Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
An overview of HDACs and HDAC...
Biological functions of HDACs...
Clinical implications of HDAC...
Concluding remarks and...
Acknowledgements
Abbreviation
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(7):757-770. doi:10.7150/ijbs.9067 This issue Cite
Review
Targeting Histone Deacetylases for Cancer Therapy: From Molecular Mechanisms to Clinical Implications
Zhiming Li1,2*, Wei-Guo Zhu1,2,3 ![]()
1. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Beijing 100191, China.
2. Department of Biochemistry and Molecular Biology, Peking University Health Science Center, Beijing, 100191, China.
3. Peking-Tsinghua University Center for Life Sciences, Peking University, Beijing 100871, China.
*Current address: Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China.
Received 2014-3-10; Accepted 2014-4-2; Published 2014-7-2
Abstract
Genetic abnormalities have been conventionally considered as hallmarks of cancer. However, studies over the past decades have demonstrated that epigenetic regulation also participates in the development of cancer. The fundamental patterns of epigenetic components, such as DNA methylation and histone modifications, are frequently altered in tumor cells. Acetylation is one of the best characterized modifications of histones, which is controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDACs are a group of enzymes which catalyze the removal of the acetyl groups of both histones and non-histone proteins. HDACs are involved in modulating most key cellular processes, including transcriptional regulation, apoptosis, DNA damage repair, cell cycle control, autophagy, metabolism, senescence and chaperone function. Because HDACs have been found to function incorrectly in cancer, various HDAC inhibitors are being investigated to act as cancer chemotherapeutics. The primary purpose of this paper is to summarize recent studies of the links between HDACs and cancer, and further discuss the underlying mechanisms of anti-tumor activities of HDAC inhibitors and clinical implications.
Keywords: HDAC, HDAC inhibitor, epigenetic therapy, cancer.
Introduction
In order to carry out cellular functions, histones are subject to about sixteen types of post-translational modifications, such as acetylation, methylation and phosphorylation [1-3]. The enzymes responsible for these reversible modifications include histone acetyltransferases (HATs) and histone deacetylases (HDACs), methyltransferases (KMTs) and demethylases (KDMs), kinases and phosphatases, and so on. Different types of modifications may have different outcomes depending on the biological contexts. For example, trimethylation of H3K4 is generally associated with transcription activation [4], while trimethylation of H3K9 is associated with transcription inactivation [5]. As regards acetylation, it is the first modification identified and one of the best characterized modifications of histones [6]. Reversible acetylation and deacetylation of histones regulated by the opposing effects of HATs and HDACs perturb genetic information flow through interruption of chromosomal structure and the availability of transcription factors to DNA [7]. By removing the negatively charged acetyl groups, HDACs generally act as transcriptional repressors by stabilizing the nucleosomal DNA-histone interaction. HDACs can also bind to various co-repressors to recruit other histone modifiers, thus regulating other chromatin-based processes. In addition, regulation of non-histone substrates expands the function repertoire of acetylation. The regulatory network of HDACs has now extended to induction of apoptosis, DNA damage repair, cell cycle control, autophagy, metabolism, senescence and so on [8-11].
Cancer is now considered as a disorder of altered genetic and epigenetic regulation [12]. Aberrant epigenome including dysregulatory expression and/or activity of HDACs has been characterized in different tumors [13]. In general, HDACs are cancer permissive despite that certain types of class III HDACs may function as tumor suppressors [14].
The regulatory mechanisms of HDACs in critical cellular properties, with a particular emphasis on classical HDACs will be discussed in this review. The role of this regulatory network in cancer development and the clinical relevance of HDAC inhibitors in cancer treatment will also be reviewed.
An overview of HDACs and HDAC inhibitors
Eighteen distinct HDACs have been identified so far and they are classified into four groups based on their structural divergence, namely class I, II, III and IV HDACs [15, 16]. Class I and II HDACs are considered as 'classical' HDACs while class III is a family of nicotinamide adenine dinucleotide (NAD+)-dependent proteins. Class IV HDAC is an atypical category of its own, based solely on its DNA sequence similarity to the others (Table 1).
An overview of HDACs. Listed below are the cytogenetic location, subcellular location and tissue distribution of HDACs. Part of the non-histone substrates of different HDACs are also listed including cancer-associated genes like p53 and Rb1. Transcription factors such as E2F1, NF-κB and STAT3, which are also related to cancer, are demonstrated to be catalytic substrates of HDACs. Metabolic enzymes like AMPK and GDH are also found to be regulated by HDACs, especially sirtuins. Non-histone substrates carry out the multiple cellular functions regulated by HDACs.
| Classification | HDAC | Cytogenetic location | Subcellular localization | Non-histone substrates (partly shown) |
|---|---|---|---|---|
| I | HDAC1 | 1p35.1 | nucleus | RB1, SHP, p53, MyoD, E2F1, STAT3, NF-κB, CtIP, AMPK |
| HDAC2 | 6q21 | nucleus | GCCR, BCL6, STAT3, YY1 | |
| HDAC3 | 5q31.3 | nucleus | SHP, YY1, GATA1, p65, STAT3, MEF2D | |
| HDAC8 | Xq13.1 | nucleus | SMC3, actin | |
| IIa | HDAC4 | 2q37.3 | nucleus/cytoplasm | GATA1, HP1 |
| HDAC5 | 17q21.31 | nucleus/cytoplasm | SMAD7, HP1 | |
| HDAC7 | 12q13.11 | nucleus/cytoplasm | PLAG1, PLAG2 | |
| HDAC9 | 7p21.1 | nucleus/cytoplasm | --- | |
| IIb | HDAC6 | Xp11.23 | mostly cytoplasm | α-tubulin, HSP90, SHP, SMAD |
| HDAC10 | 22q13.31-q13.33 | nucleus/cytoplasm | --- | |
| III | SIRT1 | 10q21.3 | nucleus/cytoplasm | p53, β-catenin, Ku70, E2F1, Rb, NF-κB, PGC1α, PPARγ, MyoD, PCAF, FOXO3, HIF1α |
| SIRT2 | 19q13.2 | cytoplasm | α-tubulin, FOXO1 | |
| SIRT3 | 11p15.5 | nucleus/mitochondria | IDH2, SDH, CypD, p53, FOXO3A, MRPL10, GDH, LCAD, Ku70, LKB1, NDUFA9 | |
| SIRT4 | 12q24.31 | mitochondria | IDE, ANT2/3, GDH | |
| SIRT5 | 6p23 | mitochondria | CPS1, Cytochrome C | |
| SIRT6 | 19p13.3 | nucleus | NF-κB, CtBP, DNA PK, PARP1, HIF1α | |
| SIRT7 | 17q25.3 | nucleolus | p53 | |
| IV | HDAC11 | 3p25.2 | nucleus/cytoplasm | --- |
*See text for references.
