Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):807-816. doi:10.7150/ijbs.9143 This issue Cite
Research Paper
SAMD9L Inactivation Promotes Cell Proliferation via Facilitating G1-S Transition in Hepatitis B Virus-associated Hepatocellular Carcinoma
Qun Wang1,2 ![]() , Yang-Yang Zhai2, Ji-Hong Dai2, Kun-Yu Li2, Qing Deng2, Ze-Guang Han1,2,3
, Yang-Yang Zhai2, Ji-Hong Dai2, Kun-Yu Li2, Qing Deng2, Ze-Guang Han1,2,3 ![]()
1. Key Laboratory of Systems Biomedicine (Ministry of Education) of Rui-Jin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China
2. Shanghai-MOST Key Laboratory for Disease and Health Genomics, Chinese National Human Genome Center at Shanghai, Shanghai 201203, China
3. Shanghai Center for systems Biomedicine, Shanghai Jiao Tong University, 800 Dong Chuan Road, Shanghai 200240, China
Received 2014-3-17; Accepted 2014-5-19; Published 2014-7-17
Abstract
Hepatocellular carcinoma (HCC) is a highly malignant cancer with poor prognosis, and driver genes harboring genetic lesions and/or expression dysregulation contribute to hepatocarcinogenesis. Sterile Alpha Motif Domain-containing 9-like (SAMD9L) was a novel identified mutated gene in our previous study on exome sequencing of hepatitis B virus (HBV)-associated HCC, but its expression and role in HCC remain unknown. Here, we demonstrated that SAMD9L was frequently inactivated by somatic mutations, and that its expression was deregulated in HCC patients with hepatitis B virus (HBV) infection. SAMD9L knockdown significantly promoted cell proliferation, colony formation of SK-hep-1, QGY-7701, BEL-7721 and MHCC-97H HCC cells. Furthermore, SK-hep-1 and MHCC-97H cells with stable SAMD9L knockdown exhibited enhanced tumorigenicity in athymic mice. Interestingly, SAMD9L silence facilitated G1-S transition of cell cycle progression and led to the elevated activity of Wnt/β-catenin pathway. Collectively, these findings highlight a novel tumor-suppressive role of SAMD9L inactivation by somatic mutation and decreased expression in human HBV-related HCC.
Keywords: SAMD9L, hepatocellular carcinoma, somatic mutation, cell cycle, Wnt pathway
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide and the second major cause of cancer-related deaths worldwide [1]. Commonly, it is a highly malignant cancer typically developing in the liver with chronic hepatitis and cirrhosis. Theoretically, hepatocarcinogenesis depends on two major principles: (1) the activation of oncogenes (e.g., c-myc, β-catenin), growth factor (e.g., IGF-II, TGF-α) and telomerase enzyme inducing cellular immortalization and (2) the inactivation of tumor suppressor genes (e.g., p53 and Rb) and decreased expression by different modifications such as promoter methylation, mutations, biallelic loss and loss of heterozygosity (LOH) [2]. Multiple risk factors for developing HCC including hepatitis B virus (HBV), hepatitis C virus (HCV), aflatoxin B1 (AFB1) contamination, alcohol abuse, ionizing radiation and metabolic products can cause DNA damage in hepatocytes [3-5]. Therefore, those accumulated DNA damages occurred on tumor suppressor genes and oncogenes are important genetic events of HCC oncogenesis and progression. A few inactivated tumor suppressor genes such as TP53 and activated oncogenes such as β-catenin, gp130 resulted from somatic mutations play critical roles during hepatocarcinogenesis, as demonstrated by previous investigations [6-8]. Recent sequencing analysis of exomes and genomes in multiple HCC cohorts has defined not only common events that target well-known cancer pathways including β-catenin/Axin, TP53, and RB/CDKN2A, as well as frequent aberrations in chromatin remodeling factors, but also a large amount of moderate and low frequency genetic lesions which may account for tumor heterogeneity and malignancy [9-13]. Among these novel identified mutated genes, SAMD9L was mutated in 5.6% of HBV-associated HCC specimens in our previous study [9], suggesting that SAMD9L plays a crucial role in a subset of HCC.
SAMD9L is located in chromosome 7q21.2 of the human genome and adjacent to its close paralogue SAMD9 in a head-to-tail orientation [14]. SAMD9 and SAMD9L may function redundantly or in related pathways, since these two proteins share considerable sequence similarity. Nonetheless, the evidence that patients with NFTC (Normophosphatemic Familial Tumoral Calcinosis) possess mutations only in SAMD9 suggests that the two proteins possibly perform non-identical roles in humans [15]. They have important roles in multiple cellular processes, such as cell proliferation, neoplastic phenotype and the immune responses to viral pathogens [14, 16]. The deleterious mutations or deletions of the two genes frequently occurred in several human disorders, such as inherited inflammatory calcified tumors and acute myeloid leukemia, suggesting their pathological importance [17-20]. Both SAMD9 and SAMD9L exhibited significantly decreased expression in T and B cell populations of patients with metastatic melanoma as compared with those from healthy control individuals [21]. Moreover, the human SAMD9L gene exhibits ubiquitous expression in human tissues and lower expression in breast cancer tissue than in normal breast tissue from the same patient [14]. SAMD9L could be induced by type I IFNs, and its function is correlated with its IFN-induced inhibitory effects on cell migration in activated human T cells [22]. Significantly, the observations in the Samd9l-deficient mice model demonstrated that Samd9l contributed to myeloid transformation through facilitating endosome fusion [23]. Those investigations suggest that SAMD9L plays an important role in human malignancies. In this study, we evaluate somatic mutations and expression of SAMD9L in HCC specimens, and investigate the roles of SAMD9L in HCC cell proliferation.
