Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):882-894. doi:10.7150/ijbs.8737 This issue Cite
Research Paper
Inhibition of Elastase-Pulmonary Emphysema in Dominant-Negative MafB Transgenic Mice
Yasuko Aida, Yoko Shibata ![]() , Shuichi Abe, Sumito Inoue, Tomomi Kimura, Akira Igarashi, Keiko Yamauchi, Keiko Nunomiya, Hiroyuki Kishi, Takako Nemoto, Masamichi Sato, Michiko Sato-Nishiwaki, Hiroshi Nakano, Kento Sato, Isao Kubota
, Shuichi Abe, Sumito Inoue, Tomomi Kimura, Akira Igarashi, Keiko Yamauchi, Keiko Nunomiya, Hiroyuki Kishi, Takako Nemoto, Masamichi Sato, Michiko Sato-Nishiwaki, Hiroshi Nakano, Kento Sato, Isao Kubota
Department of Cardiology, Pulmonology, and Nephrology, School of Medicine, Yamagata University, 2-2-2 Iida-Nishi, Yamagata 990-9585, Japan.
Received 2014-2-1; Accepted 2014-7-30; Published 2014-8-13
Abstract
Alveolar macrophages (AMs) play important roles in the pathogenesis of chronic obstructive pulmonary disease (COPD). We previously demonstrated upregulation of the transcription factor MafB in AMs of mice exposed to cigarette smoke. The aim of this study was to elucidate the roles of MafB in the development of pulmonary emphysema. Porcine pancreatic elastase was administered to wild-type (WT) and dominant-negative (DN)-MafB transgenic (Tg) mice in which MafB activity was suppressed only in macrophages. We measured the mean linear intercept and conducted cell differential analysis of bronchoalveolar lavage (BAL) cells, surface marker analysis using flow cytometry, and immunohistochemical staining using antibodies to matrix metalloproteinase (MMP)-9 and MMP-12. Airspace enlargement of the lungs was suppressed significantly in elastase-treated DN-MafB Tg mice compared with treated WT mice. AMs with projected pseudopods were decreased in DN-MafB Tg mice. The number of cells intermediately positive for F4/80 and weakly or intermediately positive for CD11b, which are considered cell subsets of matured AMs, decreased in the BAL of DN-MafB Tg mice. Furthermore, MMP-9 and -12 were significantly downregulated in BAL cells of DN-MafB Tg mice. Because MMPs exacerbate emphysema, MafB may be involved in pulmonary emphysema development through altered maturation of macrophages and MMP expression.
Keywords: Pulmonary emphysema, MafB, alveolar macrophage, matrix metalloproteinase, gene targeted mouse.
Introduction
Chronic obstructive pulmonary disease (COPD) is the fourth cause of death worldwide [1]. Pulmonary emphysema is a representative type of COPD and pathologically characterised by the enlargement of alveoli in the lung. Pulmonary emphysema is thought to be caused by chronic inflammation induced by increased numbers of inflammatory cells releasing potentially destructive mediators, namely matrix metalloproteinases (MMPs) [2,3]. Of inflammatory cells, the numbers of alveolar macrophages (AMs) increase in the lungs of COPD patients [4]. AMs play important roles in the pathogenesis and pathophysiology of pulmonary emphysema [5,6].
We previously demonstrated that the transcription factor MafB was upregulated in AMs in the lungs of mice with cigarette smoke-induced pulmonary emphysema [7]. In addition, we reported that the intensity of immunostaining of MafB in AMs correlated with the degree of airflow limitation in human smokers [8]. MafB, also known as V-maf musculoaponeurotic fibrosarcoma oncogene homolog B, is a member of the Maf protein family and possesses a basic leucine zipper structure at the carboxyl terminus. MafB is expressed only in monocytes and macrophages among hematopoietic cells, and it plays an important role in the terminal differentiation of macrophages [9]. MafB forms a homodimer and binds Maf recognition element (MARE) to transactivate the transcription of MARE regulated genes [10]. A truncated form of MafB lacking an N-terminal acidic domain reportedly acts in a dominant negative (DN) manner [9]. DN MafB forms heterodimers with endogenous MafB and inhibits the binding of endogenous MafB homodimers with MARE. In addition, it was demonstrated that MafB binds the MARE motif in the promoter region of the MMP-9 gene and up-regulates MMP-9 in macrophages [11].
MafB knockout mice die immediately after birth due to developmental anomalies of neurons in the respiratory centre [12,13]. Thus, the role of MafB in the pathogenesis of pulmonary emphysema has not been fully investigated in vivo due to the lack of an adequate animal model. To explore the role of MafB in COPD, we generated transgenic mice that express DN MafB capable of suppressing endogenous MafB transcription activity only in macrophages [14]. These transgenic mice had a similar survival rate compared with WT mice and enabled us to generate an adequate emphysema mouse model.
The elastase-induced emphysema model is a conventional experimental emphysema model. Elastase is a serine protease that has the capability of digesting elastin. Elastin is one of the major component proteins of the lung, and intratracheal administration of elastase is known to cause airspace enlargement in the lungs of experimental animals [15,16]. It takes for 3 or 4 weeks to induce pulmonary emphysema in the lungs of mice, and inflammatory mediators and endogenous MMPs from accumulated macrophages are thought to play a principal role in that emphysematous change [17-19]. Administration of macrophage-colony stimulating factor, which induces MMP-9 and -12 expression in AMs, following elastase intratracheal administration augments the development of pulmonary emphysema more than saline administration following elastase treatment, which suggests that MMPs play important roles in animal models of elastase-induced pulmonary emphysema [18]. Because this model is suited to investigate the function of macrophages in emphysema, we applied the elastase-induced emphysema model to DN-MafB Tg mice and investigated the role of MafB in the pathogenesis of pulmonary emphysema.
Methods
Mice
As described previously, we have established the macrophage scavenger receptor enhancer-promoter dominant-negative MafB transgenic (DN-MafB Tg) mice on the C57/BL6 background in which the activity of MafB was suppressed only in macrophages [14]. Eight- to twelve-week-old male mice and wild-type (WT) control mice, purchased from CLEA Japan (Tokyo, Japan), were used in these experiments. The study was approved by the Committee for Animal Experimentation, Yamagata University School of Medicine and was carried out in accordance with the Declaration of Helsinki.