The function and activity of HDACs vary depending on their structure and intracellular localization. Classical HDACs remove the acetyl groups of lysine residues with the presence of a Zn2+ ion and a conserved deacetylase core domain, producing an unacetylated lysine and acetate. Class I HDACs (including HDAC1, 2, 3 and 8) are usually located in the nucleus and are found to catalyze a set of non-histone substrates including transcription factors besides histones. Class II HDACs (including HDAC4, 5, 6, 7, 9, 10) show different sequence homology and domain organizations compared with class I HDACs and therefore conducting different downstream functions [17]. They are further divided into two subgroups, namely IIa and IIb. Members of subgroup IIa (including HDAC4, 5, 7 and 9) are localized in both nucleus and cytoplasm. A variety of cytoplasmic proteins are regulated by class IIa HDACs such as structural proteins. HDAC6 and 10 are mostly confined to cytoplasm, and HDAC6 contain two catalytic domains while HDAC10 contains one active deacetylase and one incomplete domain with some similarity to the deacetylase domain [18-22]. Class III HDACs are more generally called sirtuins, which are named after the yeast homologous gene 'silent mating-type information regulation 2'. Sirtuins function as lysine deacetylases with the presence of NAD+. The functions of sirtuins have been mainly focused on metabolic and senescent regulation [23]. Previously, four of the seven sirtuins (SIRT4, 5, 6 and 7) have been reported to have very weak or even no detectable deacetylase activity towards histones. For example, SIRT5 has desuccinylase and demalonylase activity other than deacetylase and poly-ADP-ribosylase activity shared by most other sirtuins [24], which have variable functions. SIRT1 interacts with KMT Set7/9 to regulate p53 activity [25], and SIRT2 interacts with and deacetylates FoxO1 to regulate autophagy [26]. Recently, SIRT6 has been demonstrated to catalyze histone H3K9 and H3K56 deacetylation [27, 28]. SIRT7 promotes tumor progression by deacetylation of H3K18 at the promoters of genes related to tumor repression [29], which reflects site specificity for class III HDAC enzymatic activity on histones. SIRT3, a mitochondira-localized sirtuin, is a master regulator of reactive oxidative species (ROS) scavenge and mitochondrial integrity [30]. The regulatory network of sirtuins has been recently reviewed elsewhere [31], and it will not be intensively discussed in this review. Class IV HDAC (HDAC11) contains nine deacetylase motifs shared by both class I and II HDACs [32].
Based on the characterization of HDACs and their unique functions in cancer development, scientists have developed many HDAC inhibitors as a therapeutic strategy to cure cancer. HDAC inhibitors are a group of chemical compounds which reverse the activities of HDACs and are emerging as a class of promising anti-tumor drugs for the treatment of various solid and hematological malignancies [10]. HDAC inhibitors can be categorized into hydroxamic acids, cyclic tetrapeptides, benzamides, aliphatic acids and electrophilic ketones according to their chemical structures (Table 2). These agents exert their inhibitory activities via distinct mechanisms with varying efficiency and specificity. For example, trichostatin A (TSA), the first demonstrated broad-spectrum HDAC inhibitor, functions in a Zn2+-dependent manner [33, 34]. Suberoylanilide hydroxamic acid (SAHA), the first FDA-approved HDAC inhibitor to treat cancer, is a synthetic pan-HDAC inhibitor [35]. Depsipeptide (FK228) shows potent inhibitory activity towards HDAC1 and 2 [36]. Entinostat (MS-275), a benzamide HDAC inhibitor, shows stronger activity towards HDAC1 than HDAC3 and HDAC8 [37]. Butyrate, an aliphatic acid HDAC inhibitor, was found to have anti-tumor activities before HDAC was known to be the target [38]. At least two HDAC inhibitors, SAHA and depsipeptide, have been approved for the treatment of cancer, while many others are under intensive clinical trials [39, 40]. HDAC inhibitors are also implicated in other diseases, such as central nervous system diseases and inflammatory diseases [41, 42].
HDAC inhibitors currently under clinical investigation. Various HDAC inhibitors are classified into several groups according to their structural patterns. While most inhibitors are at different stages of clinical trials, SAHA and depsipeptide have been approved by FDA for cancer chemotherapeutic intervention.
| Group | Compound | HDAC target1 | Current state |
|---|---|---|---|
| Hydroxamic acid | Vorinostat (SAHA, Zolinza) | class I, II, IV | FDA approved |
| Panobinostat (LBH589) | class I, II, IV | phase III CT | |
| Belinostat (PXD101) | class I, II, IV | phase II CT | |
| Abexinostat (PCI24781) | class I, II | phase II CT | |
| Resminostat (RAS2410) | class I, II | phase II CT | |
| Givinostat (ITF2357) | class I, II | phase II CT | |
| Dacinostat (LAQ824, NVP-LAQ824) | class I, II | phase I CT | |
| Pracinostat (SB939) | class I, II | phase II CT | |
| Cyclic tetrapeptide | Romidepsin (Depsipeptide, FK228) | HDAC1, 2 | FDA approved |
| Apicidin | HDAC2, 3 | Phase II CT | |
| Trapoxin A | HDAC1, 4, 11 | ND2 | |
| Benzamide | Mocetinostat (MGCD0103) | HDAC1, 2, 11 | phase II CT |
| Entinostat (MS-275, SNDX-275) | HDAC1, 9, 11 | phase II CT | |
| Rocilinostat (ACY-1215) | HDAC6 | phase II CT | |
| Aliphatic acid | Valproic acid (VPA) | class I | phase III CT |
| Pivanex (AN-9) | ND | phase II CT | |
| Butyrate | class I, IIa | Phase II CT | |
| Electrophilic ketone | Trifluorometchylketone | ND | ND |
*Data partially from www.clinicaltrials.gov, www.cancer.gov. Also see text for references.
1Relatively stronger inhibitory effects and lower IC50 dosage than the unlisted HDACs.
2ND: no data.
Biological functions of HDACs and HDAC inhibitors
HDACs are key modulators of chromatin environment, which is the platform of all chromatin-based processes, such as transcription (Fig.1). Here we provide an insight into the roles of HDACs in regulating cellular properties and the underlying mechanisms based on accumulating evidence.
Biological functions of HDACs. Cellular functions regulated by HDACs, including apoptosis, DNA damage repair, cell cycle control, autophagy, metabolism, ageing and many others which are not listed above.
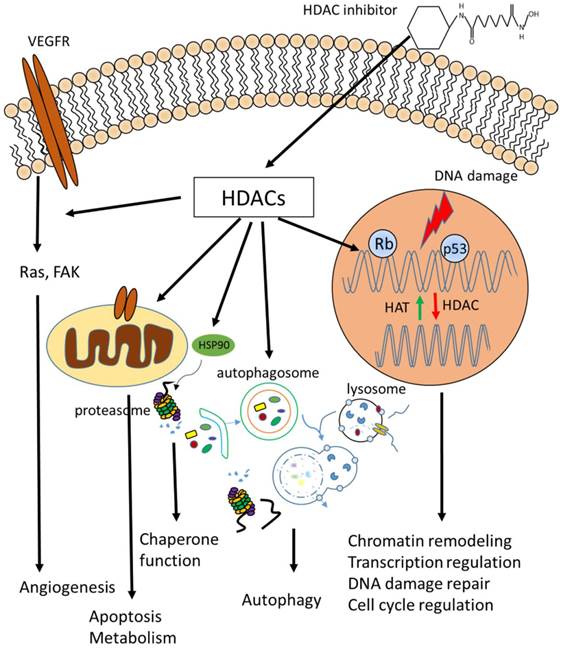
Transcriptional regulation
HDACs have been generally considered to be transcriptional repressive. On the one hand, HDACs stabilize and condense chromosome, making it less available for transcription factors. On the other hand, HDACs act as components of co-repressor complexes. This is convinced by the facts that HDACs are found to cooperate with transcription repressors, and HDAC inhibitors induce expression of certain genes [43, 44]. For example, HDAC1 forms a complex with an adaptor protein RbAp48 and a transcriptional co-repressor mSin3A to mediate gene repression in a deacetylase-dependent manner, and inhibition of HDAC activity blocks this repression [45]. Depsipeptide induces expression of p21 by induction of acetylation of p53 at lysine 373/382 [46]. It is later discovered that depsipeptide actives silenced genes such as p16 and GATA4 by inhibiting CpG and H3K9 methylation on their promoters [47]. HDACs also regulate gene expression by modulating the activity of transcription factors, such as p53 and NF-κB [48-50]. However, accumulated evidences show that HDAC inhibitors can repress certain genes. For example, TSA and sodium butyrate downregulate the expression of Bcl-2 in lymphoma cells [51]. An updated view of HDAC's role in transcriptional regulation is that HDACs function as dynamic transcriptional regulators [52, 53]. Genome-wide screen shows that HDACs are both enriched on the promoters of active and repressive genes [53]. The expression of hundreds of genes are altered when treated with TSA or SAHA using microarray analysis [54]. Although only a small proportion of genes respond to HDAC inhibitor treatment, the role of acetylation and deacetylation as transcriptional regulators should not be underestimated.