Materials and methods
Tumor tissue specimens
HCCs and adjacent non-cancerous liver specimens were obtained from the patients who were subjected to surgical liver resection and signed informed consent. These tissue specimens were immediately frozen at -80°C until DNA/RNA extraction. HCC diagnosis was confirmed by pathological examinations, and these HCC specimens were grouped according to TNM staging and Edmondson grading system. Protocols involving human samples and experimental animals were conducted in conformity with the ethical principles of research and approved by the ethic committee of the Chinese National Human Genome Center in Shanghai.
Cell lines and transfection
HCC cell lines were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 1 % penicillin/streptomycin and 10 % fetal bovine serum (Life Technologies) in a humidified chamber with 5 % CO2. Cell transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols.
Extraction of DNA and RNA
Genomic DNA was extracted from HCC specimens using DNeasy Tissue Kit (Qiagen), and total RNA was extracted from tissue or cell culture samples using the TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. The total RNA was treated by RNase-free DNase Ι (TaKaRa) to remove genomic DNA contamination.
Cell proliferation assay
HCC cells were seeded at 3000 cells per well of 96-well plates in triplicate. Cell viability was measured every day using Cell Counting Kit-8 (CCK-8, Dojindo Laboratories) according to the manufacturer's instructions.
Colony formation assay
5~10 × 104 HCC cells were cultured in the medium containing 0.6 mg/mL G418 (Life Technologies). The remaining colonies were stained with crystal violet. For soft agar colony formation assay, transfected cells were grown in medium containing 1 % base agar and 0.5 % top agar. These experiments were independently repeated at least 3 times. Colonies with more than 50 cells were counted and statistical significance was calculated by two-tailed student's t test.
RNA interference against SAMD9L
These siRNAs against SAMD9L were chemically synthesized by GenePharma (Shanghai, China) listed in Additional file 1: Supplementary Table 1. And then 2 effective siRNAs (si-1213, si-3558) confirmed by knockdown efficiency analysis were used to further function assay and their corresponding DNA nucleotides encoding short hairpin RNA (sh-1213, sh-3558) were subcloned into pSUPER vector with neomycin-resistant gene. pSUPER carrying an irrelevant nucleotide (pSUPER-shNC) was used as a negative control.
Tumorigenicity assay
HCC cells with stable SAMD9L knockdown descendant from SK-hep-1 and MHCC-97H were applied to xenograft tumorigenicity assay in nude mice. A total of 2 × 106 cells were injected subcutaneously into the flanks of 6-week-old male BALB/c nude mice. Tumor dimensions were measured each week using Vernier calipers, and tumor volume was estimated by the formula: tumor volume =π/6 × (major axis) × (minor axis)2. The nude mice were sacrificed at the end of experiment, and excised xenograft tumors were weighed. Statistical significance was determined by two-tailed student's t test.
Cell cycle analysis
Cells were harvested at 48 h after siRNA transfection, and the cells pellets were washed twice with phosphate-buffered saline (PBS) and re-suspended in PBS containing 50mg/ml propidium iodide (PI) and 100μg/ml DNase-free RNase A. Cell suspension was incubated for 30 min at room temperature and analyzed using the Flow Cytometry (Becton Dickinson). Cell cycle analysis was analyzed with CellQuest software and ModFit LT (Verity Software House, ME, USA).
Bromodeoxyuridine (BrdU) incorporation assay
For immunofluorescence assay, the cells were cultured with 20 μM BrdU (Sigma-Aldrich) for 2 h and incubated with anti-BrdU antibody, followed by incubation with Alexa Flour® Dyes (Invitrogen). For flow cytometry analysis, the cells treated with BrdU were incubated with the fluorescein isothiocyanate (FITC)-conjugated mouse anti-BrdU monoclonal antibody and mouse IgG served as a negative control (BD PharmingenTM).
Luciferase reporter assays
4 × 104 cells were seeded in 24-well plates and were transfected with TOP/FOP-flash plasmids and the two siRNAs against SAMD9L. Luciferase activity was measured using a Luciferase reporter assay system (Promega), according to the manufacturer's instructions.
