Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(7):777-788. doi:10.7150/ijbs.9220 This issue Cite
Research Paper
A Novel Role of Exogenous Carbon Monoxide on Protecting Cardiac Function and Improving Survival against Sepsis via Mitochondrial Energetic Metabolism Pathway
Xu Wang*, Weiting Qin*, Xuefeng Qiu*, Jie Cao, Dadong Liu, Bingwei Sun ![]()
Department of Burn and Plastic Surgery, Affiliated Hospital, Jiangsu University, Zhenjiang, Jiangsu Province, China
* These authors contributed equally to this work.
Received 2014-3-26; Accepted 2014-6-6; Published 2014-7-10
Abstract
Septic cardiac dysfunction is the main cause of death in septic patients. Here we investigate whether exogenous carbon monoxide can protect cardiac function and improve survival against sepsis by interfering with mitochondrial energetic metabolism. Male C57BL/6 mice were subjected to cecal ligation and puncture to induce sepsis. Exogenous carbon monoxide delivered from Tricarbonyldichlororuthenium (II) dimer (carbon monoxide releasing molecule II, 8mg/kg) was used intravenously as intervention. We found that carbon monoxide significantly improved cardiac function (LVEF 80.26 ± 2.37% vs. 71.21 ± 1.37%, P < 0.001; LVFS 43.52 ± 1.92% vs. 34.93 ± 1.28%, P < 0.001) and increased survival rate of septic mice (63% vs. 25%, P < 0.01). This phenomenon might be owing to the beneficial effect of carbon monoxide on abolishing the elevation of cardiac enzyme activity, cytokines levels and apoptosis rate, then attenuating cardiac injury in septic mice. Meanwhile, carbon monoxide significantly reversed the loss of mitochondrial number, effectively inhibited cardiac mitochondrial damage in septic mice by modulating glucose uptake, adenosine triphosphate and lactate content. Furthermore upregulation of peroxisome proliferator-activated receptor-γ coactivator-1α, nuclear respiratory factor 1 and mitochondrial transcription factor A genes in cardiac tissue were revealed in septic mice treated with carbon monoxide. Taken together, the results indicate that exogenous carbon monoxide effectively modulated mitochondrial energetic metabolisms by interfering with expression of peroxisome proliferator-activated receptor-γ coactivator-1α, nuclear respiratory factor 1 and mitochondrial transcription factor A genes, consequently exerted an important improvement in sepsis-induced cardiac dysfunction.
Keywords: Sepsis, Cardiac function, Carbon monoxide, Energy metabolism, Gene expression
Introduction
Sepsis is defined as the host inflammatory response to severe, life-threatening infection with the presence of organ dysfunction [1, 2]. Despite advancements in the understanding of its pathophysiology, sepsis continues to pose serious clinical challenges and is the leading cause of death in patients admitted to intensive care units (ICU) [3, 4]. Heart is an important target organ frequently affected by sepsis and always affected by septic shock [5, 6]. Septic patients suffering cardiac dysfunction showed a high mortality of 70-90% vs. 20% without cardiac dysfunction [7]. With improving awareness of the importance of cardiac dysfunction in sepsis, low cardiac index and echocardiographic evidence were added to the recent definition of severe sepsis or septic shock [8]. Among the main potential mechanisms underlying the pathophysiology of cardiac dysfunction in sepsis, mitochondrial depression may represent a key cellular event [9-12].
Endogenous carbon monoxide (CO), a by-product of inducible heme oxygenase (HO-1), can exert various physiological effects including modulating inflammation [13-15]. Recently, transition metal carbonyls have been identified as novel CO-releasing molecules (CORMs) with the potential to facilitate the pharmaceutical use of CO by delivering it to tissues and organs. Our previous studies confirmed that Tricarbonyldichlororuthenium (II) dimer (CORM-2) released CO attenuated inflammatory response and protected the function of liver, lung and small intestine of septic mice by interfering with activation of nuclear factor-κB (NF-κB) and expression of intercellular adhesion molecule-1 (ICAM-1) [16-18]. However, little is known whether CORM-2 released CO can protect cardiac mitochondria and improve cardiac function in septic mice.
Here, cecal ligation and puncture (CLP)-induced septic mice and LPS-stimulated neonatal rat ventricular myocyte cultures (NRVMCs) are employed to test whether CORM-2, one of the novel groups of CORMs, can modulate cardiac mitochondrial energetic metabolism and exert an important improvement in sepsis-induced cardiac dysfunction. Peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1α (PGC-1α), called 'master regulators' of mitochondrial biogenesis, is a potent activator of transcription factors nuclear respiratory factor 1 (NRF-1), nuclear respiratory factor 2 (NRF-2) and mitochondrial transcription factor A (Tfam). These genes collectively initiate the expression of mitochondrial genes and almost all the proteins involved in oxidative phosphorylation in mitochondria. Therefore, further studies discover the effect of CORM-2 on the expression of PGC-1α, NRF-1, NRF-2 and Tfam.
Materials and Methods
Detailed descriptions of the materials and methods are available in the Online Data Supplement (Additional file 1). A brief description of the methods used is given below.
Ethics statement
The experiments outlined in this manuscript conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All experimental protocols were approved by the Council on Animal Care at Jiangsu University on the Protection and the Welfare of Animals and followed the National Institutes of Health of China guidelines for the care and use of experimental animals.
Animal and CLP protocol
Total of two hundred and seventy eight male C57BL/6 mice (body weight 20 ± 2 g, Experimental Animal Center of the Jiangsu University, Zhenjiang, Jiangsu, China) were given free access to normal mouse diet and tap water. All mice were randomly assigned to 4 groups according to the different design of each experiment: Sham group; CLP group; CLP + CORM-2 group; and CLP + inactive CORM-2 (iCORM-2) group. In each experiment, mice in sham group underwent sham procedure, whereas mice in CLP group received cecal ligation and puncture, mice in CLP + CORM-2 group and CLP + iCORM-2 group were subjected to the same injury with immediate administration of CORM-2 (8 mg/kg, i.v.) and iCORM-2 (8 mg/kg, i.v.), respectively.
Primary neonatal rat ventricular myocyte cultures (NRVMCs)
One-day-old SD rats were sacrificed and hearts were harvested. After scalpel homogenization, ventricular myocytes were isolated following a series of collagenase/pancreatin digestions. The cells were resuspended in DMEN/F12 and maintained for 1 h in a humidified atmosphere with 5% CO2 and 95% air. Then the supernatant containing the ventricular myocyte was harvested gently. Forty-eight hours after planting, NRVMCs were washed with medium and incubated with lipopolysaccharide (LPS, 10 μg/mL) in the absence or presence of CORM-2 (50 μmol/L) for 12 h.
Survival
A total of 53 C57BL/6 male mice were randomly assigned into 4 groups: Sham group (n=8), CLP group (n=15), CLP + CORM-2 group (n=15), CLP + iCORM-2 group (n=15). All mice had normal access for water and food and were monitored every 6 h for 72 h.