Apoptosis
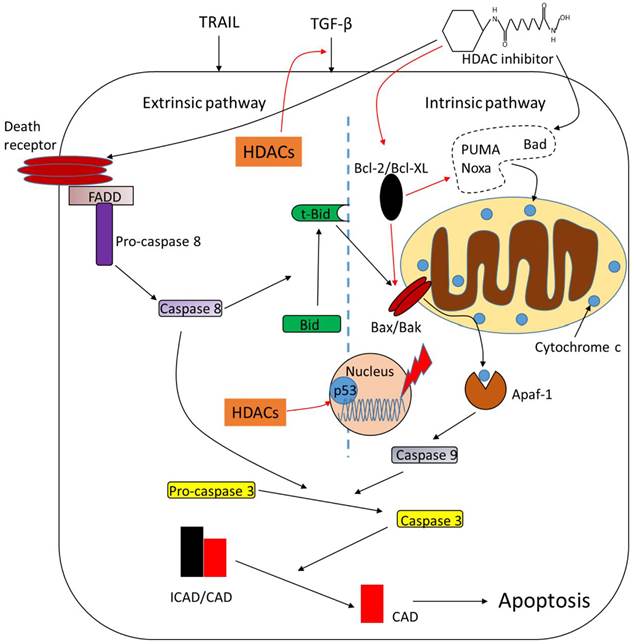
Apoptosis is the mechanism by which cells undergo programmed cell death upon intrinsic or extrinsic stimuli. This process is modulated by plenty of complex proteins including HDACs (Fig.2). Deletion of HDAC1 suppresses transforming growth factor-β1 (TGF-β1)-induced apoptosis and overexpression of HDAC1 enhances it. HDAC2, however, functions as a negative regulator of TGF-β1-induced apoptosis [55]. Targeted deletion of both HDAC1 and HDAC2 leads to increased apoptosis initiated by p53 hyperacetylation [56]. Caspase-dependent cleavage of HDAC3 leads to its accumulation in the cytoplasm and transcriptional activation of its target pro-apoptotic genes [57]. HDAC4 is also a cleavage target of caspase and the caspase-generated fragment of HDAC4 induces release of mitochondrial cytochrome c and apoptosis [58]. Runt-related transcription factor 2 (RUNX2) restrains the pro-apoptotic activity of p53 in association with HDAC6 [59]. HDAC inhibitors are potent inducers of apoptosis and pro- or anti-apoptotic factors are its modulated targets. Butyrate and TSA induce apoptosis with an activation of caspase-3 or upregulation of pro-apoptotic protein Bad in tumor cells [60, 61]. Ku70 is a DNA repair protein which represses apoptosis by sequestering Bax in the cytosol. Treatment of TSA or nicotinamide, a sirtuin inhibitor, causes Ku70 acetylation and its inability to bind and sequester Bax, resulting in apoptosis [62]. Another pro-apoptotic protein, Bak, is also upregulted by butyrate through increased binding of Sp3 [63]. Depsipeptide activates Bim to initiate apoptosis by acetylation of FoxO1 [64]. Anti-apoptotic proteins are downregulated in response to HDAC inhibitors. For example, TSA and butyrate suppress the expression of Bcl-2 [51]. HDAC inhibitors also increase the expression of Fas and Fas ligand (FasL) to induce apoptosis in various tumor cells, resulting in cytochrome c release and activation of caspase-9 and caspase-3 [65, 66]. The pro-apoptotic effects of HDAC inhibitors seem to be p53-independent because no significant difference of apoptotic cell death is observed in cells expressing wild-type or mutant p53 [67, 68]. However, there are also studies showing that HDAC inhibitors induce apoptosis in a p53-dependent way [69, 70], indicating that HDAC inhibitors may function via both pathways, namely p53-dependent and p53-independent pathways to regulate apoptotic process.
Role of HDACs in regulating apoptosis. HDACs function as apoptotic repressors, while treatment of HDAC inhibitors upregulate pro-apoptotic proteins such as Bad and downregulate anti-apoptotic proteins such as Bcl-2. HDAC inhibitors promote tumor cell apoptosis through both intrinsic and extrinsic pathways. *Arrows in black mean 'promote' while the red ones mean 'inhibit', the same goes with all the figures below.
DNA damage repair
DNA damage repair involves chromatin remodeling and employs various factors. HDACs play crucial roles in this process since HDACs are core regulators of chromatin remodeling and the acetylation level of DNA damage-related proteins (Fig.3). For example, ataxia telangiectasia mutated (ATM), an early DNA damage sensor, interacts with HDAC1 and their interplay is increased upon ionizing radiation [71]. Further evidence demonstrates that HDAC1 and HDAC2 are rapidly recruited to DNA-damage sites to promote H3K56 hypoacetylation. Depletion of HDAC1 and HDAC2 renders tumor cells more sensitive to DNA-damaging agents and dampens the ability of DNA double strand break repair [72]. HDAC3 has also been implicated in DNA damage repair [73, 74]. Deletion of HDAC3 significantly reduces DNA damage repair ability [74]. HDAC4 is recruited to DNA damage-induced foci and colocalizes with homologous recombination (HR) repair protein 53BP1 following DNA damage [75]. Inhibition of HDAC6 with isoform-specific inhibitor promotes cell death by induction of DNA damage [76]. Knockdown or inhibition of HDAC9 and HDAC10 shows impaired HR repair capacity [77]. Sirtuins are also related to DNA damage repair. SIRT1 interacts and deacetylates several DNA proteins, such as Ku70, NBS1, APE1 and XPA [78-81], reflecting a role of SIRT1 in non-homologous end joining (NHEJ) and HR repair following double strand break damage, and base excision repair and nucleotide excision repair pathway following single-strand DNA damage. Recent studies show that SIRT1 is a chromatin environment regulator at promoters of the housekeeping genes investigated, and inhibition of SIRT1 restores the transcription of genes repressed [82], suggesting an active role of SIRT1 in genomic stability regulation. SIRT6 promotes DNA end resection through deacetylation of C-terminal binding protein-interacting protein (CtIP), while depletion of SIRT6 impairs recruitment of repair proteins at DNA damage sites, leading to reduced HR repair [83]. Later studies show that SIRT6 is recruited to DNA damage sites and stimulates NHEJ and HR repair through mono-ADP-ribosylation of PARP1 on Lys 521, thereby activating PARP1 and enhancing DNA damage repair [84]. SIRT3 is found to translocate to mitochondria from nucleus upon cellular stress, deacetylating and activating Idh2, which is a key regulator of intermediary metabolism and energy production. This leads to an increase in NADPH level and ratio of reduced-to-oxidized glutathione, thus protecting cells from oxidative damage [85, 86].