Results
SAMD9L mutations are recurrent in HCC specimens
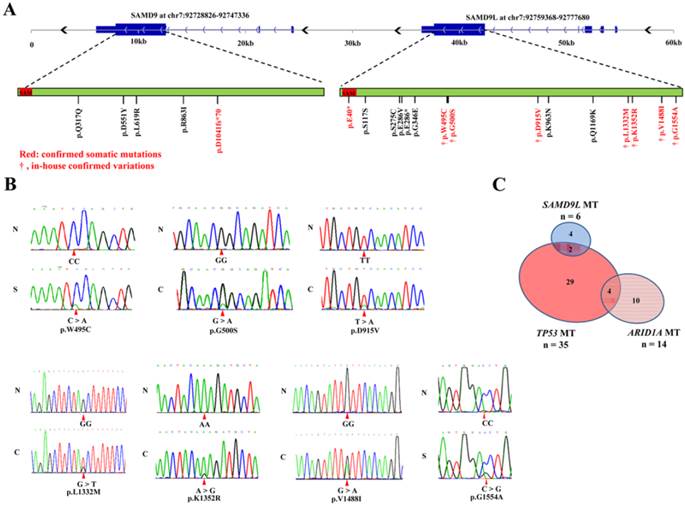
Our previous study on exome sequencing of hepatitis B virus-associated HCC identified 5 missense mutations in SAMD9L that occurred in 5 (5.6%) of 90 HCC specimens [9]. In this study, we further analyzed protein-coding exons sequences of SAMD9L in additional 40 paired HCC samples. The Sanger sequencing results showed 2 of the 40 HCC cases had missense mutations. Totally, 7 missense mutations in SAMD9L were found in 7 (5.4%) of a total of 130 HCC patients (Fig. 1A and B). Meanwhile, we also evaluated the mutation in SAMD9L and its paralogue SAMD9 in other liver carcinoma samples by means of related informations deposited on Sanger COSMIC (Catalogue Of Somatic Mutations In Cancer, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) database. Significantly, a respective confirmed somatic mutation in both SAMD9L (p.E40*) and SAMD9 (p.D1041fs*70) was identified in 2 HBV-positive HCC specimens from a published literature [24], and some genetic variants of the two genes were also found in liver carcinoma samples included in COSMIC database (Fig. 1A and Additional file 1: Supplementary Table 2). As known, bona fide tumor suppressor genes are mutated at a variety of positions throughout the coding region [25, 26]. These somatic mutations extensively locate within the coding region of SAMD9L and SAMD9 gene, suggesting that they act as HCC tumor suppressor genes. As for the mutation frequency of tumor suppressors in HBV-associated HCC, SAMD9L is next to TP53 and ARID1A [9, 11, 12]. Therefore, we next evaluated the mutation prevalence of the three tumor suppressors. The result showed that 35 (31.8%), 14 (12.7%) and 6 (5.5%) cases of 110 HCC specimens harbored somatic mutations in TP53, ARID1A and SAMD9L, respectively. Of the 30 HCC samples with TP53 mutations, only 2 exhibited SAMD9L mutations (P = 1.00, Fisher's exact test) (Fig. 1C), implying that an association between SAMD9L and TP53 mutations cannot be absolutely excluded. Overall, 5.4% of HBV-associated HCCs harbored SAMD9L mutations in China, suggesting that SAMD9L is a tumor suppressor for HBV-related HCC.
Somatic mutations of human SAMD9L and SAMD9 in HCC samples. (A) The somatic mutation distribution of human SAMD9L and SAMD9 genes in the liver cancer samples, including in-house HCC specimens and other liver carcinoma samples deposited in COSMIC (Catalogue Of Somatic Mutations In Cancer, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) database, is shown on a schematic describing genomic structures and transcripts of human SAMD9L and SAMD9 genes. The two genes contain a well conserved sterile alpha motif (SAM) domain, as indicated by red boxes in their protein-coding transcripts. The amino acid variations resulted from somatic mutations are shown for the two genes; confirmed somatic mutations were marked by red and in-house confirmed variations are indicated by symbol “†”. (B) DNA sequence chromatograms of SAMD9L mutations in HCC cohort examined. Red triangles indicate the mutation sites. N: adjacent non-cancerous liver, C: primary HCC specimen, S: PVTT (portal vein tumor thrombus) specimen. (C) The relationship between those HCC cases with TP53, ARID1A and SAMD9L mutation in 110 in-house HCC specimens, of which 35 carried TP53 mutation (Mut), 14 ARID1A Mut and 6 SAMD9L Mut.
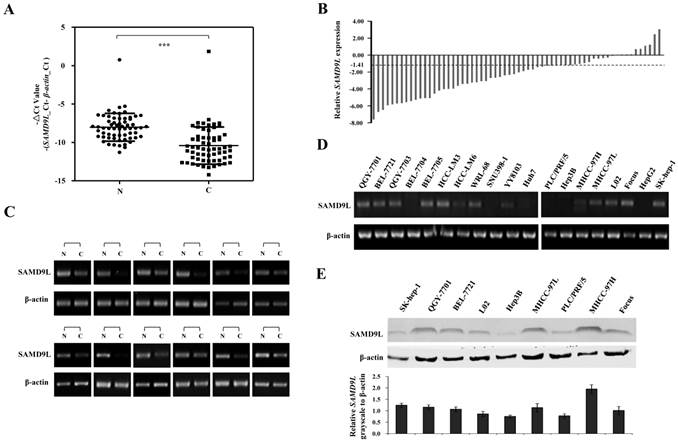
SAMD9L expression is frequently decreased in HCC specimens
To investigate the role of SAMD9L in HCC, the quantitative RT-PCR was employed to detect the mRNA level of SAMD9L in 62 HCC specimens and their adjacent non-cancerous liver tissues. The results showed that SAMD9L mRNA level was significantly decreased in HCC compared with that of the non-cancerous livers (P < 0.0001, Fig. 2A). Of the 62 HCC specimens examined, 36 (58.1%) exhibited at least a 2-fold decrease of SAMD9L expression as compared with that of the corresponding non-tumorous livers (Fig. 2B), and 12 with marked difference of -△Ct value between HCC and adjacent non-tumorous liver were confirmed by semi-quantitative PCR assay (Fig. 2C). Moreover, the relative expression fold-change of SAMD9L in the 50 paired HCC cases with available clinical information were shown in Additional file 1: Supplementary Table 3. However, SAMD9L down-regulation was not statistically correlated with the gender, age, HBV, AFP, tumor size, metastases and Edmondson (P > 0.05, Additional file 1: Supplementary Table 4). In addition, we evaluated the expression level of SAMD9L in 19 available HCC cell lines. The resulting data showed that SAMD9L exhibited low or no expression in almost half of these HCC cell lines, as shown by RT-PCR and Western blotting assay (Fig. 2D and E). To further investigate whether the epigenetic events could contribute to the down-regulation of SAMD9L, the demethylation agent DAC and histone deacetylase inhibitor TSA were employed to treat Huh7 and HepG2 cells without the endogenous SAMD9L expression. However, increased expression of SAMD9L was not observed by RT-PCR and no atypical CpG islands were identified in SAMD9L promoter region (Additional file 1: Supplementary Fig. 1), implying that SAMD9L expression is insensitive to regulation by these modifications of DNA methylation and histone acetylation in HCC cells.