Biochemical assays
Mice were anesthetized and blood samples were collected from the heart at 6, 12 and 24 h after CLP. Levels of aspartate aminotransferase (AST), lactate dehydrogenase (LDH), creatine kinase (CK) and CK myocardial isoform (CK-MB) in serum were determined by a serum autoanalyzer (AU2700, Olympus, Tokyo, Japan). The released LDH in the supernatants of NRVMCs was calculated according to the following equation: LDH release = LDH in the supernatant - LDH in the cell-free medium.
Quantitation of apoptosis
Mice were euthanized 24 h after CLP surgery. Hearts were harvested and apoptosis was quantified by flow cytometry using Annexin V and PI double staining. Annexin V is a Ca2+-dependent phospholipid-binding protein with high affinity for membranal apoptotic marker phospholipid phosphatidylserine (PS). Membranes of dead and damaged cells are permeable to PI. Cells that are positive for Annexin V but negative for PI were considered undergoing apoptosis. The apoptotic rate was calculated as the percentage of Annexin V-positive and PI-negative cells divided by the total number of cells in the gated region.
Measurement of IL-1β and TNF-α
Blood samples were collected from the heart 24 h after CLP surgery. Enzyme-linked immunosorbent assay (ELISA) kits were used to detect serum IL-1β and TNF-α concentrations following the manufacturer's recommended procedures.
Echocardiography
Transthoracic echocardiography was performed using Agilent Sonos 5500 ultrasound systems (Agilent Technologies Inc, Lexington, MA., USA). 24 h after CLP surgery mice were anesthetized with 2% inhaled isoflurane and placed on a temperature-controlled platform. After tracing left ventricular end-diastolic dimensions (LVDd) and left ventricular end-systolic dimensions (LVDs), manufacturer software automatically computed left ventricular ejection fraction (LVEF) and left ventricular fraction shortening (LVFS) according to Teichholz Formula. Analysis of the data was performed with software provided by VisualSonics (Toronto, Ont., Canada).
Mitochondria isolation
At 6, 12 and 24 h after CLP, fresh heart tissues were homogenized. The homogenates were centrifuged at 850 g for 10 min to collect supernatants, followed by centrifuging at 10,000 g for additional 10 min. The mitochondrial pellet was then resuspended in a final washing buffer. Protein concentration was determined by BCA protein assay kit.
Determination of mitochondrial ROS
Mitochondrial ROS were measured with a nonfluorescent probe DCFH-DA. All groups of isolated cardiac mitochondria were incubated with DCFH-DA at 37°C for 30 min. ROS levels were determined via a fluorescence spectrometry at an excitation wavelength of 488 nm and an emission wavelength of 535 nm.
Determination of mitochondrial membrane potential (MMP)
Mitochondrial membrane potential was measured with a fluorescent dye, JC-1. Isolated cardiac mitochondria were stained with JC-1 at 37°C for 30 min. MMP was shown as fluorescence intensity via the use of a fluorescence spectrometry at an excitation wavelength of 488 nm and an emission wavelength of 595 nm.
Determination of mitochondrial swelling
Mitochondrial swelling was assessed by measuring the absorbance at 540 nm. Cardiac mitochondria were prepared in the assay buffer. The extent of mitochondrial swelling was assayed by measuring the decrease in absorbance (A540) every 30 sec for 30 min after the addition of 50 μmol/L Ca2+ at 37°C. Results were normalized to sham group.
Transmission electron microscopic examination
Hearts were fixed in 2.5% gluteraldehyde in 0.1 M sodium cacodylate buffer and post-fixed in 1% osmium tetroxide in 0.1 M sodium cacodylate buffer with 0.3% potassium ferrocyanide at 24 h after CLP. Tissue was stained with 4% aqueous uranyl acetate, dehydrated, infiltrated, and embedded in in LX112 (Ladd Research). Ultrathin sections (80 nm) were cut and imaged using a Philips Tecnai 12 electron microscope.
mtDNA Copy Number
Total DNA from each heart was extracted using QIAamp DNA Mini Kit. Mitochondrial cytochrome c oxidase subunit II (mtCOII) gene was quantified by qPCR for mtDNA. 18S rRNA served as controls.
Confocal microscopy
Confocal microscopical examination of mitochondrial staining in heart sections was performed. Mean fluorescence intensities from at least five individual hearts were measured using ImageJ and the means of these images were used for further statistical analysis.
RNA Extraction and Real-time quantitative PCR (qPCR)
Total RNA from each heart was extracted using the TRIzol Reagent. First-strand complimentary DNA (cDNA) was synthesized using the RevertAid First Strand cDNA Synthesis Kit. Real-time qPCR was performed using Maxima SYBR Green/ROX qPCR Master Mix. Gene special primers were listed in Additional file 1: Supplementary Table 1. The relative level of gene expression was normalized to the level of GAPDH and calculated using the 2-ΔΔCT method.
Small-animal Positron Emission Tomography (PET) imaging
24 h after CLP, mice were maintained under 2% isoflurane anesthesia throughout the scanning period. PET scanning was performed 60 min after injection of 18.9 ± 1.6 MBq of FDG for 10 min. The PET images were reconstructed using the Inveon Acquisition Workplace software. Cardiac FDG uptake was calculated as standardized uptake value (SUV).
Determination of ATP and lactate content
Cardiac ATP and lactate content were assessed in homogenized tissue samples. Homogenates were centrifuged at 2,500 rpm at 4°C and the supernatants were harvested. Then, ATP and lactate content in the homogenates were measured using assay kits following the manufacturer's instructions.
Measurement of oxygen consumption
NRVMCs were harvested from culture, resuspended in 3 mL of complete medium. Out of this suspension, 2 mL was transferred into a sealed chamber connected to a Clark-type electrode, and maintained at 37°C. Oxygen consumption was recorded as the rate of decrease in oxygen tension within the chamber over the first 180 sec.
Statistical analysis
All the data were analyzed using software GraphPad Prism 5. 5-8 mice and 5 wells for each group were calculated. Values were presented as mean ± SD. Statistical analysis was performed using one-way factorial analysis of variance (ANOVA) and Student's t-test for the comparisons. Survival was analyzed with log-rank test. P < 0.05 was considered to be statistically significant.
Results
Effect of CORM-2 on survival of septic mice
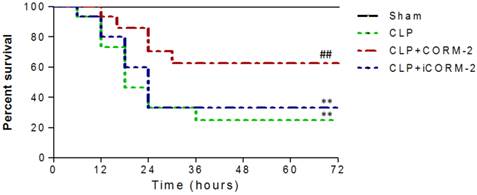
Survival of mice was analyzed during 72 h after CLP procedures. As shown in Fig 1, no deaths occurred in the sham group. Most of mice challenged with CLP died between 12 h and 24 h. CORM-2 administration dramatically increased the survival rate of septic mice from 25% to 63%. iCORM-2, which did not release carbon monoxide, had no effect on survival of septic mice.