Role of HDACs in regulating DNA damage repair. HDACs interact with DNA damage responsive factors and promote DNA damage repair. HDAC inhibitors induce DNA damage through generation of ROS or suppression of DNA repair proteins.
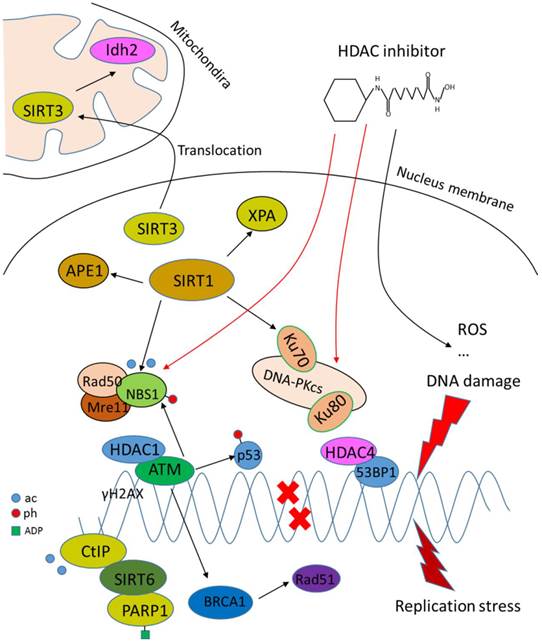
While HDACs are generally shown to potentiate DNA damage repair capacity, inhibitors that target HDACs are potent inducers of DNA damage in transformed cells. For example, depsipeptide induces DNA damage through generation of ROS [87]. LBH589, a novel broad-spectrum HDAC inhibitor, induces the expression of DNA damage repair genes including FANCG, FoxO3A and GADD45 [88]. HDAC inhibitor PCI-24781 suppresses DNA damage repair by decreasing RAD51 [89]. SAHA induces DNA damage by inhibiting DNA damage proteins such as RAD50 and MRE11 in cancer cells [90]. In a word, HDACs facilitate the DNA damage repair process by either loosening the chromatin for other factors to function or recruiting DNA repair proteins to damage sites, and HDAC inhibitors are thus considered to target HDACs to induce DNA damage.
Cell cycle control
Cell cycle progression involves a series of events that lead to replication and division of cellular contents. Duplication of nuclear materials and information needs overall chromatin reconstruction, in which HDACs are key regulators. Other cell cycle regulators, such as cyclins and CDKs, are also regulated by HDACs. Genomewide profiling in yeast reveals that HDACs function as regulators of genes involved in cell cycle [91]. The cell cycle transcription factor E2F plays a major role during G1/S transition. Retiboblastoma protein (Rb) interacts with HDAC1 to repress E2F-mediated transcription of cell cycle proteins such as cyclin E, while TSA treatment abrogates this repression [92-94]. However, later studies shows that the cell cycle inhibitory function of Rb is not necessarily dependent on the activity of HDACs [95, 96]. TSA fails to restore cyclin A and it has no significant effect on cell cycle distribution [95], indicating that HDACs may employ other mechanisms instead of Rb-mediated transcriptional repression to influence cell cycle. HDAC1 knockdown can arrest cell cycle at either G1 phase or G2/M transition [97]. Combined genetic inactivation of HDAC1 and HDAC2 causes a senescence-like G1 arrest in a p53-independent manner [98]. HDAC3 is later proved to a master regulator of mitosis. HDAC3-dependent deacetylation of histone H3 creates a hypoacetylated environment for kinase Aurora B [99]. Phosphorylation of H3S10 by Aurora B is essential for the onset of mitosis. LBH589 induces G2/M arrest through degradation of Aurora A and B [100]. HDAC3 knockdown results in spindle assembly checkpoint activation and sister chromatid dissociation, which may be related to centromeric H3K4 acetylation and loss of dimethylation at the same site [101]. Collapsed mitotic spindle is also observed in HDAC3 knockdown cells and TSA treatment results in similar defects [102]. HDAC inhibition by trapoxin arrests cell cycle at G1 and G2 phase by increasing the transcription of cyclin E [103]. TSA induces G0/G1 arrest in human liver cancer cells [104]. TSA also induces delay of G2/M transition in a transcription-dependent way [105]. Inhibition of HDAC activity can also lead to dysregulation of mitotic checkpoint activation [106]. In summary, HDACs are important elements in cell cycle regulatory machinery and HDAC inhibitors alter cell cycle progress by interacting with cell cycle regulators, resulting in cell cycle arrest at certain phases and detention of proliferation.
Autophagy
Autophagy is the process of self-degradation of unnecessary or dysfunctional cellular components through the lysosomal machinery, which ensures cell survival during starvation [107]. Autophagy has a close relationship with cancer and its role in cancer development is still under hot debate [108, 109]. It has gradually come to a common understanding that autophagy is regulated by acetylation. SAHA and butyrate can induce caspase-independent autophagic cell death [110]. SAHA activates autophagy through inactivation of mammalian target of rapamycin (mTOR) [111]. Genetic knockdown or inhibition of HDAC1 significantly induces autophagy [112]. Later in Huntington's disease model, it is reported that mutant huntington protein (Htt) is deacetylated by HDAC1 and inhibition of HDAC1 facilitates mutant Htt clearance through induction of autophagy [113]. HDAC inhibition-induced autophagy is also related to DNA damage repair [11]. However, some other studies come to an opposite conclusion. HDAC6, a microtubule-associated deacetylase, is also involved in autophagy. HDAC6 provides a link between autophagy and ubiquitin-proteasome system, and expression of HDAC6 is sufficient to induce autophagy [114]. It is later confirmed that HDAC6 is not required for autophagy activation but the fusion of autophagosomes to lysosomes [115]. It is recently found out that HDAC10 promotes autophagy-mediated survival, while inhibition of HDAC10 disrupts autophagy associated with increased sensitization to chemotherapeutic drugs in cancer cells [116]. These data indicates that HDACs and HDAC inhibitors may have pleiotropic roles in the regulation of autophagy. Sirtuins also participate in regulating autophagy. Acetylation of FoxO1 by dissociation from SIRT2 is essential for the induction of autophagy [26, 117]. SIRT1 forms a complex with critical regulators of the autophagy machinery, such as autophagy genes (Atg)5, Atg7 and Atg8 [118]. The role of HDACs in either promoting or restraining autophagy are still under investigation, and evidence supporting both sides is accumulated. The therapeutic effects of HDAC inhibitors may be explained by the dual role of autophagy in disease progress.