Expression analysis of SAMD9L in HCC tissues and cell lines. (A-B) Transcript level of SAMD9L was measured in 62 HCC specimens and their adjacent non-cancerous livers by quantitative RT-PCR. The relative mRNA level of SAMD9L was normalized based on that of an internal reference β-actin. Each plotting dot indicates the expression level of a given HCC specimen or adjacent liver tissue and lines represent the median with standard deviation of -△Ct value. P value was calculated by student's t test. N, adjacent non-cancerous livers; C, HCC specimens. ***, P < 0.001. (B) Relative expression of SAMD9L mRNA for the 62 HCC specimens as compared to their adjacent non-cancerous livers. (C) Representative results of semi-quantitative RT-PCR of SAMD9L from 12 paired samples of HCC patients. (D) The transcript level of SAMD9L was measured in 19 HCC cell lines using quantitative PCR, where β-actin was used as internal reference. (E) SAMD9L expression was evaluated in some available HCC cell lines by Western blotting assay. The grayscale intensities of western blots were measured and β-actin was used as a normalization control. Data shown are representative of three independent experiments.
SAMD9L knockdown promotes cell growth and colony formation
To determine the contribution of SAMD9L inactivation to HCC proliferation, two siRNAs against SAMD9L (si-1213, si-3558) were employed to silence endogenous SAMD9L. The two siRNAs significantly knocked SAMD9L down compared with a reference control si-Luc, demonstrated by quantitative RT-PCR in Sk-hep-1, QGY-7701, BEL-7721 and MHCC-97H cells (Fig. 3A).
SAMD9L knockdown promotes the growth and colony formation of HCC cell in vitro. (A) The efficiency of two siRNA (si-1213, si-3558) against endogenous SAMD9L was evaluated by quantitative RT-PCR assay in SK-hep-1, QGY-7701, BEL-7721 and MHCC-97H cells, where si-Luc was used as a negative control. (B) Cell growth curves were measured based on cell viability while the two siRNAs (si-1213, si-3558) were transfected into SK-hep-1, QGY-7701, BEL-7721 and MHCC-97H cells, respectively. (C) Upon SAMD9L knockdown mediated by pSUPER-sh1213 and pSUPER-sh3558 constructs, anchorage-dependent colony formation assay was performed in SK-hep-1, QGY-7701 and BEL-7721 cells. pSUPER vector carrying shLuc was used as a negative control. The histograms showed mean with standard deviation of colony numbers and statistical significance was calculated using two-tailed student's t test. *, P < 0.01; **, P < 0.01. (D) Representative microscopic photographs of anchorage-independent colony formation assay were shown in soft agar medium when SAMD9L was silenced in MHCC-97H cells. Here all experiments were performed independently 3 times.
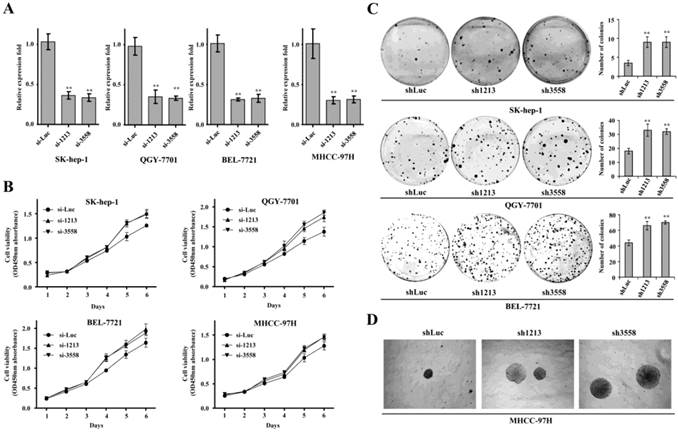
Subsequently, we performed cell growth curve assay when the two siRNAs were transfected into these HCC cell lines. The resulting data showed that SAMD9L knockdown obviously promoted proliferation of Sk-hep-1, QGY-7701, BEL-7721 and MHCC-97H cells (Fig. 3B).
Furthermore, we performed colony formation assay by transfecting recombinant pSUPER constructs carrying shRNA (sh1213, sh3558) against SAMD9L into those HCC cells. The results demonstrated that SAMD9L knockdown significantly promoted anchorage-dependent colony formation of SK-hep-1, QGY-7701 and BEL-7721 cells (Fig. 3C), and enhanced anchorage-independent colony formation of MHCC-97H cells cultured in soft agar medium (Fig. 3D). These collective data implied that loss of SAMD9L function promoted HCC proliferation in vitro, supporting that SAMD9L is a tumor suppressor.