Effect of carbon monoxide releasing molecule-2 (CORM-2) on survival of septic mice. Mice were challenged with cecal ligation and puncture (CLP) and treated with CORM-2 or inactive CORM-2 (iCORM-2). All mice had normal access to water and food. Animal survival was monitored 4 times a day for up to 72 h after CLP. Most of mice challenged with CLP died between 12 h and 24 h. CORM-2 dramatically increased the survival of septic mice from 25% to 63%. **P < 0.01 compared with sham, ##P < 0.01 compared with CLP.
Effect of CORM-2 on cardiac injury of septic mice
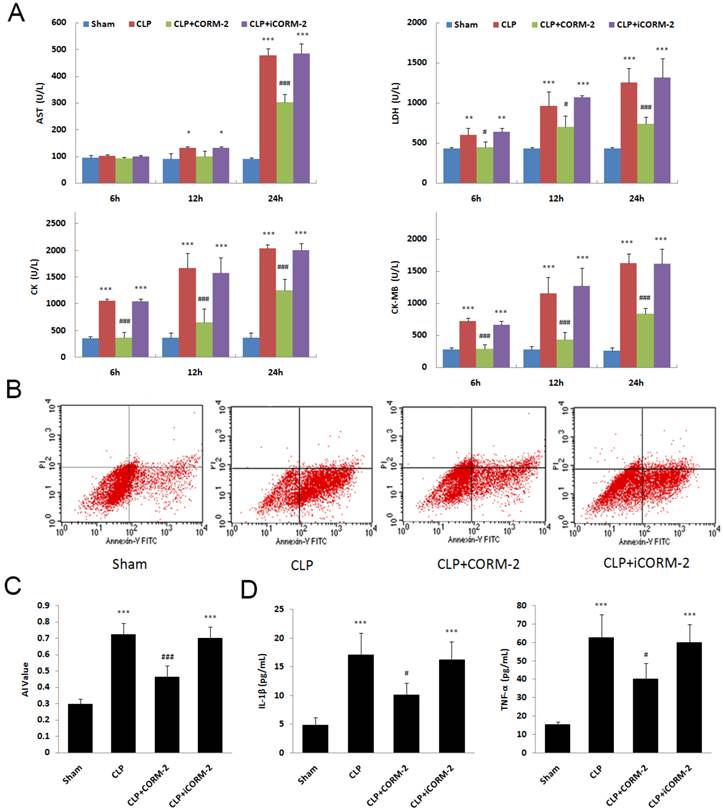
In septic mice, significant increases of myocardial enzymes in serum were seen at 6, 12 and 24 h after CLP surgery (P < 0.001. Fig 2A). The in vivo administration of CORM-2 significantly abolished this elevation (P < 0.001). Apoptosis of cardiac cells was assessed by Annexin V/PI staining. Flow cytometry (FC) results showed a significant decrease of apoptosis rate in the presence of CORM-2 at 24 h after CLP surgery (0.46 ± 0.07 vs. 0.72 ± 0.07, P < 0.001. Fig 2B). In Fig 2C, at 24 h after CLP, the levels of IL-1β and TNF-α in serum were markedly increased compared with those in sham mice (P < 0.001). After administration of CORM-2, the elevation of IL-1β and TNF-α levels were effectively abolished (P < 0.05).
Effect of carbon monoxide releasing molecule-2 (CORM-2) on cardiac injury of septic mice. Mice were challenged with CLP and treated with CORM-2 or iCORM-2. A, Myocardial enzymes, including LDH, AST, CK and CK-MB, were detected at 6, 12 and 24 h after CLP surgery. Myocardial enzymes were increased at 6, 12 and 24 h after CLP. In vivo administration of CORM-2 significantly abolished this elevation. B, 24 h following CLP surgery, apoptosis of cardiac cells was assessed by Annexin V/PI staining. Representative flow cytometry (FC) images were shown. C, FC results showed a high apoptotic rate at 24 h after CLP surgery and then a significant decrease in the presence of CORM-2. D, At 24 h after CLP, the levels of interleukin (IL)-1β and tumor necrosis factor (TNF-α) in serum were markedly increased compared with those in sham mice. After administration of CORM-2, the elevation of TNF-α and IL-1β levels were effectively abolished. Data were shown as mean ± SD, *P < 0.05 compared with sham, **P < 0.01 compared with sham, ***P < 0.001 compared with sham, #P < 0.05 compared with CLP, ##P < 0.01 compared with CLP, ###P < 0.001 compared with CLP.
Effect of CORM-2 on mitochondrial damage in the heart of septic mice
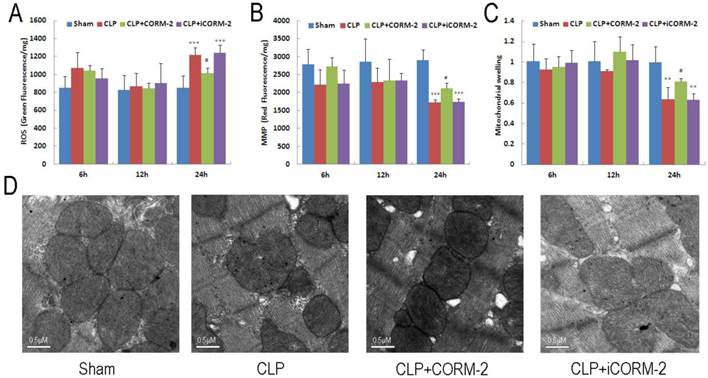
Cardiac mitochondrial function was assessed by examination of mitochondrial ROS production, mitochondrial membrane potential (MMP) and mitochondrial swelling. As shown in Fig 3A, cardiac mitochondrial ROS were significantly increased at 24 h after CLP (P < 0.001). The elevation was abolished after administration of CORM-2 (P < 0.05). Consistent results were found in MMP alternations. As shown in Fig 3B, mitochondrial depolarization was observed 24 h after surgery (P < 0.001) and this decline was prevented by CORM-2 (P < 0.05). Meanwhile, significant attenuation of Ca2+-induced mitochondrial swelling in the hearts of CLP mice was observed only at 24 h after CLP surgery (P < 0.01) (Fig 3C). Treatment with CORM-2 significantly inhibited the alterations (P < 0.05). Representative electron microscopic images (Fig 3D) showed some early changes of mitochondria like breakage of inner and outer membranes, variable swelling and lose of matrix density in CLP mice compared with sham mice. CORM-2 effectively inhibited these structural destructions.
Effect of carbon monoxide releasing molecule-2 (CORM-2) on mitochondrial damage in the heart of septic mice. Mice were challenged with CLP and treated with CORM-2 or iCORM-2. Cardiac mitochondrial ROS (A), mitochondrial membrane potential (MMP) (B) and mitochondrial swelling (C) were assessed at 6, 12 and 24 h after CLP injury. Mitochondrial damage was detected at 24 h after CLP surgery as indicated by collapse of MMP, excess generation of ROS and poor ability of swelling. Treatment with CORM-2 significantly inhibited the alterations. Data were shown as mean ± SD, **P < 0.01 compared with sham, ***P < 0.001 compared with sham, #P < 0.05 compared with CLP. (D), Representative electron microscopic images showed some early changes of mitochondria like breakage of inner and outer membranes, variable swelling and lose of matrix density in CLP mice compared with sham mice. CORM-2 inhibited these structural destructions.
