect of BK on shedding of HER ligands, cell proliferation and migration
BK, an important pro-inflammatory peptide, stimulates the kinin B1 and B2 receptors which belong to G protein coupled receptor (GPCR) family [39]. Previous studies have shown that BK induced GPCR transactivation of EGFR depends on the shedding of pro-ligands by ADAM 17 activation [40] and combined inhibition of EGFR and GPCR showed synergistic killing of HNSCC cells [41]. We therefore studied whether D1 (A12) can block the induced AREG shedding by BK in SCC9 and SCC13 cells. Seventy-two hours treatment of these two cell lines with increasing concentrations of BK (0.1-10 nM) showed that BK induced a concentration-dependent increase of AREG shedding in SCC9 (Fig. 5A) (at 10 nM, P<0.05) but not in SCC13 cells. 0.2 µM D1 (A12) showed significant inhibition of AREG shedding with 1 nM and 10 nM BK stimulation (P<0.05).
(A-B) D1(A12) inhibits BK induced the increase of AREG shedding in SCC9 cells and SCC13 cells. Cells were seeded in complete medium, followed by 48 h serum starvation and then treated with series of concentrations of BK with or without 0.2 µM D1(A12) for 72 h in SCC9 and 2 h in SCC13 cells in medium supplemented with 1% dialyzed fetal calf serum (FCS). AREG and HRG (C) were measured by ELISA in supernatants collected after treatments. Each point is the mean ± SD of 4 replicates. (D) Effect of BK on cell growth in SCC13 cells with or without the addition of D1(A12). Cells were seeded in complete medium, followed by 24 h serum starvation and then treated with series of concentrations of BK with or without 0.2 µM D1(A12) for 72 h in medium supplemented with 1% dialyzed FCS. The viability was measured by CTG and plotted as viable cells. Each point is the mean ± SD of 4 replicates. (E) Effect of BK conditioned medium (CM) on SCC13 cell migration by a wound closure assay. Cells were treated with either different inhibitor treated conditioned medium or fresh medium. Each point is the mean ± SD of 8 replicates. Tissue Inhibitor of Metalloproteinase-3 (TIMP3), a potent inhibitor of ADAM17, was used as a positive control.
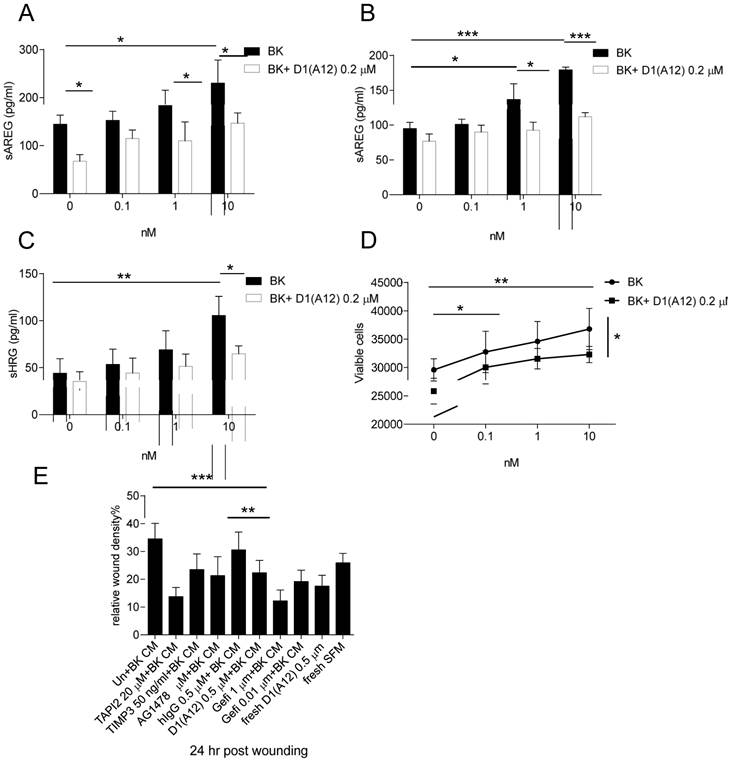
In order to examine at what time point BK could induce AREG shedding in SCC13 cells, a time course experiment was performed at 10 min to 72 h with a series of concentrations (0.1-10 nM). At 10 min and 2 h a significant increase of AREG shedding was observed with 10 nM BK and the amount of soluble AREG at 2 h was two times more than at 10 min (data not shown). Thus, we repeated the BK stimulation for 2 h with or without the addition of 0.2 µM D1 (A12) and found that the increase of soluble AREG was significantly attenuated by D1 (A12) (at 1 nM, P<0.05; 10 nM, P<0.001) (Fig. 5B). Similarly, we observed that D1 (A12) significantly decreased (P<0.05) the output of soluble HRG induced by a 2 h stimulation with 10 nM BK (Fig. 5C).