Metabolism and senescence
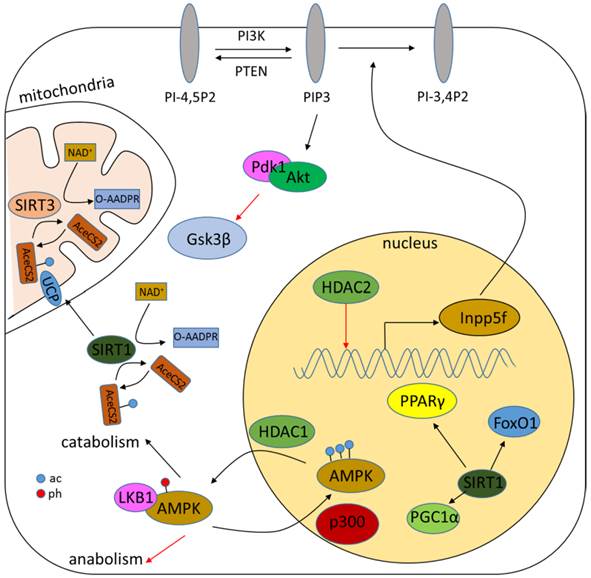
Acetylation of either histones or non-histone proteins has been demonstrated to be tightly correlated with metabolism (Fig.4) [119, 120]. The role of sirtuins in metabolism has been well characterized and reviewed elsewhere [23, 121, 122]. Classical HDACs, however, are also related to metabolism regulation. AMP-activated protein kinase (AMPK), a critical sensor and regulator of metabolism, is demonstrated to be regulated by HDAC1. HDAC1 interacts and deacetylates AMPK to increase its interaction with liver kinase B1 (LKB1), resulting in AMPK phosphorylation and activation [123]. HDAC2 transgenic mice shows increased hypertrophy associated with glycogen synthase kinase 3β (Gsk3β) inactivation, while chemical inhibition of Gsk3β activity renders HDAC2-deficient cells more sensitive to hypertrophic stimuli, suggesting a role cof HDAC2 in metabolism [124]. Genetic deletion of HDAC3 is associated with alteration of genes involved in fatty acid metabolism, glucose utilization and oxidative phosphorylation possibly through the activation of peroxisome proliferator-activated receptor-γ (PPARγ) or mTOR signaling [125-127].
Role of HDACs in regulating metabolism. HDACs regulate metabolism mainly through modulation of the acetylation level of metabolic proteins such as AMPK. Sirtuins can also regulate the NAD+/NADH ratio.
Searching of the GenAge Database reveals an amount of 288 genes analyzed for the possible association with human longevity, including HDAC1, 2, 3 and SIRT1, 3, 6 and 7. Researches show that deletion of Rpd3, homologous to class I HDAC, leads to life-span extension in yeast and calorie restriction in Drosophila, while deletion of Hda1, homologous to class II HDAC, does not [128, 129]. Butyrate and TSA treatment reduce life span of human diploid fibroblasts [130]. Whether class I HDAC is involved in human ageing, however, is still unknown. Sirtuins have been demonstrated to be a master regulator in senescence [122, 131, 132]. Sir2 mutant impairs life span extension, while an increased level of Sir2 gives rise to it in S. cerevisiae [133]. Moderate overexpression of SIRT1 suppresses the expression of senescence markers and retards ageing [134]. Increased SIRT1 level also attenuates age-dependent transcriptional changes [134, 135]. Sirtinol, a SIRT1-specific inhibitor, augments the activity of senescence-associated β-galactosidase to induce senescence-like growth arrest [136]. SIRT6-deficient mice show age-related degenerative phenotypes [137]. Other studies reveal a possible role of SIRT3 and SIRT7 in the regulation of senescence [29, 30]. The regulatory role of HDACs are largely explained by their interaction with other regulators of metabolism and senescence, while HDAC inhibitors impede this interaction.
Chaperone function
The molecular chaperone heat shock protein 90 (HSP90) facilitates structural maturation of its client proteins, which is critical for their functions and activities. HDAC6 deacetylates HSP90, while inactivation of HDAC6 results in HSP90 hyperacetylation and loss of activity in tumor cell lines [138, 139]. Inhibition or silencing of HDAC6 and HDAC10 reduces the binding of HSP90 to vascular endothelial growth factor receptor (VEGFR)1 and VEGFR2 with an increasing binding of HSP70, leading to a reduction of VEGFR1 and VEGFR2 in a proteasome-dependent pathway [140]. Depsipeptide inactivates HSP90 and disassociate RUNX1-ETO fusion oncoprotein with HSP90 to induce its proteasomal degradation [141]. The binding of mutant p53 and acetylated HSP90 is also reduced when treated with depsipeptide, leading to accelerated p53 depletion [67]. LBH589 promotes the extracellular export of HSP90α in a deacetylation-dependent manner. Extracellular HSP90α interacts with matrix metalloproteinase-2 (MMP-2) and promotes tumor cell invasion [142]. MS-275 induces HSP90 acetylation and blocks its interaction with fms-like tyrosine kinase 3 (FLT3) in leukemia cells, resulting in ubiquitination and subsequent proteasomal degradation of FLT3 [143]. Another HSP family chaperone, HSP70, is also modulated by HDAC inhibitors. Valproic acid (VPA) induces HSP70 through promoter hyperacetylation [144]. Chaperone function is critical in the stability of numerous proteins, including some important oncoproteins and tumor suppressors. HDAC inhibitors inactivate and interfere the binding of HSP90 to these client proteins, leading to their instability and degradation.
Angiogenesis
Angiogenesis is a fundamental step in the metastasis of tumors to provide nutrients for cancer overgrowth. Overexpression of HDAC1 stimulates angiogenesis, while HDAC inhibitors block angiogenesis by downregulating angiogenesis-stimulating factor hypoxia inducible factor 1α (HIF-1α) and vascular endothelial growth [145, 146]. HDAC4 and HDAC6 interact with HIF-1α directly. Inhibition or knockdown of HDAC4 and HDAC6 reduces HIF-1α level [147]. TSA and SAHA potently inhibit VEGF-induced angiogenesis by repressing VEGF-induced expression of VEGFRs or upregulating VEGF competitor [148]. Nitric oxide (NO) is a key second messenger in angiogenesis signaling. TSA also reduces NO level through downregulation of endothelial nitric oxide synthase (eNOS) [149]. HDAC7 forms a complex with HIF-1α and translocates to the nucleus to enhance the transcriptional activity of HIF-1α upon hypoxia insults, resulting in an increased level of HIF-1α target genes, including VEGF [150]. Another study shows cardiovascular abnormalities in HDAC7 mutant mice and HDAC7 represses the expression of MMP10 by interacting and repressing the activity of MEF2 [151]. Genetic silencing of HDAC7 decreases endothelial cell migration and alters the formation of capillary-like structures partly by induction of platelet-derived growth factor-B (PDGF-B) and its β receptor [152]. Depsipeptide also inhibits tumor neovascularization, possibly by suppressing pro-angiogenic factors such as EGFR or inducing anti-angiogenic factors such as von Hippel Lindau [153]. LBH589 reduces angiogenesis through inhibition of endothelial tube formation and the expression of VEGF-signaling factors, such as angiopoietin-2, survivin and CXCR4 [154]. In general, HDAC inhibitors repress neovascularization by inhibiting positive factors of angiogenesis or altering angiogenesis signaling pathway.
Other functions
In addition to the mechanisms listed above that may lead to cancer cell lethality, other pathways also contribute to the anti-tumor capacities of HDAC inhibitors. Accumulation of ROS in malignant cells is an important mechanism of HDAC inhibitor-mediated cell death [155, 156]. HDAC inhibitors alter the expression of ROS-modulated proteins such as thioredoxin-binding protein 2 and thioredoxin, the latter of which is a principal antioxidant scavenger of ROS [157]. HDAC6 is a regulatory component of aggresome, which is an intracellular storage unit of misfolded proteins [158]. Targeted inhibition of HDAC6 leads to repression of the aggresome pathway and causes autophagic cell death in tumor cells [159]. HDACs function as regulators of cytoskeletal proteins, such as tubulin and actin to modulate cell motility and migration. HDAC6 interacts and deacetylates tubulin [160, 161]. Overexpression of HDAC6 promotes cell mobility, but deletion of HDAC6 has no effect on normal development and disease progression in mice model [162, 163]. Inhibition of HDAC activity by TSA suppresses TGF-β1-induced renal tubular epithelial-to-mesenchymal transition (EMT) through upregulation of E-cadherin and downregulation of collagen type I [164], suggesting a role of HDACs in the regulation of EMT process, which is critical in the development of various diseases including cancer. Later studies confirm the role of HDACs in EMT regulation of tumor cells [165, 166]. The function spectrum of HDACs and HDAC inhibitors are rapidly expanding. In-depth studies are proceeding to explore functions and activities beyond our knowledge. Anyway, treatment of HDAC inhibitors results in an anti-tumor profile.