Effect of CORM-2 on mitochondrial number in the heart of septic mice
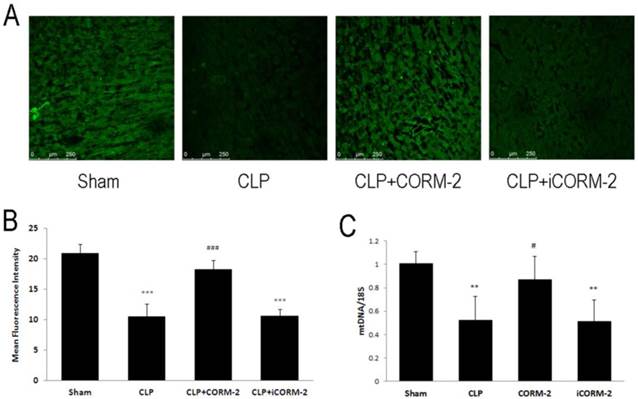
Mitochondrial number was assayed by mitotracker green staining and mtDNA copy number (Fig 4). Accumulation of mitotracker was detected by confocal microscopy. Results showed that CLP significantly reduced the accumulation of mitotracker compared with sham mice (10.46 ± 2.15 vs. 20.92 ± 1.50 MFI, P < 0.001). Administration of CORM-2 significantly reversed the loss of mitotracker accumulation (18.23 ± 1.54 vs. 10.46 ± 2.15 MFI, P < 0.001). mtDNA copy number was also reduced in CLP challenged mice while CORM-2 significantly restored it (0.87 ± 0.2 vs. 0.52 ± 0.20, P < 0.05).
Effect of carbon monoxide releasing molecule-2 (CORM-2) on mitochondrial number in the heart of septic mice. Mice were challenged with CLP and treated with CORM-2 or iCORM-2. 24 h following CLP surgery, mitochondrial number was assayed by mitotracker green staining and mtDNA copy number. Accumulation of mitotracker was detected by confocal microscopy. Representative images and mean Fluorescence Intensity were shown in A and B. Results showed that CLP significantly reduced the accumulation of mitotracker compared with sham mice. Administration of CORM-2 significantly reversed the loss of mitotracker. C, mtDNA copy number was also reduced in CLP challenged mice while CORM-2 significantly abolished this reduction. Data were shown as mean ± SD, **P < 0.01 compared with sham, ***P < 0.001 compared with sham, #P < 0.05 compared with CLP, ###P < 0.001 compared with CLP.
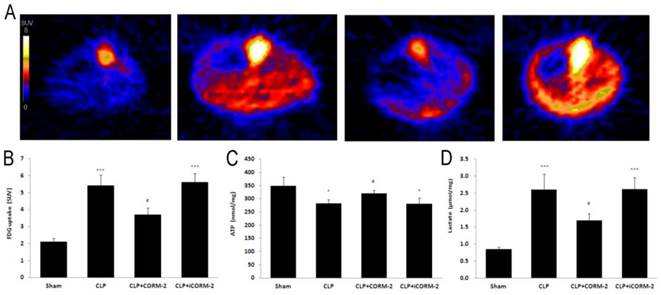
Effect of CORM-2 on FDG uptake, ATP and lactate content in the heart of septic mice
Positron emission tomography (PET) was used to assay the 18F labeled FDG uptake in mice (Fig 5A). Calculated as SUV, results (Fig 5B) displayed that CLP mice had higher FDG uptake than that in sham mice at 24 h after CLP injury (5.4 ± 0.6 vs. 2.1 ± 0.2 SUV, P < 0.001). After in vivo administration of CORM-2, FDG uptake was significantly decreased (3.7 ± 0.4 vs. 5.4 ± 0.6 SUV, P < 0.05). Cardiac ATP (Fig 5C) content was slightly but significantly lower in CLP mice when compared with sham mice at 24 h after CLP surgery (281.55 ± 14.25 vs. 348.15 ± 34.19 nmol/mg, P < 0.05). With the treatment with CORM-2, ATP content was significantly increased (319.85 ± 10.88 vs. 281.55 ± 14.25 nmol/mg, P < 0.05). Production of lactate (Fig 5D) was low in sham group. After CLP challenge, lactate levels were significantly increased (2.60 ± 0.46 vs. 0.85 ± 0.07 U/L, P < 0.001) whereas administration of CORM-2 markedly decreased lactate production (1.69 ± 0.21 vs. 2.60 ± 0.46 U/L, P < 0.05).
Effect of carbon monoxide releasing molecule-2 (CORM-2) on FDG uptake, ATP and lactate content in the heart of septic mice. Mice were challenged with CLP and treated with CORM-2 or iCORM-2. A, Representative images of Positron Emission Tomography (PET) scanning. B, Standardized uptake value (SUV) results displayed that CLP mice had higher 18F-fluorodeoxyglucose (FDG) uptake than that in sham mice at 24 h after CLP injury. After in vivo administration of CORM-2, FDG uptake was significantly decreased. C, Cardiac ATP content was slightly but significantly lower in CLP mice as compared with sham mice at 24 h after CLP surgery. With the treatment of CORM-2, ATP content was significantly increased. D, Production of lactate was low in sham group. After CLP challenge, lactate levels were significantly increased whereas administration of CORM-2 markedly decreased lactate production. Data were shown as mean ± SD, *P < 0.05 compared with sham, ***P < 0.001 compared with sham, #P < 0.05 compared with CLP.
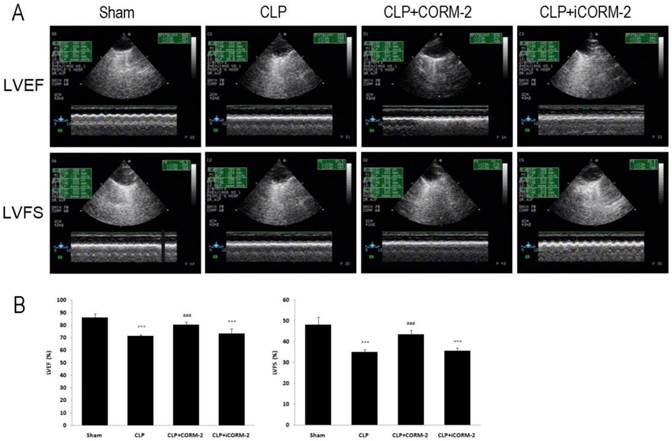
Effect of CORM-2 on cardiac function of septic mice
LVEF and LVFS were determined by echocardiography to assess cardiac function (Fig 6, Additional file 1: Supplementary Table 2). In Fig 6, at 24 h after CLP, LVEF and LVFS were found to be significantly lower in CLP mice as compared with sham mice (LVEF 71.21 ± 1.37 vs. 85.88 ± 3.31, LVFS 34.93 ± 1.28 vs. 48.01 ± 3.84, P < 0.001). Treatment with CORM-2 prevented the reductions of LVEF and LVFS (LVEF 80.26 ± 2.37 vs. 71.21 ± 1.37, LVFS 43.52 ± 1.92 vs. 34.93 ± 1.28, P < 0.001).