An emphysematous model
Mice were anaesthetised with an intraperitoneal injection of pentobarbital sodium (150 mg/kg body weight). Porcine pancreatic elastase (PPE; Sigma-Aldrich, St Louis, MO, USA) in saline was administered at a dose of 1.5 U/100 μL using a MicroSprayer aerosoliser (1A-1C; Penn-Century, Philadelphia, PA, USA) attached to a high-pressure syringe (FMJ-250; Penn-Century). Control mice were treated similarly but with 100 μl of saline only. After 7 d and 21 d of treatment, the mice were sacrificed for lung fixation and for bronchoalveolar lavage (BAL) analysis.
Lung fixation and morphometry
After intraperitoneal injection of pentobarbital, the animals were exsanguinated by cutting the distal aorta. The lungs were fixed intratracheally with buffered formalin (4%) at a constant pressure of 25 cm H2O to prepare paraffin-embedded lung blocks. These sections were subsequently stained with hematoxylin and eosin or Elastica van Gieson. In the lungs after 21 d of PPE administration, the mean linear intercept (MLI), as a measure of the interalveolar septal wall distance, was measured using a light microscope at a magnification of ×200. The MLI was obtained by dividing the length of a line drawn across the lung section by the total number of intercepts encountered in 50 lines per mouse lung, as described previously [20,21].
BAL
BAL was performed by infusing 1 mL of Hanks' balanced salt solution (HBSS) with 0.5 mM ethylenediamine tetraacetic acid five times via a 20-gauge catheter inserted into the trachea. The BAL fluid was centrifuged at 1200 rpm for 5 min. The BAL cells were fixed on slides by subjecting the BAL fluid to a cytospin of 1000 rpm for 5 min and subsequently stained with a Diff-Quick solution (International Reagents Corp., Kobe, Japan) for differential cell counts.
Evaluation of the shape of AMs
To examine whether AM shape alteration occurred in the DN-MafB Tg mice, we divided macrophage lineage cells into three types in accordance with optical microscope findings: macrophages with projected pseudopods, macrophages without projected pseudopods (not recognised with an optical microscope), and alveolar monocyte-like cells. Alveolar monocyte-like cells exhibited a smaller cellular size and less cytoplasmic granularity than differentiated macrophages but were bigger than lymphocytes. One-hundred cells were classified in each mouse.
Immunohistochemistry of AMs
Cytospun BAL cells were fixed with an acetone/methanol (1:1) solution and incubated with Mac-3 (Becton, Dickinson and Company, San Diego, CA, USA; diluted 1:100), CD11c (Abcam, Cambridge, MA, USA; diluted 1:50), metalloproteinase (MMP)-9, or MMP-12 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; diluted 1:50, 1:100, respectively) antibodies and then secondary antibodies. Nuclei were counterstained with hematoxylin in the immunohistochemical analysis of MMP-9 and MMP-12.
Flow cytometry
AM cell surface markers (F4/80, CD11b and CD11c) were analysed using flow cytometry. The BAL cells were recovered from untreated mice or mice 7 d after PPE administration. Cells were resuspended in fluorescence-activated cell sorter (FACS) buffer (PBS, 0.2% BSA, 0.01% sodium azide). Erythrocytes were haemolysed before analysis. The BAL cells were stained with FITC-labelled F4/80 antibody (AbD serotec), PerCP-cy5.5-labelled CD11b antibody (Becton, Dickinson and Company), PerCP-cy5.5-labelled CD11c antibody (Becton, Dickinson and Company), PE-labelled Gr-1 antibody (Becton, Dickinson and Company), or an isotype control (Becton, Dickinson and Company) at 4°C for 30 min. The stained cells were subjected to flow cytometry using a FACS Canto II flow cytometer (BD Biosciences). To avoid the contamination of neutrophils in the study, Gr-1 positive cells were omitted from the analysis. For the analyses of cells 7 d after PPE administration, Gr-1 negative cells were divided into three groups based on their F4/80 and CD11b fluorescence intensity. The evaluation of these surface markers was performed based on the ratio of each of the three groups to all Gr-1 negative cells. For analyses of cells from untreated mice, the fluorescence intensity of CD11c in Gr-1 negative and F4/80-positive cells were assessed in TG and control mice. Data were analysed using DIVA software (BD Bioimaging systems, MD, USA).
Biochemical analysis of BAL fluid
Murine TNF-alpha and IL-6 in the BAL fluid were measured by ELISA (Quantikine; R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Reverse transcription (RT)-PCR
Total RNA of AMs was isolated using an RNeasy Mini Kit (QIAGEN, Valencia, CA, USA) according to the manufacturer's protocol. RT was performed using SuperScript III (Invitrogen). The sequences of the specific primers were: MafB forward, CGT CCA GCA GAA ACA TCA CC; reverse, TCG CAC TTG ACC TTG TAG GC; GAPDH forward, CTT CAC CAC CAT GGA GAA GGC; reverse, GGC ATG GAC TGT GGT CAT GAG. PCR products were electrophoresed in 1.5% agarose gels containing ethidium bromide and visualized digitally on a UV illuminator (AB1500 Printgraph and AE 6905H Image Saver HR; ATTO Bioscience, Tokyo, Japan) [14].
Establishment of macrophage cell line constitutively expressing MafB short hairpin (sh)-RNA
The MafB sh-RNA plasmid and control scrambled-sequence sh-RNA plasmid vector were generated by inserting the double-strand oligonucleotide (MafB sh-RNA sense, GCC TAC AAG GTC AAG TGC GAG AAA CTC GCC; MafB sh-RNA antisense, GGC GAG TTT CTC GCA CTT GAC CTT GTA GGC; control sh-RNA sense, GCG AGA CAA CCA GTG AAC TGG TCT AAC GCC; control sh-RNA antisense, GGC GTT AGA CCA GTT CAC TGG TTG TCT CGC) into piGENETMtRNApuro (Takara, Tokyo, Japan) according to the manufacturer's protocol. Constitutive expression of MafB sh-RNA was achieved by transfection of RAW264.7 cells with a plasmid vector expressing sh-RNA MafB by GenePORTER2 (Gene Therapy Systems Inc., San Diego, CA) [7]. As a transfection control, a scrambled-sequence sh-RNA plasmid was transfected into RAW264.7 cells. Antibiotic selection with puromycin (10 μg/ml) was started 48 h after transfection. After 10-20 days in selection medium, the cells were subjected to a limiting dilution method to obtain MafB-repressing clones. Expression of MafB in each clone was examined using RT-PCR. From all 45 clones, the most suppressive clone was selected as MafB-repressing cells. These cells were designated as RAW264.7-MafB-shRNA, whereas control cells were named RAW264.7-control-shRNA.