BK is capable of stimulating proliferation via transactivation of EGFR in HNSCC [40, 41] and breast cancer cells [42]. However, we only observed that 72 h treatment with BK promoted cell growth (at 0.1 nM, P<0.05; 1 and 10 nM, P<0.01 compared to untreated cells) and the addition of 0.2 µM D1(A12) significantly attenuated the cell proliferation induced by 10 nM BK (P<0.05) in SCC13 cells (Fig. 5D). No obvious cell proliferation change was observed in SCC9 cells after 72 h BK treatment (data not shown).
To study whether BK induces SCC13 cell migration and whether treatment with D1 (A12) can counteract this effect, conditioned media from SCC13 cells treated with indicated diverse inhibitors was used in a scratch wound assay. As shown in Fig. 5E, the 24 h migration assay showed that 0.5 µM D1 (A12) reduced the migration of SCC13 cells by 36% (P<0.01) and 27% (P<0.001) compared to PBS and human IgG treated cells, respectively. Freshly added D1(A12) in PBS reduced cell migration by 33% (P<0.001). Positive controls of recombinant human TAPI 2 and TIMP 3, pharmacological inhibitors of ADAM17 and AG1478, EGFR tyrosine kinase inhibitor, inhibited SCC9 and SCC13 cell migration as expected.
Inhibition of ADAM17 blocks BK induced GPCR transactivation of HERs and downstream signalling
It was reported that BK action on its GPCR can transactivate EGFR through activation of ADAM17 to generate activating ligands in tumour cells [40]. We have shown that D1 (A12) can block BK induced AREG and HRG shedding, cell proliferation and migration in SCC13 cells. To understand how D1(A12) performs these cellular functions, SCC13 cells were first treated with D1(A12) and indicated diverse inhibitors as positive controls for 2 h in serum free medium. Human IgG used as a negative control. Cells were then challenged with BK for 10 min to transactivate EGFR. Western blotting analysis revealed that D1(A12) decreased the phosphorylation of EGFR, HER3, STAT3, AKT and ERK in SCC13 cells (Fig. 6A) and the phosphorylation of EGFR and ERK in SCC9 cells (Fig. 6B). Silencing ADAM17 expression by siRNA in SCC13 cells also diminished the phosphorylation of EGFR and ERK (Fig. 6C). In order to find out whether ADAM10 is involved in BK induced GPRC transactivation of EGFR, we knocked down ADAM10 in SCC13 cells. Fig. 6D showed that ADAM10 knockdown did not affect the phosphorylation of EGFR and ERK, suggesting the dominant role of ADAM17 in this signal transduction cascade.
Effect of D1(A12) on the phosphorylation of HER family receptors and downstream signalling molecules under BK stimulation in HNSCC cells. (A) SCC13 cells were pre-incubated with different inhibitors for 2 h then treated with 10 nM BK for 10 min, after which the cell extracts were immunoblotted with the indicated antibodies. (B) SCC9 cells were treated in the same way as SCC13 cells and analysed with indicated antibodies. (C-D) Effect of ADAM17 siRNA and ADAM10 siRNA on the phosphorylation of EGFR and ERK in SCC13 cells. Cells were transfected with 3 different siRNAs and non-targeting siRNA. 72 h later, cells were serum starved for 48 h and then challenged with 10 nM BK for 10 min. After cell lysis, equal amount of proteins were analyzed by immunoblotting with indicated antibodies. (E-F) Effect of D1(A12) on the phosphorylation of HER family receptors in SCC13 cells. (E) Cells were treated as in A and analysed by immunoblotting with indicated antibodies. The same cell extract as 6E were exposed to the RayBio human EGFR phosphorylation antibody array 1 as described in M&M. (F) On each membrane, the left upper corner and right lower corner were spotted with positive control used for signal normalisation. The left lower corner was spotted with pan HER 1, 2 and 3. Five different phosphorylation sites of HER2, HER3 and HER4 were detected in the middle part of the membrane. (G) Normalisation of D1(A12) treated signals to human IgG treated signals. Signal intensity for a particular spot was divided by the average signal intensity of 4 positive controls on the same array membrane. Each phosphorylation site was spotted duplicate.