Effect of carbon monoxide releasing molecule-2 (CORM-2) on cardiac function of septic mice. Mice were challenged with CLP and treated with CORM-2 or iCORM-2. A, Representative images of echocardiography. 24 h following CLP surgery left ventricular end-diastolic dimensions (LVDd), left ventricular end-systolic dimensions (LVDs) were measured by echocardiography and left ventricular ejection fraction (LVEF), left ventricular fraction shortening (LVFS) were calculated automatically by software. B, LVEF and LVFS were found to be significantly lower in CLP mice as compared with sham mice. Treatment with CORM-2 prevented the reductions of LVEF and LVFS. Data were shown as mean ± SD, ***P < 0.001 compared with sham, ###P < 0.001 compared with CLP.
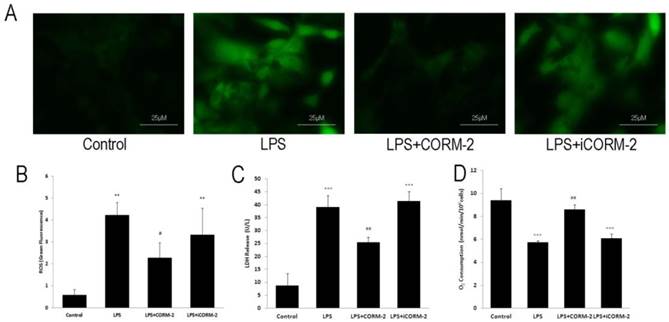
Effect of CORM-2 on LPS-stimulated NRVMCs
To assess the effect of CORM-2 on the septic cardiac myocytes, we employed the in vitro LPS-stimulated NRVMCs to mimic the septic pathophysiology. Representative fluorescence photomicrographs (Fig 7A) revealed that LPS stimulation increased ROS levels compared with control whereas CORM-2 treatment significantly reduced LPS-induced ROS generation. Fluorescence spectrometry analysis (Fig 7B) was consistent with the fluorescence spectrometry results. Damaged cells lead to the release of LDH into culture medium. LDH release (Fig 7C) was markedly higher in NRVMCs treated with LPS for 12 h than that in control group (39.00 ± 4.58 vs. 8.67 ± 4.73 U/L, P < 0.001) but was significantly decreased when NRVMCs were co-incubated with LPS plus CORM-2 (25.33 ± 2.08 vs. 39.00 ± 4.58 U/L, P < 0.01). Fig 7D showed that mitochondrial oxygen consumption was significantly reduced at 12 h following LPS stimulation as compared with the control group (2.86 ± 0.08 vs. 4.70 ± 0.51 nmol/min/106cells, P < 0.001). However, CORM-2 administration markedly improved oxygen consumption (3.63 ± 0.37 vs. 2.86 ± 0.08 nmol/min/106cells, P < 0.01).
Effect of carbon monoxide releasing molecule-2 (CORM-2) on reactive oxygen species (ROS) levels, lactate dehydrogenase (LDH) release and oxygen consumption in lipopolysaccharide (LPS)-stimulated neonatal rat ventricular myocyte cultures (NRVMCs). NRVMCs were stimulated by LPS for 12 h and treated with CORM-2 or iCORM-2. A, Representative fluorescence photomicrographs revealed that LPS stimulation increased ROS levels compared with the control, whereas CORM-2 treatment significantly reduced LPS-induced ROS generation. B, Fluorescence spectrometry analysis was consistent with the fluorescence spectrometry results. C, LDH release was markedly higher in NRVMCs treated with LPS than that in control group and LDH release was significantly decreased when NRVMCs were co-incubated with LPS and CORM-2. D, Oxygen consumption was assayed by Clark-type electrode. Oxygen consumption was significantly reduced at 12 h following LPS stimulation as compared with control group. However, CORM-2 markedly improved oxygen consumption. Data were shown as mean ± SD, **P < 0.01 compared with sham, ***P < 0.001 compared with sham, #P < 0.05 compared with CLP, ##P < 0.01 compared with CLP
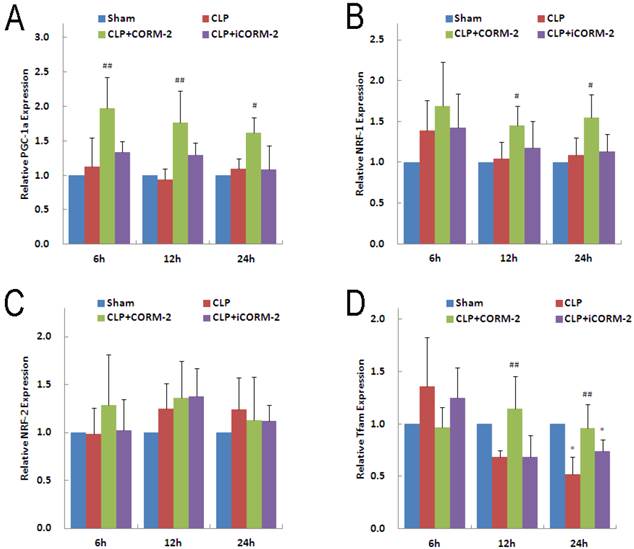
Effect of CORM-2 on mitochondrial biogenesis genes in the heart of septic mice
PGC-1α and its downstream transcriptional factor NRF-1 and NRF-2 are essential for almost all the proteins involved in oxidative phosphorylation in mitochondria. In addition, PGC1-α and NRFs are responsible for the transcription of the Tfam, a molecule controlling mitochondrial DNA replication and transcription of mitochondria-encoded genes. Results of PGC-1α mRNA (Fig 8A) showed no significant difference in CLP mice at 6, 12 and 24 h compared with sham mice. However, in vivo administration of CORM-2 significantly elevated the expression of PGC-1α mRNA (6 h 1.98 ± 0.50 vs. 1.12 ± 0.42, P < 0.01; 12 h 1.76 ± 0.46 vs. 0.93 ± 1.61, P < 0.01; 24 h 1.62 ± 0.22 vs. 1.09 ± 0.15, P < 0.05). Analogous to PGC-1α, NRF-1 mRNA (Fig 8B) was not significantly altered by CLP surgery but in vivo administration of CORM-2 effectively raised the expression of NRF-1 (12 h 1.45 ± 0.24 vs. 1.05 ± 0.20, 24 h 1.55 ± 0.28 vs. 1.09 ± 0.21, P < 0.05). No significant difference was observed for the expression of NRF-2 mRNA (Fig 8C) among all groups at 6, 12 or 24 h. Expression of Tfam mRNA (Fig 8D) in CLP mice was significantly decreased at 24 h (0.52 ± 0.17 vs. 1.00 ± 0.12, P < 0.05), CORM-2 treatment markedly abolished this decline (12 h 1.14 ± 0.31 vs. 0.69 ± 0.06, 24 h 0.96 ± 0.23 vs. 0.52 ± 0.17, P < 0.01).