Activity of nuclear factor-kappa B (NF-kB) and activator protein (AP)-1
RAW264.7-MafB-shRNA and RAW264.7-control-shRNA cells were treated with porcine pancreatic elastase at the concentration of 100 mU/mL in Dulbecco's Modified Eagle Medium containing 10% foetal bovine serum for 30 min in a 5% CO2 incubator. Nuclear protein was isolated from these cells using Nuclear Extract Kit (Active Motif, Carlsbad, CA) according to manufacturer's protocol, and 10 ng of nuclear protein was used to assess the activity of NF-kB p65 and AP-1, c-fos, using TransAM® NFκB Family Transcription Factor ELISA Kit (Active Motif, Carlsbad, CA).
Statistical analyses
The Kruskal-Wallis test for nonparametric data was used to analyse the difference between the three groups. The results are expressed as the mean ± standard deviation. Significance was inferred for P values < 0.05. Statistical analyses were performed using JMP 9.0 computer software (SAS Institute, Inc., Cary, NC, USA).
Results
Suppression of emphysematous change in the lungs of elastase-treated DN-MafB Tg mice
The airspaces of the WT mice 21 d after PPE administration were larger than those of the saline-treated control mice (Figure 1a). However, this airspace enlargement was suppressed in the lungs of PPE-treated DN-MafB Tg mice. Furthermore, the elastic fibres, including elastin, maintained their structure in the lungs of PPE-treated DN-MafB Tg mice compared with those of the PPE-treated WT mice (Figure 1b). The MLI, which represents the quantification of airspace enlargement, was reduced significantly in the lungs of the DN-MafB Tg mice compared with that of the control mice (P < 0.05) (Figure 2).
Pulmonary emphysema in wild-type mice and dominant negative-MafB transgenic mice on Day 21 after elastase administration. Porcine pancreatic elastase (PPE) or saline was administered intratracheally to wild-type (WT) mice and dominant negative-MafB transgenic (DN-MafB Tg) mice. On Day 21 after treatment, the lungs were excised and fixed with buffered formalin (4%) to prepare paraffin-embedded lung blocks. These sections were stained with hematoxylin and eosin (a) and Elastica van Gieson (b). In Elastica van Gieson staining, elastic fibres were stained purple. Original magnification: ×100 (a) and ×200 (b).
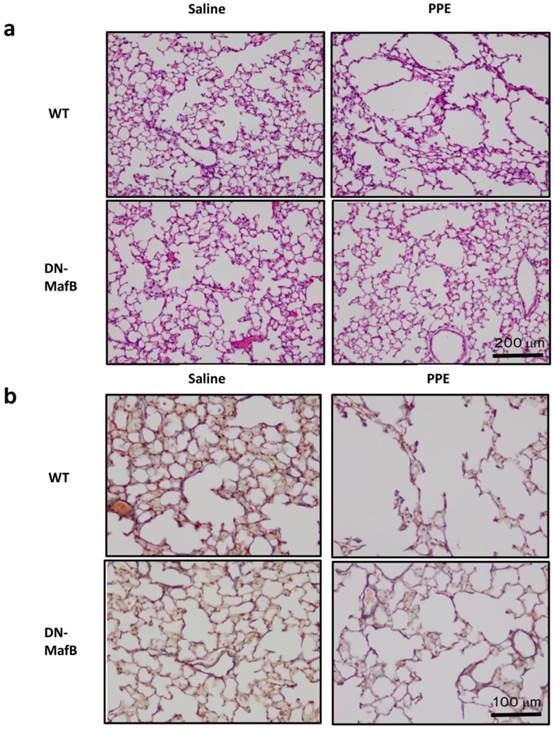
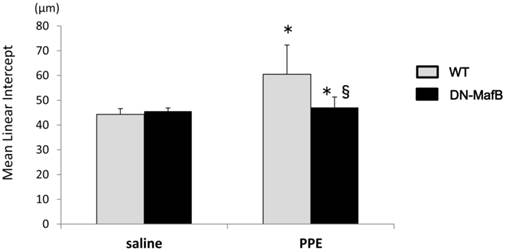
Alveolar space enlargement induced by elastase treatment was suppressed in the lungs of DN-MafB Tg mice. PPE or saline was administered intratracheally to WT mice and DN-MafB Tg mice. On Day 21 after treatment, the development of pulmonary emphysema was assessed by morphometric analysis (mean linear intercepts). Mean linear intercepts were increased in the lungs of PPE-treated WT mice. This change was reduced significantly in PPE- treated DN-MafB Tg mice. *P < 0.05 vs. saline-treated control mice; §P < 0.05 vs. PPE-treated WT mice; n= 4 for saline-treated group, n = 6 for PPE-treated group.
BAL cells of DN-MafB Tg mice
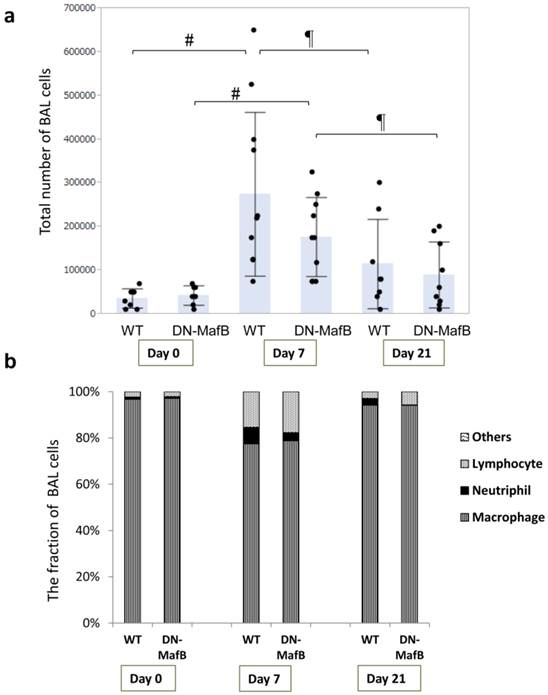
To evaluate the change in the total cell number and cell fraction, BAL was performed on Day 0, Day 7, and Day 21 after PPE treatment (Figure 3 and Table 1). On Day 7, total cell count, as well as the number of macrophages, neutrophils, and lymphocytes increased in both WT and Tg mice (Figure 3a and 3b and Table 1). In both WT and Tg mice, the total cell number on Day 21 was lower than that on Day 7, and the cell fraction almost returned to the basal level. However, the total cell number and cell fraction in the BAL cells of the DN-MafB Tg mice were not significantly different from those of the WT mice (Table 1 and Figure 3b).