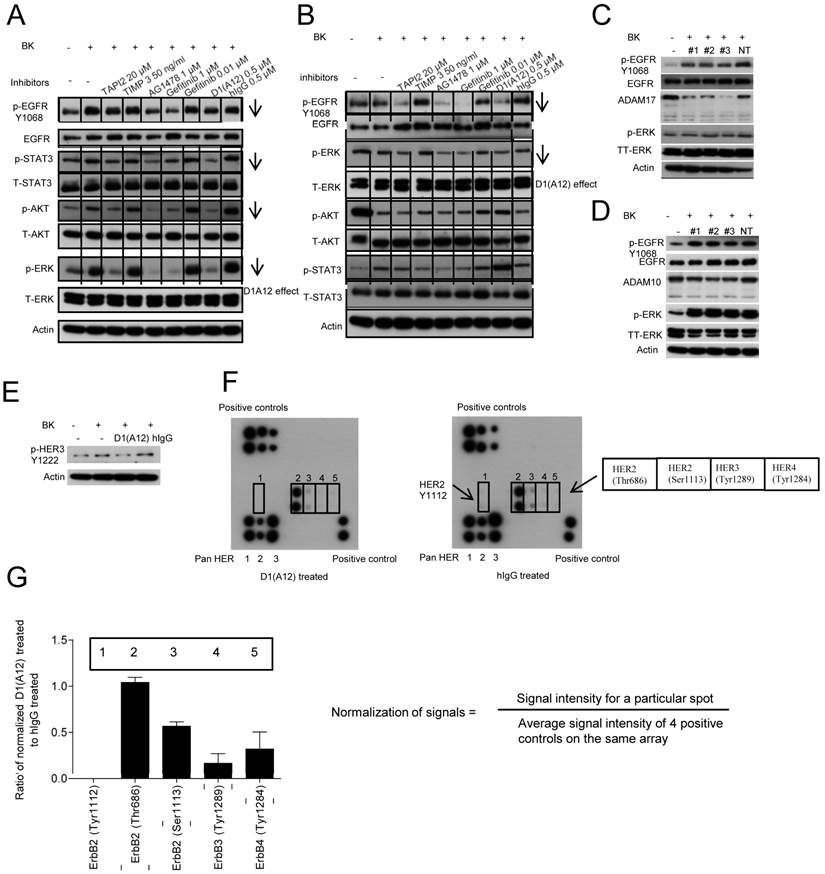
To investigate whether BK can transactivate HER2, HER3 and HER4 along with EGFR and whether D1(A12) can prevent this induction in SCC13 cells, the RayBio human EGFR phosphorylation antibody array 1 was used to detect the relative levels of phosphorylation at specific sites of human HER family proteins. Fig. 6E shows that the phosphorylation of HER 3 (Y1222) was decreased by D1 (A12) compared to untreated or human IgG treated cells. Next, immunbolotting analysis of protein array showed that D1(A12) reduced the autophosphorylation of HER 2 at Tyr1112/Ser1113, HER3 at Tyr1222/Tyr1289 and HER4 at Tyr1284 sites (Fig. 6F) compared to human IgG treated cell lysate. Signals were normalized as signal intensity for a particular spot divided by average signal intensity of 4 positive controls on the same membrane. Those specific sites were decreased to different ratio of intensity compared to human IgG control (Fig. 6G). Thus, D1(A12) can reduce the phosphorylation of specific sites on HER-family proteins.