Effect of carbon monoxide releasing molecule-2 (CORM-2) on mitochondrial biogenesis genes in the heart of septic mice. Mice were challenged with CLP and treated with CORM-2 or iCORM-2. Expression of PGC-1α, NRF-1, NRF-2 and Tfam mRNAs were detected by RT-qPCR. A, Results showed that CLP surgery did not changed the expression of PGC-1α, however CORM-2 treatment significantly up-regulated the expression of PGC-1α at 6, 12 and 24 h after CLP surgery. B, NRF-1 genes were not changed in CLP challenged group but significantly up-regulated at 12 and 24 h after CLP surgery in the presence of CORM-2. C, NRF-2 genes were not alterated among all groups. D, Tfam genes were down-regulated at 24 h after CLP challenge, and CORM-2 treatment significantly reversed this decline. Data were shown as mean ± SD, #P < 0.05 compared with CLP, ##P < 0.01 compared with CLP.
Discussion
Sepsis is the most leading cause of death in patients admitted to ICU [3]. In a recent study, annual number of cases in America exceeded 750,000 with 215,000 associated deaths [19]. Cardiac dysfunction is a major factor to the mortality of patients suffering sepsis [20]. It was reported that the mortality of septic patients with cardiac dysfunction reached up to 70-90% compared with 20% without cardiac dysfunction [7]. Mitochondria are crucial for maintaining cardiac function. A growing body of evidence indicates that mitochondria contribute to the pathogenesis of septic cardiac dysfunction [21-23]. In the present study, we provide the evidence that exogenous CO liberated from CORM-2 prevents cardiac dysfunction against sepsis and this beneficial effect may be owing to the improvement in mitochondrial energetic metabolism.
CO is a metabolic product of heme oxygenase (HO) with remarkable anti-inflammatory capacity [24, 25]. Recently, transition metal carbonyls have been identified as the potential to facilitate the pharmaceutical use of CO by delivering it to the tissues and organs of interest [13, 26]. Studies elucidated that CORM-2 suppressed LPS-induced inflammatory responses in human umbilical vein endothelial cells (HUVECs), peripheral blood mononuclear cells (PBMCs) and macrophages [27, 28]. Similarly, the results reported in our laboratory and others confirmed that CO derived from CORMs rescued mice from lethal sepsis induced by LPS or CLP [16-18]. Until now, however, little is known whether CORM-2 can protect cardiac function in septic mice.
CLP, golden standard model for polymicrobial sepsis, was employed to mimic the septic pathophysiologic conditions [29, 30]. Survival assay revealed that only 25% of mice survived from CLP while in vivo administration of CORM-2 markedly increased it to 63%. Levels of myocardial enzymes were dramatically downregulated by CORM-2. Cardiac enzymes, including AST, LDH, CK and CK-MB, are proteins from heart muscle cells that are released into the bloodstream when heart muscle is damaged. The levels of serum cardiac enzymes dramatically increased with the prolonged time of CLP injury and multiplied several times at 24 h which indicated the severe cardiac injury during sepsis. Cardiac enzymes were released due to the damage of cardiac cells so we further examined the apoptotic rate of cardiac cells by Annexin V/PI staining at 24 h after CLP surgery. Flow cytometry results proved the high apoptotic rate in CLP-challenged mice and CORM-2 significantly suppressed the apoptosis. Evidenced by the decreased myocardial enzymes levels and apoptosis rate, administration of CORM-2 significantly alleviated the cardiac injury in septic mice. IL-1β and TNF-α were potent mediators of cardiac injury in septic cardiac injury [31, 32]. After application of CORM-2, the elevation of serum IL-1β and TNF-α was markedly abolished which suggested the role of CORM-2 in the prevention of CLP-induced tissue damage might involve in downregulation of the production of cytokines.
It is well-known that mitochondrial damage directly deteriorates energy supply for maintaining normal cell functions, especially when it goes to cardiac cells [33-35]. Extensive researches highlighted the fact of mitochondrial damage in septic patients and animal models [36, 37]. To estimate the mitochondrial damage, MMP, mitochondrial ROS and mitochondrial swelling were detected at 6, 12 and 24 h after CLP surgery. Results revealed that collapse of MMP, excess generation of ROS and poor ability to swell occurred at 24 h after CLP surgery. These data were in accordance with the previous studies that short-term (< 6 h) laboratory models of sepsis showed variable changes in mitochondrial respiration, whereas longer term models (>12 h) consistently revealed impaired mitochondrial function [38]. The lengthy course of sepsis and extended exposure to reactive nitrogen species (RNS) might be the cause of irreversible inhibition or permanent damage to mitochondria [39]. After in vivo administration of CORM-2, mitochondrial damage was greatly inhibited. Not only damaged mitochondria but also decreased mitochondrial number was proved to affect cardiac function in sepsis. Thus we assayed the mitochondrial number by mitotracker green staining and mtDNA copy number at 24 h after CLP surgery. We found that CLP significantly reduced the mitochondrial number compared with sham mice. As expected, the extent of decrease of mitochondrial number was slight in CLP mice treated with CORM-2.
To determine the potential consequence of the above phenomena, we investigated the in vivo ability of mice for cardiac glucose metabolism using 18F-FDG/micro-PET. To date, the application of PET technology has enabled remarkable research observations in cancer and neurobiology. However, the application of PET technology in the field of inflammation is constrained, and this is primarily due to the limitations of tracers and resolution [40]. An increasing number of radiolabels have been applied for in vivo imaging of small animals. 18F-FDG is a glucose-like radioactive tracer, which has been applied for the in vivo study of changes in glucose metabolic ability of local tissue and organs in humans and animals, as influenced by various diseases or drugs [40]. 18F-FDG is transported into cells by the glucose transporter that is located on the cell membrane and then phosphorylated to 18F-2'-FDG-6 by hexokinase (HK). Unlike G-6-P, 18F-2'-FDG-6 does not continue glucose metabolism but is retained in cells. Therefore, the 18F-FDG levels represent the ability of cells to transport and metabolize glucose. Evidence shows that high FDG uptake not only occurs in malignancies, but also is commonly present in benign inflammatory responses [40]. In this study, 18F-FDG/micro-PET was applied for the first time to investigate changes in cardiac glucose metabolism in septic mice after CORM-2 intervention. Results showed that the level of cardiac glucose metabolism in septic mice was significantly increased, and exceeded normal levels. By contrast, CORM-2-treated septic mice showed no significant difference when compared with the sham group. Despite sepsis-induced high glucose uptake, significantly impaired production of ATP and excessive accumulation of lactate were observed in septic mice as the result of mitochondrial damage and loss of mitochondrial number. Owing to the protective role of CORM-2 on mitochondria, ATP production was preserved and lactate accumulation was abolished.