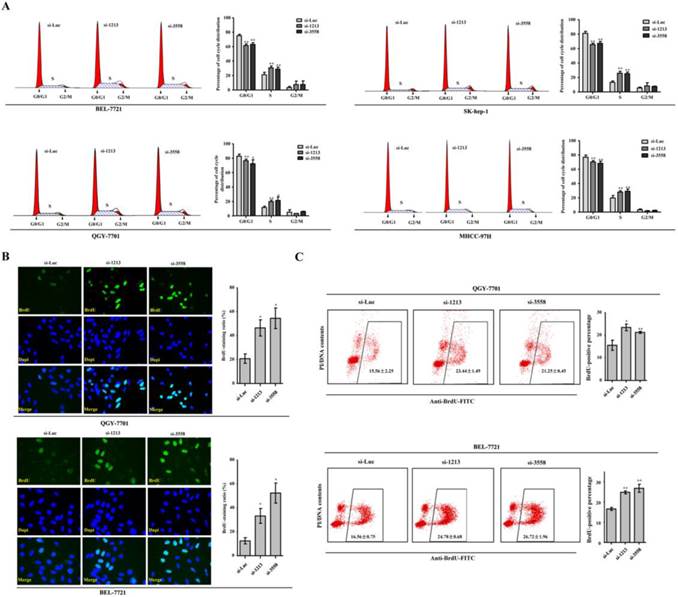
RNA interference against SAMD9L facilitates G1-S transition
In search of the cellular events involved in the promoting effects of SAMD9L knockdown on HCC proliferation, we analyzed cell cycle when the two siRNAs against SAMD9L were transfected into HCC cells. Significantly, the proportion of cell population in S phase increased, whereas the proportion of cell population in G0/G1 phase decreased in Sk-hep-1, QGY-7701, BEL-7721 and MHCC-97H cells (Fig. 4A). To further verify the observation that SAMD9L knockdown facilitated G1-S transition of cell cycle progression, de novo DNA synthesis was evaluated by BrdU incorporation assay. Expectedly, SAMD9L knockdown led to the significant increase of BrdU-incorporating ratios in QGY-7701 and BEL-7721 cells, as demonstrated by immunofluorescence assay with BrdU antibody (Fig. 4B). Furthermore, flow cytometry analysis also supported the similar results in QGY-7701 and BEL-7721 cells (Fig. 4C). SAMD9L knockdown facilitates entry into S phase of cell cycle and this cellular event could account for increased HCC cell proliferation.
SAMD9L knockdown leads to accelerated G1-S transition of cell cycle progression. (A) Cell cycle analysis by DNA content (Propidium Iodide) was performed using flow cytometry when the two siRNAs against SAMD9L were transfected into BEL-7721, QGY-7701, SK-hep-1 and MHCC-97H cells for 24 h. Cell cycle models fit by ModFit LT software (left) and the average percentages of G0/G1, S and G2/M phases (right) were shown. (B) Representative vision fields of immunofluorescence assay with anti-BrdU antibody were shown, where BrdU-incorporated signal were green and nucleus were stained blue with Dapi in QGY-7701, BEL-7721 cells with SAMD9L knockdown. The histograms demonstrated mean with standard deviation of BrdU-incorporated ratio. (C) The scattergrams of FACS analysis present the BrdU-incorporated population of QGY-7701, BEL-7721 cells, upon SAMD9L knockdown. The histograms indicates mean with standard deviation of BrdU-positive percentage. All experiments were performed with three replicates and data are shown as mean ± SD. Statistical significance was determined by two-tailed student's t test. *, P < 0.01; **, P < 0.01.
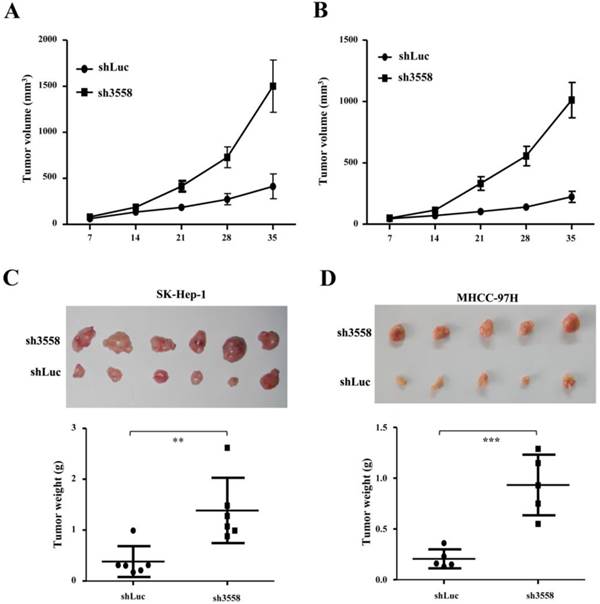
SAMD9L knockdown promoted tumorigenicity in nude mice
To further address the effect of SAMD9L knockdown on tumorigenicity in vivo, offspring cells with endogenous SAMD9L knockdown were established by transfecting pSUPER carrying sh3558 against SAMD9L into SK-hep-1 and MHCC-97H cells. The transfected cells were subjected to resistant selection for a week, and then were inoculated subcutaneously into a flank of athymic mice, while control cells transfected with pSUPER-shLuc were injected into the opposite side of the same mice. Intriguingly, SAMD9L knockdown increased tumor growth in the two groups of inoculated mice, where the tumor size under observation (Fig. 5A and B) and the wet weight (Fig. 5C and D) of xenograft tumors formed from cells transfected with pSUPER-sh3558 were significantly increased as compared with that of the control. Subsequently, SAMD9L expression levels in these excised xenograft tumors were evaluated by quantitative RT-PCR (Additional file 1: Supplementary Fig. 2).