Discussion
Despite the success of EGFR inhibition therapy in HNSCC, the acquisition of drug resistance is common and leads to tumour recurrence. Thus, there is a pressing need to identify new avenues to overcome this roadblock. In this study, we use a human inhibitory antibody against ADAM17 and a siRNAs approach to reveal a key role for ADAM17 in driving proliferation and motility of HNSCC cells. We show that an ADAM17 inhibitory antibody D1 (A12) can block the shedding of HER ligands, as well as BK-induced cell proliferation and migration (Fig. 2 and 3). Intriguingly, treatment with D1 (A12) can reduce the activation of all four HER family receptors and their downstream signalling pathways (Fig. 6).
A potential mechanism of resistance to EGFR inhibition is the activation of other HER family receptors, which mediate proliferation, migration and cell survival [4]. We observed that D1 (A12) inhibited phosphorylation of multiple tyrosine residues for EGFR, HER 2, HER3 and HER4 under the stimulation of BK (Fig. 6). Given that these HER family proteins are emerging as potential therapeutic targets in diverse cancer types, the ability of D1 (A12) to act as a broad HER TKI may be therapeutically valuable in HER-driven tumours [43].
Resistance to EGFR inhibition may also result from the activation of EGFR downstream signalling pathways in squamous cell cancer. Of note, STAT3 is overexpressed in HNSCC [44] and AKT has been linked to poor prognosis [45]. Moreover, an toxin specifically targeting ERK/MAPK-dependent cells showed anti-tumour effects in HNSCC cell lines [46]. Interestingly, our anti-ADAM17 inhibitory antibody supresses the activation of STAT3, AKT and ERK in HNSCC cells, suggesting that blockage of ADAM17 can sensitize cells to EGFR inhibition.
Because considerable evidence suggests that particular ADAMs are causally involved in cancer formation and progression, it might be expected that inhibition of these proteases could be beneficial for cancer treatment. Despite the high therapeutic potential of ADAM inhibition, all clinical trials using broad-spectrum metalloprotease inhibitors have failed so far due to poor bioavailability, pharmacokinetic problems, non-selectivity or musculoskeletal side effects [47]. Zhou and colleagues [48] demonstrated a low molecular weight molecule INCB3619, a dual ADAM10/17 inhibitor, prevents the processing of multiple HER ligands and activation of EGFR and HER3 signalling and has been shown to inhibit tumour cell growth in several different models such as non-small cell lung cancer and breast cancer. Similarly, the proliferation of breast cancer cell lines could be inhibited using anti-ADAM17 antibodies [18]. Compared to other available metalloprotease inhibitors, D1 (A12) is a highly specific human ADAM17 inhibitory antibody and not active against other ADAM family metalloproteases and therefore it could provide a far superior drug safety profile and less side effects in preclinical models.
Additional signalling cross-talk between EGFR and other growth factor receptors might play a role in HNSCC, although the impact of these signalling interactions for pathway activation and resistance to therapy are not fully understood. A number of studies have shown the enhanced effects of combined targeted agents in various types of cancers, such as the combination of cMET and gefitinib in non-small-cell lung cancer (NSCLC) [49], the combination of MEK inhibitor and gefitinib in lung cancer cells [50], the combination of the mammalian target of rapamycin (mTOR) and gefitinib in breast cancer cells [51]. Therefore, exploring the combination of D1(A12) with other targeted therapies to achieve anti-tumour effects in HNSCC is worthy of investigation.