Septic cardiac dysfunction is characterized by impaired left ventricular systolic functions [5, 20]. To evaluate the effects of CORM-2 on cardiac function, transthoracic echocardiography was performed in the mice at 24 h after CLP surgery. In the present study, left ventricular systolic function in CLP mice was significantly impaired as evidenced by decreased LVEF and LVFS. However, in vivo administration of CORM-2 significantly improved LVEF and LVFS, in other words, the cardiac systolic function during sepsis. As a consequence, survival rate of CLP-induced septic mice was found to be markedly increased by CORM-2 intervention.
LPS, a constituent of the outer membrane of Gram-negative bacteria, is the leading cause of sepsis and experimental administrated to NRVMCs can mimic the same inflammatory response [41]. After LPS administration, ROS generation and LDH release in NRVMCs were markedly increased while this elevation was abolished when co-incubated with CORM-2 which indicated a cytoprotective role of CORM-2. Since mitochondria consumed approximately 90% oxygen of cell oxygen uptake, decreased oxygen consumption directly reflected the poor mitochondrial function. Mitochondrial dysfunction evidenced by decreased oxygen consumption was found in LPS-stimulated NRVMCs whereas CORM-2 significantly improved it.
Cells do not generate mitochondria de novo, but instead identify and dispose of defective mitochondria while stimulating healthy mitochondria to proliferate through mitochondrial biogenesis [42]. Mitochondrial biogenesis protects metabolism from mitochondrial dysfunction produced by activation of innate immunity by LPS or other bacterial products [43, 44]. PGC family, called 'master regulators' of mitochondrial biogenesis, are potent activators of transcription factors NRF-1 and NRF-2 which are responsible for almost all the proteins involved in oxidative phosphorylation in mitochondria [45, 46]. Moreover, PGC-1α cooperates with NRF-1 and NRF-2 on Tfam controlling mitochondrial DNA replication and transcription of mitochondria-encoded genes [47]. RT-qPCR results showed that CLP slightly increased the expression of PGC-1α and NRF-1 genes with no significance compared with those in sham mice. However, these increases were important for supporting mitochondrial function in response to CLP challenges in early phase of sepsis [43]. Interestingly, CORM-2 administration significantly up-regulated the expression of PGC-1α and NRF-1 genes after CLP, leading to exerting more effective protection of mitochondrial function. On the contrary, CLP challenge obviously reduced the expression of Tfam, resulting in impaired mitochondrial DNA replication. Expectantly, the expression of Tfam was significantly up-regulated in septic mice after CORM-2 intervention. Therefore, we speculated that activation and upregulation of mitochondrial biogenesis (PGC-1α, NRF-1 and Tfam genes) were the potential mechanism of CORM-2 on improving mitochondrial energetic metabolism and protecting mitochondrial function during sepsis. Previous studies confirmed that cyclic GMP (cGMP), as the second messenger effector produced by soluble guanylyl cyclase (sGC), was generally believed to mediate the effect of CO in other well-established cardiovascular and immune systems [48-50]. Mitochondrial biogenesis was regulated by cGMP through increasing the expression of PGC-1α, NRF-1 and Tfam genes [51, 52]. Thus the sGC/cGMP pathway might be involved in the protective effect of CO on mitochondrial biogenesis in septic animals.
In summary, data from the present study consisting of in vivo and in vitro experiments provide a basis for the use of CORM-2 as an effective strategy for improvement in sepsis-induced cardiac dysfunction. Mitochondrial energetic metabolic involvement may underlie the potential mechanisms. However, there is still a great gap in the respect to applying CORM-2 to treatment in clinical sepsis. Murine CLP and LPS-challenged sepsis model can't fully mimic the pathophysiology in humans and the positive results of CORM-2 in our study need further experimental investigations. Besides, although CO gas has already passed safety evaluation in Phase I testing in healthy humans, possessed a backbone carrier moiety, CORM-2 needs to be stringently characterized from a metabolical and toxicological standpoint [13]. Further studies are required to assist for more comprehensive understanding of the pharmacokinetics and biology of CO and CORMs.
Abbreviations
CORM-2: carbon monoxide-releasing molecule-2; CLP: cecal ligation and puncture; LPS: lipopolysaccharide; NF-κB: nuclear factor-κB; ELISA: enzyme-linked immunosorbent assay; NRVMCs: neonatal rat ventricular myocyte cultures; LVEF: left ventricular ejection fraction; LVFS: left ventricular fraction shortening; LVDd: left ventricular end-diastolic dimensions; LVDs: left ventricular end-systolic dimensions; RT-qPCR: real time-quantitative polymerase chain reaction; ROS: reactive oxygen species; PET: positron emission tomography; FDG: 18F-fluorodeoxyglucos; PGC-1α: peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1α; NRF-1: nuclear respiratory factor-1; NRF-2: nuclear respiratory factor-2; Tfam: transcription factor A, mitochondrial.
Supplementary Material
Additional File 1Materials, Supplementary Tables and Figures.
Acknowledgements
This study was supported by the National Natural Science Foundation of China, No. 30772256, No. 81071546 and No. 81272148; by the Jiangsu Natural Science Foundation, No. BK2012703.