Clinical implications of HDAC inhibitors in cancer treatment
HDAC inhibitors are under intensive investigation for cancer therapy. Tens of structurally disparate HDAC inhibitors are under different stages of clinical trials while some of them have shown promising effects for various cancers, especially in hematological malignancies (Table 2). Depsipeptide, first identified as a natural produrg, induces a complete or partial clinical response in cutaneous T cell lymphoma (CTCL) patients [167]. SAHA can induce growth arrest and kill various types of cancer cells with little cytotoxicity to normal cells [168]. Both depsipeptide and SAHA have now been approved to manage advanced CTCL [40]. LBH589, another potent hydroxamic acid-based HDAC inhibitor, shows clinical effects in CTCL patients with a rapid change of the expression of genes involved in apoptosis, immune regulation and angiogenesis [169]. HDAC inhibitors have also been implicated in the treatment of solid tumors such as VPA [170]. VPA used as a single agent or in combination with other cytotoxic drugs may have positive effects across several different types of cancer including lung, breast, pancreas and prostate cancer [170, 171].
A major barrier for the development of HDAC inhibitors is that most of them have side effects because of their cytotoxicity to normal host cells although they are much more favorable than many other traditional cancer chemotherapeutics [40]. Common adverse effects including nausea, vomiting, anorexia, and fatigue are mostly manageable. However, some specific HDAC inhibitors may cause serious adverse events. For example, depsipeptide has been proved to be associated with life-threatening cardiac arrhythmia in cancer patients [172]. The different cytotoxicity profiles of HDAC inhibitors compared to conventional chemotherapeutic agents have led to the abortion of a large number of clinical trials [39]. This arouses considerable doubts about the clinical safety of HDAC inhibitors. Combination with other therapies or development of more specific inhibitors may be a way out as many combination trials are being pursued [173]. Second-generation HDAC inhibitors with more specific inhibitory selectivity and greater potency are under clinical development. Although HDAC inhibitors have shown potent anti-tumor activities when used alone, experimental studies and clinical trials support the synergistic combination of HDAC inhibitors and other anti-cancer therapies [173]. For example, doxorubicin (DAC), a DNA demethylating drug and cancer chemotherapeutic agent, enhances HDAC inhibitor-induced tumor cell apoptosis [68]. Later studies demonstrate that DAC also cooperates with depsipeptide to inhibit tumor cell proliferation in a methylation-independent way [174].
Another issue concerning cancer treatment is resistance to HDAC inhibitors. The underlying mechanisms are not well elucidated and a better understanding will surely improve their clinical efficacy [175]. Previous studies have proposed the mechanisms of drug efflux, altered expression and mutations of HDACs, protection from oxidative stress by antioxidants and altered expression of apoptosis-related proteins as determinants of resistance [175].
Concluding remarks and perspective
Over the past few decades, the growing understanding of epigenetics has led to a rapid expansion of our knowledge on its role in cancer. The execution of HDAC functions depends largely on the modifications of non-histone substrates although it has been widely accepted that histone modifications play an important role in this process. However, this also raises the question whether the modification of non-histone proteins is epigenetic or not. Epigenetics has been defined as the study of inheritable phenotypic changes that do not involve the alteration of DNA sequences. While protein is the effector of the genetic information flow, its role in epigenetic regulation cannot be ignored. By this token, the modifications of non-histone proteins can be at least considered as part of the epigenetic regulatory network. The contrary roles of HATs and HDACs in regulating the acetylation levels of histones and non-histone proteins make it possible that acetyl groups can be added or erased whenever necessary. The consistence of epigenome including acetylation patterns indicates that the acetylation of non-histone proteins may be inheritable along generations. This motivates scientists to find and characterize the determinants of the transmission of acetylation patterns. The roles of HDACs, especially sirtuins, in regulating non-histone proteins have arouse intense interests and should be explored more deeply. Further elucidation of this network would provide insights into the mechanisms of a great number of cellular processes.
The established network of HDACs in the regulation of cellular behaviors under physiological conditions or upon external insults makes HDACs critical modulators of disorders such as cancer and inflammatory diseases. The development of HDAC inhibitors as anti-tumor drugs opens a completely new window for cancer therapeutics. While the underlying mechanisms are under intensive investigation, there are quite a few questions that need to be answered regarding to the clinical application of HDAC inhibitors. How come that HDAC inhibitors specifically target tumor cells, while normal non-malignant cells remained largely unaltered? Although there has been some clues to explain this selectivity, further research is still required to elucidate this issue. Meanwhile, the specificity of HDAC inhibitors in cancer therapy should also be questioned. Clinical responses diversify between different HDAC inhibitors which target the same HDACs. To address this problem, more isoform-specific HDAC inhibitors may be needed. Along with the selectivity and specificity is the resistance to HDAC inhibitors. How and why does the tumor cells respond to HDAC inhibitors differently remain poorly understood, and the mechanistic basis of the resistance needs to be illuminated for the development of better HDAC inhibitors.
Epigenetic therapy has emerged as a hot issue in cancer research. Inhibitors of deregulated chromatin modifiers are now under different stages of investigation [1]. Inhibitors of HDACs, DNA-methyltransferases (DNMTs), and JAK2 have shown great therapeutic benefits while a large number of epigenetic drugs are in development. Although there is still a long way to go in fighting cancer, epigenetic therapy may provide a bright future for us to follow.
Acknowledgements
We sincerely apologize for unable to include many other valuable papers in this review due to the space limitation. The studies mentioned in this review from the author's group have been supported by grants from National Natural Science Foundation of China(No.81321003, No.91319302 and No.31070691), Ministry of Science and Technology of China(2011CB504200), Ministry of Education of China (111 project) and Peking University-Tsinghua University Center for Life Science. We also sincerely appreciate the helpful advices provided by other members of our group during the writing of this review.
Abbreviation
AMPK, AMP-activated protein kinase; EMT, epithelial-to-mesenchymal transition; eNOS, endothelial nitric oxide synthase; FK228, depsipeptide; FoxO, forkhead box protein O; HAT, histone acetyltransferase; HDAC, histone deacetylase; HIF-1α, hypoxia inducible factor-1α; HR, homologous recombination; HSP, heat shock protein; KMT, histone methyltransferase; KDM, histone demethylase; MS-275, entinostat; NAD+, nicotinamide adenine dinucleotide; NF-κB, nuclear factor-κB; NHEJ, non-homologous end joining; NO, nitric oxide; Rb, retinoblastoma protein; ROS, reactive oxygen species; SIRT, sirtuin; TSA, trichostatin A; SAHA, suberoylanilide hydroxamic acid; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; VPA, valproic acid
Competing Interests
No competing interest exists.