SAMD9L knockdown promotes tumor growth of subcutaneous xenograft of HCC cells in nude mice. (A) SK-hep-1 and (B) MHCC-97H cells with stable SAMD9L silence were established, and a total of 2 ×106 cells were subcutaneously injected into a flank of nude mice, while cells stably transfected with pSUPER-shLuc as controls were subcutaneously injected into the opposite flanks of the same mice. Tumor dimensions were measured once every week using a digital caliper for 5 weeks, and then observed mean tumor volume (± SEM) was shown. (C and D) The two groups of nude mice were sacrificed at the end of 5-week experiment. These excised tumors are shown and tumor weights were statistically analyzed using two-tailed Student's t test.
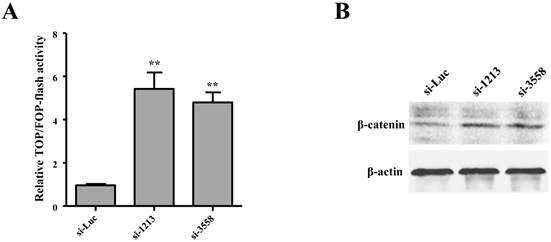
SAMD9L knockdown activates Wnt/β-catenin pathway
Wnt/β-catenin pathway is often activated but with a low incidence of β-catenin mutations in HBV-related HCC [27]. We attempted to investigate the role of SAMD9L in regulating the activity of Wnt/β-catenin pathway by employing TOPflash and TOPflash reporters, which are widely applied to reflect β-catenin signaling activity. In order to determine the relationship between SAMD9L down-regulation and the activity of the Wnt/β-catenin pathway, we first evaluated the effects of SAMD9L knockdown on Wnt/β-catenin activity. Upon SAMD9L knockdown, the activity of the Wnt/β-catenin pathway was significantly elevated in SK-hep-1 cells, as shown by the luciferase activity assay (Fig. 6A). Furthermore, Western blot assay demonstrated the expression level of β-catenin increased when SAMD9L was silenced in SK-hep-1 cells (Fig. 6B). Taken together, the results suggest SAMD9L plays an important role in regulating the activity of Wnt/β-catenin signaling pathway.
Wnt/β-catenin signal pathway is involved in the role of SAMD9L in HBV-associated HCC. (A) Effect of SAMD9L knockdown on TOP/FOP flash activity in SK-hep-1 cells. Statistical significance was calculated by the two-tailed student's t test. **, P < 0.01. (B) The protein expression level of β-catenin was examined using western blot assay when the two siRNAs (si-1213, si-3558) against SAMD9L were transfected into SK-hep-1 cells.
Discussion
This work followed a previous finding about recurrent SAMD9L mutations in HBV-related HCC patients, and defined SAMD9L as a novel HCC tumor suppressor. We demonstrated that somatic mutation in SAMD9L and down-regulated expression of SAMD9L were implicated with more than 60% of cases in this HCC cohort. Compared with SAMD9L, occasional mutations in SAMD9 were also found in several cohorts of liver carcinomas including our in-house HCC cohort (data not shown). SAMD9L down-regulation could be attributed to various abnormal genetic and/or epigenetic events such as loss of heterozygosity (LOH), deletion, promoter methylation, histone modifications and chromatin remodeling. However, chromosome 7q21.3 locus containing SAMD9L was amplified in HCC [28], and SAMD9L gene promoter had no typical CpG islands, a key regulatory DNA element for methylation modification. More importantly, SAMD9L expression exhibits no response to DAC and/or TSA treatment in HCC cells. This result suggests that additional genetic and/or epigenetic alterations lead to SAMD9L dysregulation in the development of HCC.
SAMD9/SAMD9L genes were localized on a common microdeletion cluster of 7q21.3 in patients with juvenile myelomonocytic leukemia and Samd9l-deficient mice with developed myeloid diseases confirm their roles as tumor suppressors in myeloid malignancy [17, 23]. Moreover, the expression correlation of SAMD9 and SAMD9L in HCC tissue specimens was evaluated through analyzing public microarray databases. The results show that there is a high expression correlation between SAMD9L and its paralogue SAMD9 in those liver tissues from the two independent datasets (Additional file 1: Supplementary Fig. 3). The study demonstrated their tumor-suppressive roles in HBV-associated HCC, based on the observed effects of SAMD9L knockdown on HCC cell lines. We also constructed the plasmid vector carrying the full-length SAMD9L-coding sequence and then transfected into HCC cells. However, we didn't observe significant effect of SAMD9L overexpression on cell proliferation due to low transfection efficiency (data not shown). Hence, further investigation is needed to evaluate the effects of SAMD9L overexpression on HCC cells by means of some advanced approaches to improve gene expression efficiency, such as retrovirus system and CRISPR/Cas9-mediated genome editing system [29, 30].