Comparison of bronchoalveolar cell differentials between wild-type (WT) and dominant negative-MafB (DN-MafB) mice after elastase treatment
| Cell type | Day | WT (n=8-11) | DN-MafB (n=7-10) | Pa |
|---|---|---|---|---|
| Macrophages, × 104/body | 0 | 3.47±2.12 | 4.17±2.18 | 0.5448 |
| 7 | 21.6±16.9* | 13.2±6.17* | 0.1347 | |
| 21 | 10.9±9.96 | 8.21±6.67 | 0.5212 | |
| Neutrophils, × 103/body | 0 | 0.50±1.41 | 0.14±0.37 | 0.5113 |
| 7 | 24.9±25.6* | 8.57±6.89* | 0.1237 | |
| 21 | 1.60±4.79# | 0.16±0.29# | 0.3929 | |
| Lymphocytes, × 103/body | 0 | 0.97±1.66 | 1.00±0.78 | 0.9705 |
| 7 | 37.5±22.4* | 36.5±32.4* | 0.941 | |
| 21 | 3.12±4.21# | 7.73±10.0# | 0.2924 |
Data are expressed as the mean ± standard deviation. a: Statistical difference between WT and DN-MafB mice. *: P < 0.05 vs. Day 0 for same cell type, #: P < 0.05 vs. Day 7 for same cell type.
Total number and fraction of BAL cells in WT and DN-MafB Tg mice. PPE was administered intratracheally to WT and DN-MafB Tg mice. On Day 0, Day 7, and Day 21 after treatment, BAL was performed by infusing 1 mL of HBSS 5 times via a catheter. BAL fluid was centrifuged, and the total number of BAL cells was counted under a light microscope using a haemocytometer (a). Fraction of BAL cells in cytospun BAL was examined (b). Total cell number and cell fractions of DN-MafB Tg mice were not significantly different from those of WT mice. Results are shown as columns representing mean values with error bars (standard deviation) for each group. Dots indicate individual values of total cell numbers. # P < 0.05 compared with cell number on Day 0, ¶ P < 0.05 compared with cell number on Day 7; n = 7-11 for each group.
Morphological changes in the AMs of DN-MafB Tg mice
We previously demonstrated that the shapes of pseudopods on the AMs of DN-MafB Tg mice were altered and that alveolar monocyte-like cells were increased in the BAL cells of untreated DN-MafB Tg mice [14]. Therefore, we divided macrophage lineage cells into three types based on their shapes and sizes: macrophages with projected pseudopods, macrophages without projected pseudopods, and alveolar monocyte-like cells (Figure 4a). First, we subjected cytospun BAL cells to immunostaining using a Mac-3 antibody to distinguish alveolar monocyte-like cells from lymphocytes according to their positivity for Mac-3 (Figure 4b). All three types of cells were positive for Mac-3, confirming that these cells are macrophages. To determine whether these cells have characteristics similar to those to alveolar macrophages, immunohistochemistry for CD11c, a surface marker for alveolar macrophages, was performed. As shown in Figure 4c, most alveolar macrophages in TG mice maintained CD11c expression. To confirm this finding, we performed FACS analysis. As shown in the Figure 4d, the immunoreactivity of CD11c in alveolar macrophages (Gr-1 negative- and F4/80 positive-cells) was not different between TG and wild-type mice, suggesting that these alveolar macrophages in DN-MafB mice maintain one of the typical characteristics of alveolar macrophages [22,23].
Subtype of alveolar macrophages. In the bronchoalveolar lavage (BAL) analysis, cytospun BAL cells were obtained and stained with a Diff-Quick solution and immunohistochemical methods using Mac-3 and CD11c antibodies. Using an optical microscope, macrophage lineage cells were divided into three types: macrophages with projected pseudopods, macrophages without projected pseudopods (not recognised with an optical microscope), and alveolar monocyte-like cells (a). Alveolar monocyte-like cells exhibited smaller cellular size and less cytoplasmic granularity than differentiated macrophages but were bigger than lymphocytes. All three types of macrophage lineage cells were Mac-3 positive (b). Small macrophages were distinguished from lymphocytes due to their positivity for Mac-3 and their size. Most alveolar macrophages in DN-MafB Tg mice were positive for CD11c (c, right panel), similar to those of control mice (c, left panel). FACS analysis confirmed that the fluorescent intensity of CD11c in Gr-1 negative- and F4/80 positive-cells was not different between DN-MafB TG and wild-type mice (d). Original magnification: ×400; counterstain: haematoxylin. The image for the FACS analysis is representative of 3 independent experiments.