References
1. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12-27
2. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41-45
3. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693-705
4. Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NT. et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407-411
5. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC. et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120-124
6. Pogo B, Allfrey V, Mirsky A. RNA synthesis and histone acetylation during the course of gene activation in lymphocytes. Proc Natl Acad Sci U S A. 1966;55:805
7. Yang X-J, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143-153
8. Xu W, Parmigiani R, Marks P. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541-5552
9. Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194-202
10. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38-51
11. Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R. et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74-9
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
13. Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168-176
14. Bosch-Presegué L, Vaquero A. The dual role of sirtuins in cancer. Genes Cancer. 2011;2:648-662
15. De Ruijter A, Van Gennip A, Caron H, Kemp S, van Kuilenburg A. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737-749
16. Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417-435
17. Yang X-J, Grégoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol. 2005;25:2873-2884
18. Guardiola AR, Yao T-P. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J Biol Chem. 2002;277:3350-3356
19. Tong JJ, Liu J, Bertos NR, Yang X-J. Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 2002;30:1114-1123
20. Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A. et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455-458
21. Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96:4868-4873
22. Kao H-Y, Lee C-H, Komarov A, Han CC, Evans RM. Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J Biol Chem. 2002;277:187-193
23. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
24. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806-9
25. Liu X, Wang D, Zhao Y, Tu B, Zheng Z, Wang L. et al. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc Natl Acad Sci U S A. 2011;108:1925-1930
26. Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S. et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665-75
27. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M. et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492-496
28. Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009;8:2662-2663
29. Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z. et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487:114-118
30. Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD. et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41-52
31. Yao Y, Yang Y, Zhu WG. Sirtuins: nodes connecting aging, metabolism and tumorigenesis. Curr Pharm Des. 2013 In press
32. Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002;277:25748-55
33. Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174-17179
34. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188-193
35. Grant S, Easley C, Kirkpatrick P. Vorinostat. Nat Rev Drug Discov. 2007;6:21-22
36. Furumai R, Matsuyama A, Kobashi N, Lee K-H, Nishiyama M, Nakajima H. et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916-4921
37. Hu E, Dul E, Sung C-M, Chen Z, Kirkpatrick R, Zhang G-F. et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720-728
38. Riggs M, Neumann RWJ, Ingram V. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462-4
39. Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenet. 2010;1:117-136
40. Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:1-13
41. Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854-868
42. Halili MA, Andrews MR, Sweet MJ, Fairlie DP. Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem. 2009;9:309-319
43. Hassig CA, Schreiber SL. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr Opin Chem Biol. 1997;1:300-308
44. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349-52
45. Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG, Ayer DE. et al. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc Natl Acad Sci U S A. 1998;95:3519-3524
46. Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y. et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol Cell Biol. 2006;26:2782-90
47. Wu LP, Wang X, Li L, Zhao Y, Lu S, Yu Y. et al. Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Mol Cell Biol. 2008;28:3219-35
48. Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377-81
49. Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS. et al. Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem. 2000;275:20436-43
50. Ashburner BP, Westerheide SD, Baldwin AS Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065-77
51. Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25:1608-19
52. Nusinzon I, Horvath CM. Histone deacetylases as transcriptional activators? role reversal in inducible gene regulation. Sci STKE. 2005;2005:re11
53. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W. et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019-1031
54. Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA. et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697-3702
55. Lei WW, Zhang KH, Pan XC, Wang DM, Hu Y, Yang YN. et al. Histone deacetylase 1 and 2 differentially regulate apoptosis by opposing effects on extracellular signal-regulated kinase 1/2. Cell Death Dis. 2010;1:e44
56. Ma P, Pan H, Montgomery RL, Olson EN, Schultz RM. Compensatory functions of histone deacetylase 1 (HDAC1) and HDAC2 regulate transcription and apoptosis during mouse oocyte development. Proc Natl Acad Sci U S A. 2012;109:E481-E489
57. Escaffit F, Vaute O, Chevillard-Briet M, Segui B, Takami Y, Nakayama T. et al. Cleavage and cytoplasmic relocalization of histone deacetylase 3 are important for apoptosis progression. Mol Cell Biol. 2007;27:554-67
58. Paroni G, Mizzau M, Henderson C, Del Sal G, Schneider C, Brancolini C. Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol Biol Cell. 2004;15:2804-18
59. Ozaki T, Wu D, Sugimoto H, Nagase H, Nakagawara A. Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis. 2013;4:e610
60. Medina V, Edmonds B, Young GP, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57:3697-3707
61. Sawa H, Murakami H, Ohshima Y, Sugino T, Nakajyo T, Kisanuki T. et al. Histone deacetylase inhibitors such as sodium butyrate and trichostatin A induce apoptosis through an increase of the bcl-2-related protein Bad. Brain Tumor Pathol. 2001;18:109-14
62. Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C. et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627-38
63. Chirakkal H, Leech SH, Brookes KE, Prais AL, Waby JS, Corfe BM. Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene. 2006;25:7192-200
64. Yang Y, Zhao Y, Liao W, Yang J, Wu L, Zheng Z. et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11:313-24
65. Glick RD, Swendeman SL, Coffey DC, Rifkind RA, Marks PA, Richon VM. et al. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 1999;59:4392-4399
66. Kwon SH, Ahn SH, Kim YK, Bae G-U, Yoon JW, Hong S. et al. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J Biol Chem. 2002;277:2073-2080
67. Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA. et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94:504-513
68. Zhu WG, Lakshmanan RR, Beal MD, Otterson GA. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2001;61:1327-33
69. Bandyopadhyay D, Mishra A, Medrano EE. Overexpression of histone deacetylase 1 confers resistance to sodium butyrate-mediated apoptosis in melanoma cells through a p53-mediated pathway. Cancer Res. 2004;64:7706-10
70. McCormack E, Haaland I, Venas G, Forthun RB, Huseby S, Gausdal G. et al. Synergistic induction of p53 mediated apoptosis by valproic acid and nutlin-3 in acute myeloid leukemia. Leukemia. 2012;26:910-7
71. Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem. 1999;274:31127-30
72. Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S. et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144-51
73. Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW. et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61-72
74. Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S. et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436-47
75. Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003;160:1017-27
76. Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A. 2010;107:20003-8
77. Kotian S, Liyanarachchi S, Zelent A, Parvin JD. Histone deacetylases 9 and 10 are required for homologous recombination. J Biol Chem. 2011;286:7722-6
78. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B. et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390-2
79. Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149-62
80. Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K. et al. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010;38:832-45
81. Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. 2010;39:247-58
82. Velez-Cruz R, Zadorin AS, Coin F, Egly JM. Sirt1 suppresses RNA synthesis after UV irradiation in combined xeroderma pigmentosum group D/Cockayne syndrome (XP-D/CS) cells. Proc Natl Acad Sci U S A. 2013;110:E212-20
83. Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348-53
84. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A. et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443-6
85. Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21:920-8
86. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C. et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802-12
87. Wang H, Zhou W, Zheng Z, Zhang P, Tu B, He Q. et al. The HDAC inhibitor depsipeptide transactivates the p53/p21 pathway by inducing DNA damage. DNA Repair. 2012;11:146-56
88. Scuto A, Kirschbaum M, Kowolik C, Kretzner L, Juhasz A, Atadja P. et al. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 2008;111:5093-100
89. Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104:19482-19487
90. Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010;107:14639-44
91. Bernstein BE, Tong JK, Schreiber SL. Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci U S A. 2000;97:13708-13
92. Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597-601
93. Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP. et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601-5
94. Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463-73
95. Siddiqui H, Solomon DA, Gunawardena RW, Wang Y, Knudsen ES. Histone deacetylation of RB-responsive promoters: requisite for specific gene repression but dispensable for cell cycle inhibition. Mol Cell Biol. 2003;23:7719-31
96. Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX. et al. Exit from G1 and S phase of the cell cycle Is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79-89
97. Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A. et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27:4784-4795
98. Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J. et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29:2586-2597
99. Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J. et al. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006;20:2566-2579
100. Cha T-L, Chuang M-J, Wu S-T, Sun G-H, Chang S-Y, Yu D-S. et al. Dual degradation of Aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res. 2009;15:840-850
101. Eot-Houllier G, Fulcrand G, Watanabe Y, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase 3 is required for centromeric H3K4 deacetylation and sister chromatid cohesion. Genes Dev. 2008;22:2639-2644
102. Ishii S, Kurasawa Y, Wong J, Yu-Lee L-y. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci U S A. 2008;105:4179-4184
103. Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N. et al. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274:34940-34947
104. Yamashita Y-i, Shimada M, Harimoto N, Rikimaru T, Shirabe K, Tanaka S. et al. Histone deacetylase inhibitor trichostatin a induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int J Cancer. 2003;103:572-576
105. Noh EJ, Lim D-S, Jeong G, Lee J-S. An HDAC inhibitor, trichostatin A, induces a delay at G2/M transition, slippage of spindle checkpoint, and cell death in a transcription-dependent manner. Biochem Biophys Res Commun. 2009;378:326-331
106. Shin HJ, Baek KH, Jeon AH, Kim SJ, Jang KL, Sung YC. et al. Inhibition of histone deacetylase activity increases chromosomal instability by the aberrant regulation of mitotic checkpoint activation. Oncogene. 2003;22:3853-8
107. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67-93
108. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961-967
109. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308-5316
110. Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101:18030-18035
111. Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A. 2012;109:6561-5
112. Oh M, Choi IK, Kwon HJ. Inhibition of histone deacetylase1 induces autophagy. Biochem Biophys Res Commun. 2008;369:1179-83
113. Jeong H, Then F, Melia Jr TJ, Mazzulli JR, Cui L, Savas JN. et al. Acetylation targets mutant Huntingtin to autophagosomes for degradation. Cell. 2009;137:60-72
114. Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB. et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:860-864
115. Lee J-Y, Koga H, Kawaguchi Y, Tang W, Wong E, Gao Y-S. et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969-980
116. Oehme I, Linke J-P, Böck BC, Milde T, Lodrini M, Hartenstein B. et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A. 2013;110:E2592-E2601
117. Zhao Y, Wang L, Yang J, Zhang P, Ma K, Zhou J. et al. Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death. Autophagy. 2010;6:988-990
118. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374-3379
119. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076-80
120. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T. et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000-4
121. Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104-12
122. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225-38
123. Lin YY, Kiihl S, Suhail Y, Liu SY, Chou YH, Kuang Z. et al. Functional dissection of lysine deacetylases reveals that HDAC1 and p300 regulate AMPK. Nature. 2012;482:251-5
124. Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T. et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324-31
125. Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB. et al. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002;3:903-10
126. Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM. et al. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588-97
127. Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008;27:1017-28
128. Kim S, Benguria A, Lai CY, Jazwinski SM. Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:3125-36
129. Rogina B, Helfand SL, Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science. 2002;298:1745
130. Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol. 1996;16:5210-5218
131. Haigis MC, Guarente LP. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913-2921
132. Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257-268
133. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570-80
134. Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X. et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512-1521
135. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK. et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907-918
136. Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M. et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2005;25:176-185
137. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L. et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315-329
138. Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV. et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601-7
139. Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F. et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729-34
140. Park J-H, Kim S-H, Choi M-C, Lee J, Oh D-Y, Im S-A. et al. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun. 2008;368:318-322
141. Yang G, Thompson M, Brandt S, Hiebert S. Histone deacetylase inhibitors induce the degradation of the t (8; 21) fusion oncoprotein. Oncogene. 2006;26:91-101
142. Yang Y, Rao R, Shen J, Tang Y, Fiskus W, Nechtman J. et al. Role of acetylation and extracellular location of heat shock protein 90α in tumor cell invasion. Cancer Res. 2008;68:4833-4842
143. Nishioka C, Ikezoe T, Yang J, Takeuchi S, Phillip Koeffler H, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leuk Res. 2008;32:1382-1392
144. Marinova Z, Ren M, Wendland JR, Leng Y, Liang M-H, Yasuda S. et al. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem. 2009;111:976-987
145. Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW. et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437-43
146. Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019-28
147. Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P. et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66:8814-21
148. Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N. et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21:427-436
149. Rössig L, Li H, Fisslthaler B, Urbich C, Fleming I, Förstermann U. et al. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837-844
150. Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1α and increases transcriptional activity. J Biol Chem. 2004;279:41966-41974
151. Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126:321-34
152. Mottet D, Bellahcène A, Pirotte S, Waltregny D, Deroanne C, Lamour V. et al. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101:1237-1246
153. Kwon HJ, Kim MS, Kim MJ, Nakajima H, Kim KW. Histone deacetylase inhibitor FK228 inhibits tumor angiogenesis. Int J Cancer. 2001;97:290-296
154. Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HMW, Salumbides B. et al. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634-642
155. Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R. et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001;98:10833-8
156. Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1. Cancer Res. 2003;63:3637-45
157. Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA. et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A. 2002;99:11700-5
158. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727-38
159. Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. Role of the aggresome pathway in cancer: Targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008;68:2557-2560
160. Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A. et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455-8
161. Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S. et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168-79
162. Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M. et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688-701
163. Bobrowska A, Paganetti P, Matthias P, Bates GP. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington's disease. PLoS One. 2011;6:e20696
164. Yoshikawa M, Hishikawa K, Marumo T, Fujita T. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-β1 in human renal epithelial cells. J Am Soc Nephrol. 2007;18:58-65
165. Nagathihalli NS, Massion PP, Gonzalez AL, Lu P, Datta PK. Smoking induces epithelial-to-mesenchymal transition in non-small cell lung cancer through HDAC-mediated downregulation of E-cadherin. Mol Cancer Ther. 2012;11:2362-72
166. Roy SS, Gonugunta VK, Bandyopadhyay A, Rao MK, Goodall GJ, Sun LZ. et al. Significance of PELP1/HDAC2/miR-200 regulatory network in EMT and metastasis of breast cancer. Oncogene. 2013 In press
167. Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L. et al. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood. 2001;98:2865-2868
168. Marks P. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351-1356
169. Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R. et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14:4500-4510
170. Munster P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R. et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol. 2007;25:1979-85
171. Atmaca A, Al-Batran S, Maurer A, Neumann A, Heinzel T, Hentsch B. et al. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer. 2007;97:177-182
172. Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M. et al. Cardiotoxicity of Histone Deacetylase Inhibitor Depsipeptide in Patients with Metastatic Neuroendocrine Tumors. Clin Cancer Res. 2006;12:3997-4003
173. Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res. 2009;15:3970-7
174. Chai G, Li L, Zhou W, Wu L, Zhao Y, Wang D. et al. HDAC inhibitors act with 5-aza-2′-deoxycytidine to inhibit cell proliferation by suppressing removal of incorporated abases in lung cancer cells. PLoS One. 2008;3:e2445
175. Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res. 2007;13:7237-42
Author contact
![]() Corresponding author: Wei-Guo Zhu, Key Laboratory of Carcinogenesis and Translational Research, Department of Biochemistry and Molecular Biology, Peking-Tsinghua University Center for Life Sciences, Peking University, Beijing, 100871 Tel: 86-10-82202235 Fax: 86-10-82805079 E-mail: zhuweiguoedu.cn.
Corresponding author: Wei-Guo Zhu, Key Laboratory of Carcinogenesis and Translational Research, Department of Biochemistry and Molecular Biology, Peking-Tsinghua University Center for Life Sciences, Peking University, Beijing, 100871 Tel: 86-10-82202235 Fax: 86-10-82805079 E-mail: zhuweiguoedu.cn.