Furthermore, the specific mechanism by which SAMD9L contributes to HBV-related HCC remains unclear. Interestingly, the two proteins of SAMD9 and SAMD9L share considerable sequence similarity suggesting they may function redundantly in related pathways. And recently human SAMD9 was identified as a novel innate antiviral factor against poxviruses [31] and as one of host proteins involved in Japanese encephalitis virus infection [32], indicating that SAMD9/SAMD9L possess anti-viral properties and play crucial role in host defense against viral pathogens. Since deleterious mutations in the SAMD9 led to a rare autosomal recessive disease, normophosphatemic familial tumoral carcinosis (NFTC) characterized by abnormal inflammation of the skin and gingiva induced by excessive signaling via tumor necrosis factor α and/or interferon-γ pathways [18, 20], thus it could be suggested that SAMD9/SAMD9L suppress these inflammatory pathways. As known, inflammation as the hallmark of chronic hepatitis is a major trigger in the development of HCC [33, 34]. Chronic inflammation might activate cell proliferation and deregulate mechanisms of cell death [35]. Hence, we proposed a model to demonstrate the possible mechanisms by which SAMD9/SAMD9L contribute to the development of HBV-associated HCC (Additional file 1: Supplementary Fig. 4). In the proposed model, SAMD9/SAMD9L may play physiological roles in anti-infection of hepatitis B viral and/or inhibition on inflammation response, whereas their inactivation caused by genetic lesions and down-regulation could contribute to the initiation and development of HBV-associated HCC. Besides the Wnt/β-catenin signaling pathway, the JAK-STAT3 and NF-κB pathways, which closely modulate the inflammatory response [36-38], may be implicated with the tumor-suppressive roles of SAMD9/SAMD9L in hepatitis-related HCC.
Supplementary Material
Additional File 1Supplementary Figures and tables.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (81101875 and 81201690), Shanghai Natural Science Foundation (11ZR1425300), the Chinese National Key Program on Basic Research (2010CB529204, 2010CB529206 and 2012CB722308), the China National Key Projects for Infectious Disease (2012ZX10002012-008 and 2013ZX10002010-006). We thank Dr. Edward J Richards for English manuscript editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11-30 doi:10.3322/caac.21166
2. Martin J, Dufour JF. Tumor suppressor and hepatocellular carcinoma. World journal of gastroenterology: WJG. 2008;14:1720-33
3. Tirnitz-Parker JE, Glanfield A, Olynyk JK, Ramm GA. Iron and hepatic carcinogenesis. Critical reviews in oncogenesis. 2013;18:391-407
4. McKillop IH, Schrum LW. Role of alcohol in liver carcinogenesis. Seminars in liver disease. 2009;29:222-32 doi:10.1055/s-0029-1214377
5. Moudgil V, Redhu D, Dhanda S, Singh J. A review of molecular mechanisms in the development of hepatocellular carcinoma by aflatoxin and hepatitis B and C viruses. Journal of environmental pathology, toxicology and oncology: official organ of the International Society for Environmental Toxicology and Cancer. 2013;32:165-75
6. Staib F, Hussain SP, Hofseth LJ, Wang XW, Harris CC. TP53 and liver carcinogenesis. Human mutation. 2003;21:201-16 doi:10.1002/humu.10176
7. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O. et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:8847-51
8. Rebouissou S, Amessou M, Couchy G, Poussin K, Imbeaud S, Pilati C. et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature. 2009;457:200-4 doi:10.1038/nature07475
9. Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N. et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nature genetics. 2012;44:1117-21 doi:10.1038/ng.2391
10. Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA. et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nature genetics. 2011;43:828-9 doi:10.1038/ng.903
11. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature genetics. 2012;44:694-8 doi:10.1038/ng.2256
12. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH. et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature genetics. 2012;44:760-4 doi:10.1038/ng.2291
13. Knudsen ES, Gopal P, Singal AG. The Changing Landscape of Hepatocellular Carcinoma: Etiology, Genetics, and Therapy. The American journal of pathology. 2014;184:574-83 doi:10.1016/j.ajpath.2013.10.028
14. Li CF, MacDonald JR, Wei RY, Ray J, Lau K, Kandel C. et al. Human sterile alpha motif domain 9, a novel gene identified as down-regulated in aggressive fibromatosis, is absent in the mouse. BMC genomics. 2007;8:92. doi:10.1186/1471-2164-8-92
15. Hershkovitz D, Gross Y, Nahum S, Yehezkel S, Sarig O, Uitto J. et al. Functional characterization of SAMD9, a protein deficient in normophosphatemic familial tumoral calcinosis. The Journal of investigative dermatology. 2011;131:662-9 doi:10.1038/jid.2010.387
16. Lemos de Matos A, Liu J, McFadden G, Esteves PJ. Evolution and divergence of the mammalian SAMD9/SAMD9L gene family. BMC evolutionary biology. 2013;13:121. doi:10.1186/1471-2148-13-121