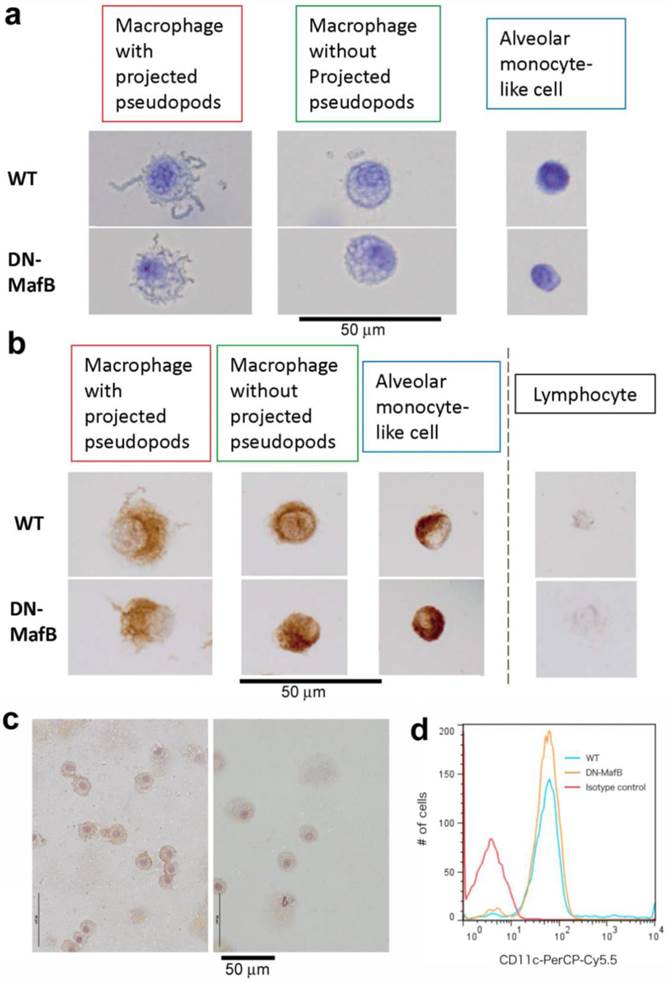
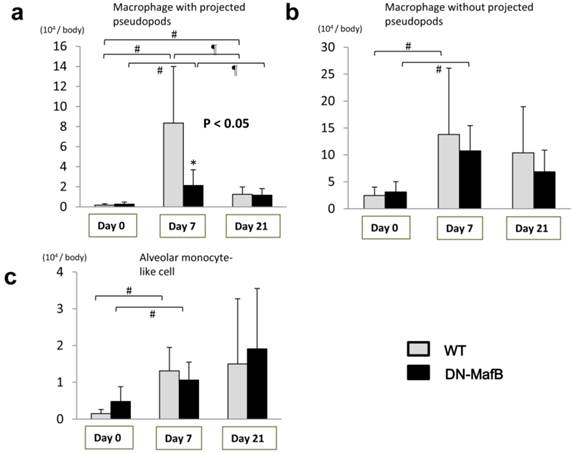
Figure 5 displays the results of the classification of the macrophage lineage cells. On Day 0, the macrophage lineage cells were primarily macrophages without projected pseudopods (Figure 5b). On Day 7, all of the macrophage lineage cells had accumulated in the lungs of both the Tg and control mice. However, the number of macrophages with projected pseudopods in the Tg mice was significantly lower than those of the control mice on Day 7 (Figure 5a). The number of macrophages with projected pseudopods was not different between the two groups on Day 0 and Day 21 (Figure 5a). The number of macrophages without projected pseudopods was not different between the WT mice and the DN-MafB Tg mice during the observation period (Figure 5b). The number of alveolar monocyte-like cells on Day 7 increased in both the WT mice and the DN-MafB Tg mice, but no difference was observed between the two groups (Figure 5c).
Morphological changes in the AMs of DN-MafB Tg mice. We classified macrophage lineage cells according to Figure 4. One-hundred cells were classified in each mouse. The number of macrophages with projected pseudopods decreased significantly in DN-MafB mice by Day 7 (a). The number of macrophages without projected pseudopods was not different between WT and DN-MafB Tg mice (b). The number of monocyte-like cells was not significantly different between WT mice and DN-MafB Tg mice (c). *P < 0.05 compared with cell number in WT mice, # P < 0.05 compared with cell number on Day 0, ¶ P < 0.05 compared with cell number on Day 7; n = 5-7 for each group.
Alteration of macrophage surface markers in DN-MafB Tg mice
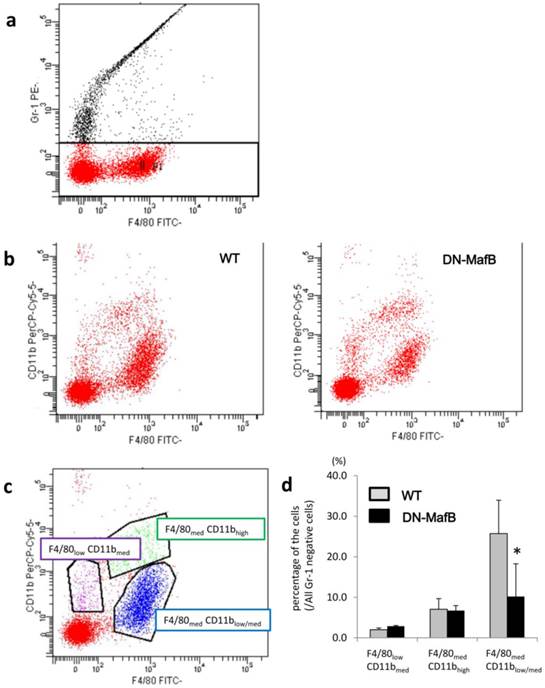
To assess whether the maturation of AMs was altered in the DN-MafB Tg mice, we examined the expression of CD11b and F4/80 on AMs on Day 7 after treatment using flow cytometric analysis. Figure 6 displays the flow cytometric observations of surface markers (F4/80 and CD11b) on AMs from the WT mice and the DN-MafB Tg mice. The analysis of the AMs was performed based on their F4/80 and CD11b fluorescence intensity (Figure 6b), which was used to divide the AMs into three groups: F4/80-weakly positive (F4/80low) and CD11b-intermediately (CD11bmed) positive cells, F4/80med-positive and CD11b-highly positive (CD11bhigh) cells, and F4/80med-positive and CD11b-weakly or -intermediately (CD11blow/med) positive cells (Figure 6c). The number of F4/80med and CD11blow/med cells significantly decreased in the AMs of the DN-MafB Tg mice compared with the WT mice (Figure 6d). The ratio of the other two groups was not different between the AMs of the WT mice and the DN-MafB Tg mice.