Acknowledgements
We thank Dr. Frances Richards for useful discussions and Prof. Fiona Watt for providing HNSCC cells. This study was fully supported by a grant from Cancer Research UK. Dr. Hang Fai Kwok (joint corresponding and senior author in this manuscript) is currently supported by a grant from the Faculty of Health Sciences, University of Macau (SRG2014-00006-FHS).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chin D, Boyle GM, Porceddu S. et al. Head and neck cancer: past, present and future. Expert Review of Anticancer Therapy. 2006;6:1111-18
2. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nature Reviews: Cancer. 2011;11:9-22
3. Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumour development: EGFR gene and cancer. FEBS Journal. 2010;277:301-8
4. Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocrine-related Cancer. 2005;12:S17-27
5. Hegymegi-Barakonyi B, Eros D, Szantai-Kis C. et al. Tyrosine kinase inhibitors - small molecular weight compounds inhibiting EGFR. Current Opinion in Molecular Therapeutics. 2009;11:308-21
6. Astsaturov I, Cohen RB, Harari P. EGFR-targeting monoclonal antibodies in head and neck cancer. Current Cancer Drug Targets. 2007;7:650-65
7. Zimmermann M, Zouhair A, Azria D. et al. The epidermal growth factor receptor (EGFR) in head and neck cancer: its role and treatment implications. Radiation Oncology. 2006;1:11
8. Rabinowits G, Haddad RI. Overcoming resistance to EGFR inhibitor in head and neck cancer: a review of the literature. Oral Oncology. 2012;48:1085-89
9. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nature Reviews: Clinical Oncology. 2010;7:493-507
10. Box C, Zimmermann M, Eccles S. Molecular markers of response and resistance to EGFR inhibitors in head and neck cancers. Front Biosci. (Landmark Ed). 2013;18:520-42
11. Zaczek A, Brandt B, Bielawski KP. The diverse signaling network of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histology and Histopathology. 2005;20:1005-15
12. Huang Z, Brdlik C, Jin P. et al. A pan-HER approach for cancer therapy: background, current status and future development. Expert Opinion on Biological Therapy. 2009;9:97-110
13. Wheeler DL, Huang S, Kruser TJ. et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944-56
14. Barnea I, Haif S, Keshet R. et al. Targeting ErbB-1 and ErbB-4 in irradiated head and neck cancer: results of in vitro and in vivo studies. Head & Neck. 2013;35:399-407
15. Black RA. Tumour necrosis factor-alpha converting enzyme. International Journal of Biochemistry & Cell Biology. 2002;34:1-5
16. Lee DC, Sunnarborg SW, Hinkle CL. et al. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann. New York Acad. Sci. 2003;995:22-38
17. Zheng X, Jiang F, Katakowski M. et al. ADAM17 promotes breast cancer cell malignant phenotype through EGFR-PI3K-AKT activation. Cancer Biol. Ther. 2009;8:1045-54
18. Lendeckel U, Kohl J, Arndt M. et al. Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. Journal of Cancer Research and Clinical Oncology. 2005;131:41-8
19. Blanchot-Jossic F, Jarry A, Masson D. et al. Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J. Pathol. 2005;207:156-63
20. Ringel J, Jesnowski R, Moniaux N. et al. Aberrant expression of a disintegrin and metalloproteinase 17/tumour necrosis factor-alpha converting enzyme increases the malignant potential in human pancreatic ductal adenocarcinoma. Cancer Res. 2006;66:9045-53
21. Karan D, Lin FC, Bryan M. et al. Expression of ADAMs (a disintegrin and metalloproteases) and TIMP-3 (tissue inhibitor of metalloproteinase-3) in human prostatic adenocarcinomas. Int. J. Oncol. 2003;23:1365-71
22. Schafer B, Marg B, Gschwind A. et al. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J. Biol. Chem. 2004;279:47929-38
23. Duffy MJ, Mullooly M, O'Donovan N. et al. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clinical Proteomics. 2011;8:9
24. Grandis JR, Chakraborty A, Zeng Q. et al. Downmodulation of TGF-alpha protein expression with antisense oligonucleotides inhibits proliferation of head and neck squamous carcinoma but not normal mucosal epithelial cells. J. Cell. Biochem. 1998;69:55-62
25. Wang F, Liu R, Lee SW. et al. Heparin-binding EGF-like growth factor is an early response gene to chemotherapy and contributes to chemotherapy resistance. Oncogene. 2007;26:2006-16
26. Castillo J, Erroba E, Perugorria MJ. et al. Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res. 2006;66:6129-38