17. Asou H, Matsui H, Ozaki Y, Nagamachi A, Nakamura M, Aki D. et al. Identification of a common microdeletion cluster in 7q21.3 subband among patients with myeloid leukemia and myelodysplastic syndrome. Biochemical and biophysical research communications. 2009;383:245-51 doi:10.1016/j.bbrc.2009.04.004
18. Chefetz I, Ben Amitai D, Browning S, Skorecki K, Adir N, Thomas MG. et al. Normophosphatemic familial tumoral calcinosis is caused by deleterious mutations in SAMD9, encoding a TNF-alpha responsive protein. The Journal of investigative dermatology. 2008;128:1423-9 doi:10.1038/sj.jid.5701203
19. Dereure O. [SAMD9 mutation in normophosphatemic familial tumoral calcinosis]. Annales de dermatologie et de venereologie. 2007;134:505
20. Topaz O, Indelman M, Chefetz I, Geiger D, Metzker A, Altschuler Y. et al. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. American journal of human genetics. 2006;79:759-64 doi:10.1086/508069
21. Critchley-Thorne RJ, Yan N, Nacu S, Weber J, Holmes SP, Lee PP. Down-regulation of the interferon signaling pathway in T lymphocytes from patients with metastatic melanoma. PLoS medicine. 2007;4:e176. doi:10.1371/journal.pmed.0040176
22. Pappas DJ, Coppola G, Gabatto PA, Gao F, Geschwind DH, Oksenberg JR. et al. Longitudinal system-based analysis of transcriptional responses to type I interferons. Physiological genomics. 2009;38:362-71 doi:10.1152/physiolgenomics.00058.2009
23. Nagamachi A, Matsui H, Asou H, Ozaki Y, Aki D, Kanai A. et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer cell. 2013;24:305-17 doi:10.1016/j.ccr.2013.08.011
24. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z. et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome research. 2013;23:1422-33 doi:10.1101/gr.154492.113
25. Campbell RM, Tummino PJ. Cancer epigenetics drug discovery and development: the challenge of hitting the mark. The Journal of clinical investigation. 2014;124:64-9 doi:10.1172/JCI71605
26. Choi JD, Lee JS. Interplay between Epigenetics and Genetics in Cancer. Genomics & informatics. 2013;11:164-73 doi:10.5808/GI.2013.11.4.164
27. Moeini A, Cornella H, Villanueva A. Emerging Signaling Pathways in Hepatocellular Carcinoma. Liver cancer. 2012;1:83-93 doi:10.1159/000342405
28. Dong H, Zhang H, Liang J, Yan H, Chen Y, Shen Y. et al. Digital karyotyping reveals probable target genes at 7q21.3 locus in hepatocellular carcinoma. BMC medical genomics. 2011;4:60. doi:10.1186/1755-8794-4-60
29. Diez-Villasenor C, Guzman NM, Almendros C, Garcia-Martinez J, Mojica FJ. CRISPR-spacer integration reporter plasmids reveal distinct genuine acquisition specificities among CRISPR-Cas I-E variants of Escherichia coli. RNA biology. 2013;10:792-802 doi:10.4161/rna.24023
30. Djordjevic M, Djordjevic M, Severinov K. CRISPR transcript processing: a mechanism for generating a large number of small interfering RNAs. Biology direct. 2012;7:24. doi:10.1186/1745-6150-7-24
31. Liu J, Wennier S, Zhang L, McFadden G. M062 is a host range factor essential for myxoma virus pathogenesis and functions as an antagonist of host SAMD9 in human cells. Journal of virology. 2011;85:3270-82 doi:10.1128/JVI.02243-10
32. Zhang LK, Chai F, Li HY, Xiao G, Guo L. Identification of host proteins involved in Japanese encephalitis virus infection by quantitative proteomics analysis. Journal of proteome research. 2013;12:2666-78 doi:10.1021/pr400011k
33. Sun B, Karin M. Inflammation and liver tumorigenesis. Frontiers of medicine. 2013;7:242-54 doi:10.1007/s11684-013-0256-4
34. Lata J. Chronic liver diseases as liver tumor precursors. Dig Dis. 2010;28:596-9 doi:10.1159/000320057
35. Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clinical reviews in allergy & immunology. 2009;36:4-12 doi:10.1007/s12016-008-8091-0
36. Martin M, Herceg Z. From hepatitis to hepatocellular carcinoma: a proposed model for cross-talk between inflammation and epigenetic mechanisms. Genome medicine. 2012;4:8. doi:10.1186/gm307
37. Gao B, Wang H, Lafdil F, Feng D. STAT proteins - key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. Journal of hepatology. 2012;57:430-41 doi:10.1016/j.jhep.2012.01.029
38. Cairo S, Buendia MA. How transient becomes stable: an epigenetic switch linking liver inflammation and tumorigenesis. Journal of hepatology. 2012;57:910-2 doi:10.1016/j.jhep.2012.05.017
Author contact
![]() Corresponding author: Qing Deng PhD, Shanghai-MOST Key Laboratory for Disease and Health Genomics, Chinese National Human Genome Center at Shanghai, 351 Guo Shou-Jing Road, Shanghai 201203, China. E-mail: dengqsh.cn; Ze-Guang Han MD, Key Laboratory of Systems Biomedicine (Ministry of Education) of Rui-Jin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China. E-mail: hanzgsh.cn; Tel: +86-21-50801325; Fax: +86-21-50800402.
Corresponding author: Qing Deng PhD, Shanghai-MOST Key Laboratory for Disease and Health Genomics, Chinese National Human Genome Center at Shanghai, 351 Guo Shou-Jing Road, Shanghai 201203, China. E-mail: dengqsh.cn; Ze-Guang Han MD, Key Laboratory of Systems Biomedicine (Ministry of Education) of Rui-Jin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China. E-mail: hanzgsh.cn; Tel: +86-21-50801325; Fax: +86-21-50800402.