Flow cytometric analysis of AM surface markers. AM cell surface markers (F4/80 and CD11b) were analysed using flow cytometry. BAL cells 7 d after PPE administration were stained with FITC-labelled F4/80 antibody, PerCP-by5.5-labelled CD11b antibody, and PE-labelled Gr-1 antibody and subjected to flow cytometry using the FACS Canto II flow cytometer. To avoid the contamination of neutrophils, Gr-1-positive cells were omitted from the analysis (a). Next, the analysis of Gr-1 negative cells was performed based on their F4/80 and CD11b fluorescence intensity (a). Representative FACS images for WT (b, left) and Tg (b, right) mice are shown. The Gr-1 negative cells were divided into three groups: F4/80-weakly positive (F4/80low) and CD11b-intermediately positive (CD11bmed) cells, purple dots; F4/80med positive and CD11b-highly positive (CD11bhigh) cells, green dots; and F4/80 med positive and CD11b-weakly or -intermediately (CD11blow/med) positive cells, blue dots (c). The ratio of each groups was examined (d). The number of F4/80 med and CD11blow/med cells decreased significantly in the AMs of DN-MafB Tg mice compared with those of WT mice. n = 3 for each group. *: P < 0.05.
Suppression of immunostaining intensity of MMPs in AMs of DN-MafB Tg mice
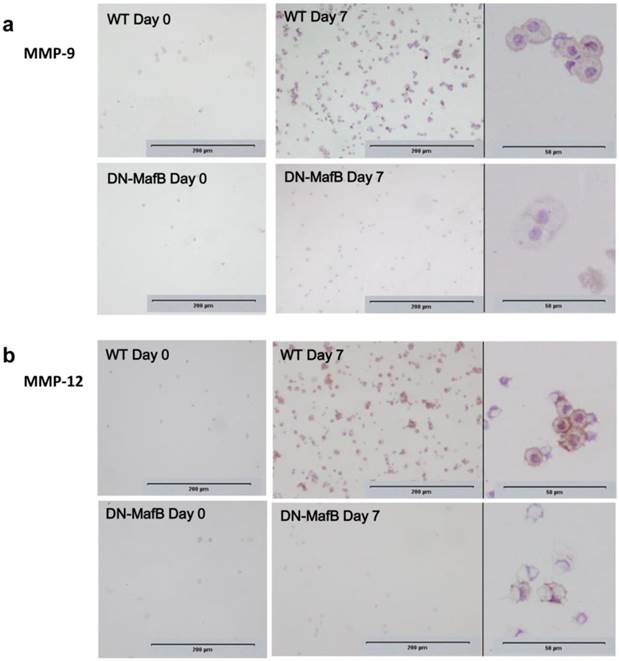
Cytospun BAL cells were immunohistochemically stained using antibodies to MMP-9 and MMP-12 on Day 0 and Day 7 (Figure 7). On Day 0, there were few MMP-9- and MMP-12-positive cells in both WT and DN-MafB Tg mice. On Day 7, MMP-9-positive and MMP-12-positive cells were observed commonly in the WT mice; however, these positive cells were decreased in the DN-MafB Tg mice.
Suppression of immunostaining intensity of matrix metalloproteinase in AMs of DN-MafB Tg mice. BAL cells were immunohistochemically stained using antibodies to matrix metalloproteinase (MMP)-9 (a) and MMP-12 (b) at on Day 0 and Day 7. MMP-9- and MMP-12-positive cells were observed commonly in WT mice, while there were very few in DN-MafB Tg mice. Intensity of staining against MMP-9 and MMP-12 were weaker in positive cells of DN-MafB Tg mice than WT mice. Original magnification: ×100 and ×400.
TNF-alpha and IL-6 production in DN-MafB Tg mice
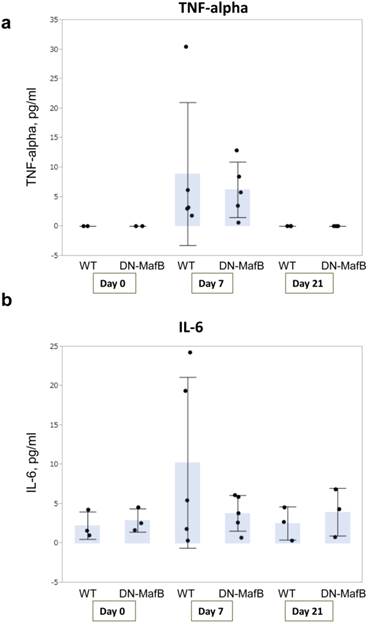
To assess the degree of inflammation in this mouse model, the concentrations of TNF-alpha and IL-6, which are both important for pathogenesis of COPD clinically and experimentally [19,24-28], were measured in the BAL fluid obtained from the WT and DN-MafB Tg mice on Day 0, Day 7, and Day 21 (Figure 8). Both the TNF-alpha and IL-6 concentrations were elevated on Day 7. However, the levels of TNF-alpha and IL-6 remained low, and there was no significant difference between the WT and DN-MafB Tg mice.
TNF-alpha and IL-6 production in DN-MafB Tg mice. TNF-alpha and IL-6 concentrations were measured in the BAL fluid of WT and DN-MafB Tg mice on Day 0, Day 7, and Day 21 (a and b). Both TNF-alpha and IL-6 concentrations were elevated by Day 7. However, the levels of TNF-alpha and IL-6 remained low, and no significant difference between WT and DN-MafB Tg mice was observed. n = 3 for Day 0 group, n = 6 for Day 7 after PPE treatment, n = 3 for Day 21 after PPE treatment. Results are shown as columns representing mean values with error bars (standard deviation) for each group. Dots indicate individual values for TNF-alpha and IL-6.