Authors' contributions
XW, WQ and XQ contributed equally to the work. XW performed the experiments of mitochondrial assay and in vitro studies. WQ and XQ performed the cardiac function and molecular genetic studies, assisted with the interpretation of data and helped to draft the manuscript. JC and DL performed animal surgeries. FL participated in the design of the study and performed the statistical analysis. BS and XW conceived the initial concept for the experimental aims and design, contributed to the systematic review and data extraction, performed the analysis and interpreted the results. BS drafted the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Angus DC, van der Poll T. Severe sepsis and septic shock. The New England journal of medicine. 2013;369(9):840-851
2. Charles PE, Gibot S. Predicting outcome in patients with sepsis: new biomarkers for old expectations. Critical care. 2014;18(1):108
3. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Critical care medicine. 2001;29(7):1303-1310
4. Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: A trend analysis from 1993 to 2003. Critical care medicine. 2007;35(5):1244-1250
5. Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Critical care medicine. 2007;35(6):1599-1608
6. Rudiger A, Singer M. The Heart in Sepsis: From Basic Mechanisms to Clinical Management. Curr Vasc Pharmacol. 2013;11(2):187-195
7. Court O, Kumar A, Parrillo JE, Kumar A. Clinical review: Myocardial depression in sepsis and septic shock. Critical care. 2002;6(6):500-508
8. Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365(9453):63-78
9. Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4(5-6):729-741
10. Garrabou G, Moren C, Lopez S, Tobias E, Cardellach F, Miro O, Casademont J. The effects of sepsis on mitochondria. The Journal of infectious diseases. 2012;205(3):392-400
11. Li L, Hu BC, Chen CQ, Gong SJ, Yu YH, Dai HW, Yan J. Role of mitochondrial damage during cardiac apoptosis in septic rats. Chinese Medical Journal. 2013;126(10):1860-1866
12. Grundler K, Angstwurm M, Hilge R, Baumann P, Annecke T, Crispin A, Sohn HY, Massberg S, Kraemer BF. Platelet mitochondrial membrane depolarization reflects disease severity in patients with sepsis and correlates with clinical outcome. Critical care. 2014;18(1):R31
13. Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nature reviews Drug discovery. 2010;9(9):728-743
14. Koulouras VP, Li R, Chen L, Hedenstierna GG. Effects of inhaled carbon monoxide and glucocorticoids in porcine endotoxin sepsis. International Journal of Clinical and Experimental Medicine. 2011;4(1):53-66
15. Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, Nakahira K, Ryter SW, Choi AM. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxidants & redox signaling. 2014;20(3):432-442
16. Wang X, Cao J, Sun BW, Liu DD, Liang F, Gao L. Exogenous carbon monoxide attenuates inflammatory responses in the small intestine of septic mice. World journal of gastroenterology: WJG. 2012;18(40):5719-5728
17. Sun BW, Sun Y, Sun ZW, Chen X. CO liberated from CORM-2 modulates the inflammatory response in the liver of thermally injured mice. World journal of gastroenterology: WJG. 2008;14(4):547-553
18. Sun B, Sun H, Liu C, Shen J, Chen Z, Chen X. Role of CO-releasing molecules liberated CO in attenuating leukocytes sequestration and inflammatory responses in the lung of thermally injured mice. The Journal of surgical research. 2007;139(1):128-135
19. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Critical care medicine. 2001;29(7):1303-1310
20. Merx MW, Weber C. Sepsis and the heart. Circulation. 2007;116(7):793-802
21. Ruggieri AJ, Levy RJ, Deutschman CS. Mitochondrial dysfunction and resuscitation in sepsis. Critical care clinics. 2010;26(3):567-575
22. Sayeed MM. Mitochondrial dysfunction in sepsis: a familiar song with new lyrics. Critical care medicine. 2002;30(12):2780-2781
23. Singer M. Mitochondrial function in sepsis: acute phase versus multiple organ failure. Critical care medicine. 2007;35(9 Suppl):S441-448
24. Song R, Kubo M, Morse D, Zhou Z, Zhang X, Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS. et al. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. The American journal of pathology. 2003;163(1):231-242
25. Otterbein LE. Carbon monoxide: innovative anti-inflammatory properties of an age-old gas molecule. Antioxidants & redox signaling. 2002;4(2):309-319
26. Caumartin Y, Stephen J, Deng JP, Lian D, Lan Z, Liu W, Garcia B, Jevnikar AM, Wang H, Cepinskas G. et al. Carbon monoxide-releasing molecules protect against ischemia-reperfusion injury during kidney transplantation. Kidney international. 2011;79(10):1080-1089
27. Maruyama K, Morishita E, Yuno T, Sekiya A, Asakura H, Ohtake S, Yachie A. Carbon monoxide (CO)-releasing molecule-derived CO regulates tissue factor and plasminogen activator inhibitor type 1 in human endothelial cells. Thromb Res. 2012;130(3):e188-193
28. Tsoyi K, Ha YM, Kim YM, Lee YS, Kim HJ, Kim HJ, Seo HG, Lee JH, Chang KC. Activation of PPAR-gamma by Carbon Monoxide from CORM-2 Leads to the Inhibition of iNOS but not COX-2 Expression in LPS-Stimulated Macrophages. Inflammation. 2009;32(6):364-371
29. Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends in microbiology. 2011;19(4):198-208
30. Bastarache JA, Matthay MA. Cecal ligation model of sepsis in mice: new insights. Critical care medicine. 2013;41(1):356-357
31. Mitchell MD, Laird RE, Brown RD, Long CS. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. American journal of physiology Heart and circulatory physiology. 2007;292(2):H1139-1147
32. Meador BM, Krzyszton CP, Johnson RW, Huey KA. Effects of IL-10 and age on IL-6, IL-1beta, and TNF-alpha responses in mouse skeletal and cardiac muscle to an acute inflammatory insult. J Appl Physiol. 2008;104(4):991-997
33. Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a Therapeutic Target in Heart Failure. Journal of the American College of Cardiology. 2013;61(6):599-610
34. Walters AM, Porter GA, Brookes PS. Mitochondria as a Drug Target in Ischemic Heart Disease and Cardiomyopathy. Circulation research. 2012;111(9):1222-1236
35. Moslehi J, DePinho RA, Sahin E. Telomeres and Mitochondria in the Aging Heart. Circulation research. 2012;110(9):1226-1237
36. Garrabou G, Moren C, Lopez S, Tobias E, Cardellach F, Miro O, Casademont J. The Effects of Sepsis on Mitochondria. Journal of Infectious Diseases. 2012;205(3):392-400
37. Balestra GM, Legrand M, Ince C. Microcirculation and mitochondria in sepsis: getting out of breath. Current Opinion in Anesthesiology. 2009;22(2):184-190
38. Singer M, Brealey D. Mitochondrial dysfunction in sepsis. Biochemical Society symposium. 1999;66:149-166
39. Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219-223
40. Mittra E, Quon A. Positron Emission Tomography/Computed Tomography: The Current Technology and Applications. Radiologic Clinics of North America. 2009;47(1):147
41. Gong J, Jing L. Glutamine induces heat shock protein 70 expression via O-GlcNAc modification and subsequent increased expression and transcriptional activity of heat shock factor-1. Minerva Anestesiol. 2011;77(5):488-495
42. Kowald A, Kirkwood TBL. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(25):10237-10242
43. MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. American journal of respiratory and critical care medicine. 2012;185(8):851-861
44. Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem. 2011;286(18):16374-16385
45. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends in Endocrinology and Metabolism. 2012;23(9):459-466
46. Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93(4):884S-890
47. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiological reviews. 2008;88(2):611-638
48. Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, Yoshida K, Nagai R. Carbon monoxide protects against cardiac ischemia--reperfusion injury in vivo via MAPK and Akt--eNOS pathways. Arteriosclerosis, thrombosis, and vascular biology. 2004;24(10):1848-1853
49. Liu H, Song D, Lee SS. Role of heme oxygenase-carbon monoxide pathway in pathogenesis of cirrhotic cardiomyopathy in the rat. American journal of physiology Gastrointestinal and liver physiology. 2001;280(1):G68-74
50. Otterbein LE, Bach FH, Alam J, Soares M, Lu HT, Wysk M, Davis RJ, Flavell RA, Choi AMK. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nature medicine. 2000;6(4):422-428
51. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S. et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299(5608):896-899
52. Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M. et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(47):16507-16512
Author contact
![]() Corresponding author: Professor Bingwei Sun, MD, PhD, Department of Burns and Plastic Surgery of Affiliated Hospital, Jiangsu University, Zhenjiang 212001, Jiangsu Province, China. Telephone: +86-511-85082258; Fax: +86-511-85029089; Email: sunbinwecom
Corresponding author: Professor Bingwei Sun, MD, PhD, Department of Burns and Plastic Surgery of Affiliated Hospital, Jiangsu University, Zhenjiang 212001, Jiangsu Province, China. Telephone: +86-511-85082258; Fax: +86-511-85029089; Email: sunbinwecom