27. Kakiuchi S, Daigo Y, Ishikawa N. et al. Prediction of sensitivity of advanced non-small cell lung cancers to gefitinib (Iressa, ZD1839). Human Molecular Genetics. 2004;13:3029-43
28. Kenny PA. Tackling EGFR signaling with TACE antagonists: a rational target for metalloprotease inhibitors in cancer. Expert Opinion on Therapeutic Targets. 2007;11:1287-98
29. Kornfeld JW, Meder S, Wohlberg M. et al. Overexpression of TACE and TIMP3 mRNA in head and neck cancer: association with tumour development and progression. Br. J. Cancer. 2011;104:138-45
30. Tape CJ, Willems SH, Dombernowsky SL. et al. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA. 2011;108:5578-83
31. Richards FM, Tape CJ, Jodrell DI. et al. Anti-tumour effects of a specific anti-ADAM17 antibody in an ovarian cancer model in vivo. PLoS One. 2012;7(e):40597
32. Lin P, Sun X, Feng T. et al. ADAM17 regulates prostate cancer cell proliferation through mediating cell cycle progression by EGFR/PI3K/AKT pathway. Mol. Cell Biochem. 2012;359:235-43
33. An Z, Wang X, Willmott N. et al. Conversion of highly malignant colon cancer from an aggressive to a controlled disease by oral administration of a metalloproteinase inhibitor. Clin. Exp. Metastasis. 1997;15:184-95
34. Kondo N, Ishiguro Y, Kimura M. et al. Antitumour effect of gefitinib on head and neck squamous cell carcinoma enhanced by trastuzumab. Oncology Reports. 2008;20:373-78
35. Hinsley EE, Hunt S, Hunter KD. et al. Endothelin-1 stimulates motility of head and neck squamous carcinoma cells by promoting stromal-epithelial interactions. International Journal of Cancer. 2012;130:40-7
36. Yang Z, Bagheri-Yarmand R, Wang RA. et al. The epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 (Iressa) suppresses c-Src and Pak1 pathways and invasiveness of human cancer cells. Clinical Cancer Research. 2004;10:658-67
37. Johnson NR, Wang Y. Controlled delivery of heparin-binding EGF-like growth factor yields fast and comprehensive wound healing. Journal of Controlled Release. 2013;166:124-9
38. Sahin U, Weskamp G, Kelly K. et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. Journal of Cell Biology. 2004;164:769-79
39. Lerner UH, Modéer T. et al. Bradykinin B1 and B2 receptor agonists synergistically potentiate interleukin-1-induced prostaglandin biosynthesis in human gingival fibroblasts. Inflammation. 1991;15:427-36
40. Gschwind A, Hart S, Fischer OM. et al. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO Journal. 2003;22:2411-21
41. Thomas SM, Bhola NE, Zhang Q. et al. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Research. 2006;66:11831-39
42. Greco S, Muscella A, Elia MG. et al. Mitogenic signalling by B2 bradykinin receptor in epithelial breast cells. Journal of Cellular Physiology. 2004;201:84-96
43. Nam HJ, Ching KA, Kan J. et al. Evaluation of the antitumour effects and mechanisms of PF00299804, a pan-HER inhibitor, alone or in combination with chemotherapy or targeted agents in gastric cancer. Molecular Cancer Therapeutics. 2012;11:439-451
44. Bonner JA, Yang ES, Trummell HQ. et al. Inhibition of STAT-3 results in greater cetuximab sensitivity in head and neck squamous cell carcinoma. Radiotherapy and Oncology. 2011;99:339-343
45. Yu Z, Weinberger PM, Sasaki C. et al. Phosphorylation of Akt (Ser473) predicts poor clinical outcome in oropharyngeal squamous cell cancer. Cancer Epidemiology, Biomarkers & Prevention. 2007;16:553-8
46. Schafer JM, Peters DE, Morley T. et al. Efficient targeting of head and neck squamous cell carcinoma by systemic administration of a dual uPA and MMP-activated engineered anthrax toxin. PLoS One. 2011;6:e20532
47. Murumkar PR, DasGupta S, Chandani SR. et al. Novel TACE inhibitors in drug discovery: a review of patented compounds. Expert Opinion on Therapeutic Patents. 2010;20:31-57
48. Zhou BB, Peyton M, He B. et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39-50
49. Zucali PA, Ruiz MG, Giovannetti E. et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Annals of Oncology. 2008;19:1605-12
50. Huang MH, Lee JH, Chang YJ. et al. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Molecular Oncology. 2013;7:112-120
51. Dragowska WH, Weppler SA, Qadir MA. et al. The combination of gefitinib and RAD001 inhibits growth of HER2 overexpressing breast cancer cells and tumours irrespective of trastuzumab sensitivity. BMC Cancer. 2011;11:420
Author contact
![]() Corresponding authors: yh345ac.uk (Y.H) / hfkwokmo (H.F.K).
Corresponding authors: yh345ac.uk (Y.H) / hfkwokmo (H.F.K).