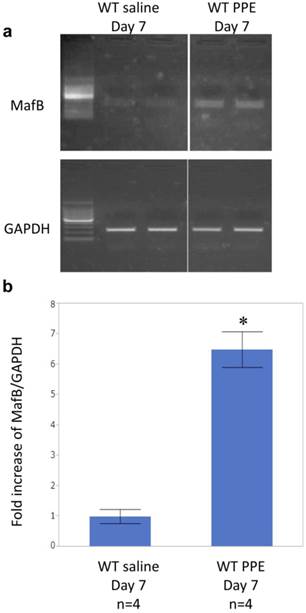
Intratracheal administration of pancreatic elastase induced MafB gene expression in alveolar macrophages of wild type mice
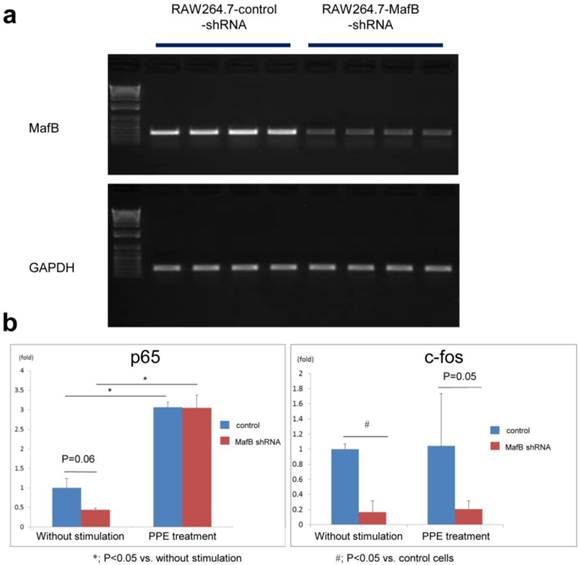
We investigated whether intratracheal administration of pancreatic elastase induces MafB gene expression in alveolar macrophages of wild-type mice by RT-PCR. MafB was upregulated in wild-type mice 7 days after intratracheal administration of elastase (Figure 9). Because MMP gene expression is known to be regulated by transcription factors such as nuclear factor kappa B (NF-kB) and activated protein (AP)-1 [29-33], we investigated whether MafB is associated with the activity of these transcription factors using a macrophage cell line in which MafB gene expression is constitutively supressed by MafB sh-RNA (RAW264.7 MafB-shRNA; Figure 10a). Elastase treatment significantly increased NF-kB p65 activity in both control (R264.7-control-shRNA) and MafB-shRNA cells, but there was no difference in the level of p65 activity between the two groups. In contrast, the activity of c-fos, a component of AP-1, was not enhanced by elastase treatment in both cell types. Under the resting condition, the c-fos activity of RAW264.7-MafB-shRNA cells was significantly supressed relative to that in control cells. After stimulation by elastase, the difference in c-fos activity between RAW264.7 MafB-shRNA and control cells was not statistically significant, but c-fos activity tended to be suppressed in elastase-stimulated RAW264.7 MafB-shRNA cells relative to control cells (P =0.052, Figure 10b).
Intratracheal administration of porcine elastase enhanced the expression of MafB in bronchoalveolar lavage (BAL) cells recovered from wild-type mice. a) Porcine pancreatic elastase (PPE) or saline was administered intratracheally to wild-type mice. BAL was performed at 7 days after PPE administration, and the total RNA was isolated from BAL cells. The expression of MafB was examined by RT-PCR and found to be enhanced by intratracheal administration of elastase. b) Band intensity was measured by computer software (Scion Image). The fold increase of MafB/GAPDH is shown. Data are expressed as the mean ± standard deviation. *; P <0.01 vs. saline treatment group.
Generation of MafB short hairpin (sh) RNA-expressing cell line and assessment of nuclear factor-kappa B (NF-kB) p65 and c-fos activity in resting and elastase-stimulated conditions. A) RAW264.7-MafB-shRNA and RAW264.7-control-shRNA cells were generated as described in the Methods. In RAW264.7-MafB-shRNA cells, MafB gene expression was one fifth of that in RAW264.7-control-shRNA cells. b) Elastase treatment significantly enhanced NF-kB p65 activity in both control (R264.7-control-shRNA) cells and MafB-shRNA cells, but there was no difference in the level of p65 activity between the two cell types. In contrast, activity of c-fos, a component of AP-1, was not enhanced by elastase treatment in both cell types. In the resting condition, the c-fos activity in RAW264.7-MafB-shRNA cells was significantly supressed relative to that in control cells. After stimulation by elastase, the difference in c-fos activity between RAW264.7 MafB-shRNA and control cells was not statistically significant, but c-fos activity tended to be suppressed in elastase-stimulated RAW264.7 MafB-shRNA cells relative to control cells (P =0.052).
Discussion
AMs play an important role in the pathogenesis of COPD [4], release inflammatory mediators, and induce prolonged inflammation and alveolar destruction in the lungs. The inflammatory mediators include TNF-alpha, IL-1beta, IL-6, and MMPs. We previously demonstrated that MafB was upregulated in AMs in the lungs of mice with cigarette smoke-induced emphysema, suggesting a relationship between cigarette smoking and MafB in AMs [7]. In addition, we reported that the intensity of MafB immunostaining in AMs was correlated with the degree of airflow limitation in human smokers [8]. In the present study, we generated an elastase-induced emphysema model in DN-MafB Tg mice, which exhibit reduced macrophage MafB activity, and demonstrated that an emphysematous change was suppressed significantly in the DN-MafB Tg mice (Figure 1 and 2). In addition, elastase instillation into the lungs of wild-type mice increased the expression of MafB (Figure 9). Therefore, the present study indicates an important role of MafB for the development of emphysema induced by intratracheal administration of elastase.
MafB is known as a regulator of macrophage differentiation [9] and other various developmental processes, such as segmentation in the hindbrain [34], islet beta-cell differentiation [35], and chondrocyte differentiation [36]. Zhang et al. reported that retinoic acid-induced MMP-3 and MMP-13 gene expression was attenuated in MafB-knockdown chondrocytes [36]. MMP-3 and MMP-13 are induced in terminally differentiated chondrocytes, and MMP-12 is produced by differentiated macrophages [37]. Because the production of MMPs may partly depend on cellular differentiation and maturation, which MafB can regulate, we examined the expression of F4/80 and CD11b to evaluate the maturation of AMs in DN-MafB Tg mice. F4/80 is a specific cell surface marker for murine macrophages [38,39]. CD11b is widely expressed on the surface of monocytes, macrophages, NK cells, and granulocytes. The number of F4/80med CD11blow/med cells was decreased in the BAL cells of DN-MafB Tg mice compared to the BAL cells of WT mice by Day 7 after PPE administration (Figure 6). AMs present lower expression of CD11b than other macrophage-lineage cells, such as interstitial macrophages, dendritic cells, and monocytes [40,41]. Thus, F4/80med CD11blow/med cells were considered to be mature AMs. Our data suggest that the number of maturated AMs after elastase-induced inflammation is reduced in DN-MafB Tg mice.
Pseudopods on AMs of DN-MafB Tg mice have been reported to have a 'lamellipodium-like' shape, as determined by electron microscopy. In addition, actin filament formation was shown to be attenuated in the pseudopods of AMs of DN-MafB Tg mice [14]. Although no morphological changes were observed in the BAL cells of untreated DN-MafB Tg mice with an optical microscope, the number of macrophages with projected pseudopods decreased significantly in the DN-MafB Tg mice by Day 7 after elastase administration. Macrophages with projected pseudopods are considered to be activated AMs [42]. Therefore, our results suggest that the number of activated AMs by Day 7 after elastase administration was decreased in DN-MafB Tg mice relative to WT mice.
Pulmonary emphysema is pathologically characterized by the enlargement of alveoli in the lung. Among inflammatory mediators from AMs, MMPs are believed to play important roles in the development of emphysema by degrading the extracellular matrix in the lung. MMPs are a family of zinc-dependent proteinases capable of degrading the extracellular matrix. Even in the murine model of pulmonary emphysema induced by a single instillation of porcine pancreatic elastase, subsequent production of MMPs from AMs is considered to be important for disease development [18]. MMP-12 is believed to play an important role in the development of emphysema by degrading the extracellular matrix in the lung. Elastin, the major target protein of MMP-12, is downregulated in the lung parenchyma of emphysema patients [43]. It has been reported that the expression of MMP-12 is higher in COPD patients than in control subjects, both in BAL samples and bronchial biopsies [44], and that MMP-12-deficient mice do not develop air space enlargement in response to smoke exposure [45].
In the present study, the expression of MMP-12 in BAL cells was reduced significantly in DN-MafB Tg mice (Figure 7), and elastase-induced pulmonary emphysema was suppressed in DN-MafB Tg mice (Figure 2). Based on previous reports, the decrease of MMP-12 levels in DN-MafB Tg mice could attenuate the airspace enlargement of the lung in this elastase-induced emphysema. MafB directly regulates the expression of MMP-9 through binding to the MARE motif in its promoter region [11]. In addition, the results using macrophage cell line in this study (Figure 9) suggests that MafB is associated with the activity of AP-1 that is important for MMP expression [29,31]. Thus, MafB may be indirectly associated with the development of elastase-induced pulmonary emphysema through regulation of the activity of AP-1. However, the mechanisms underlying suppression of MMP expression by DN-MafB have not been fully elucidated. Future experiments should therefore investigate how the transcription factor MafB regulates the expression of MMPs.
The changes in the extent of inflammation after elastase administration in the lungs of DN-MafB Tg mice compared to wild-type mice may be another conceivable reason for the decrease of MMP-12 levels. The activity of MMP-12 is known to be stimulated by inflammatory cytokines such as TNF-alpha and IL-6 [46,47]. We assessed the concentrations of TNF-alpha and IL-6 in elastase-administered DN-MafB Tg mice, and the concentrations were not different from those in the elastase-administered WT mice (Figure 8). In addition, the levels of proinflammatory cytokines remained low even by Day 7 of PPE treatment. One limitation of our study is that we did not assess these cytokines in the acute inflammatory phase. However, the majority of airspace enlargement in this model is believed to occur in the chronic inflammatory phase [19]. Therefore, inhibition of inflammatory cytokines does not seem to play an important role in the suppression of MMPs in elastase-treated DN-MafB Tg mice.
Another limitation of the present study is that we did not evaluate the effect of cigarette smoke exposure in the Tg mice. Because animal models using cigarette smoke exposure are thought to be more similar to human pulmonary emphysema, future studies should use animal smoking models. In addition, an experiment transplanting mature macrophages from wild-type mice to DN-MafB mice by adoptive transfer may shed light on the mechanism underlying the inhibition of elastase-emphysema in the Tg mice. However, the efficacy of adoptive transfer does not seem sufficient for such an experiment [40].
In conclusion, the present study demonstrated that DN-MafB suppressed elastase-induced pulmonary emphysema development by altering the production of MMPs; therefore, MafB may play an important role in the pathogenesis of pulmonary emphysema via alveolar macrophage maturation.
Acknowledgements
We thank Emiko Nishidate, Junko Higuchi, Miyuki Tsuda, Taiko Aita, Emiko Nakamura, and Eiji Tsuchida (Department of Cardiology, Pulmonology and Nephrology, Yamagata University School of Medicine) for their excellent technical assistance.
Funding
This study was supported by the Japan Society for the Promotion of Science (19590880, 20590892, and 23390220).
Ethics committee approval
This study was approved by the Committee for Animal Experimentation, Yamagata University School of Medicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA. et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532-55
2. Sharafkhaneh A, Hanania NA, Kim V. Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc. 2008;5:475-7
3. Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL. et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27:397-412
4. Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:S29-32
5. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672-88
6. Shapiro SD. COPD unwound. N Engl J Med. 2005;352:2016-9
7. Machiya J, Shibata Y, Yamauchi K, Hirama N, Wada T. et al. Enhanced expression of MafB inhibits macrophage apoptosis induced by cigarette smoke exposure. Am J Respir Cell Mol Biol. 2007;36:418-26
8. Sato M, Shibata Y, Kimura T, Yamauchi K, Abe S. et al. Immunohistochemical staining for transcription factor MafB in alveolar macrophages is correlated with spirometric measures of airflow limitation in smokers. Respirology. 2011;16:124-30
9. Kelly LM, Englmeier U, Lafon I, Sieweke MH, Graf T. MafB is an inducer of monocytic differentiation. Embo J. 2000;19:1987-97
10. Kataoka K, Fujiwara KT, Noda M, Nishizawa M. MafB, a new Maf family transcription activator that can associate with Maf and Fos but not with Jun. Mol Cell Biol. 1994;14:7581-91
11. Gemelli C, Zanocco Marani T, Bicciato S, Mazza EM, Boraschi D. et al. MafB is a downstream target of the IL-10/STAT3 signaling pathway, involved in the regulation of macrophage de-activation. Biochim Biophys Acta. 2014;1843:955-64
12. Blanchi B, Kelly LM, Viemari JC, Lafon I, Burnet H. et al. MafB deficiency causes defective respiratory rhythmogenesis and fatal central apnea at birth. Nat Neurosci. 2003;6:1091-100
13. Blanchi B, Sieweke MH. Mutations of brainstem transcription factors and central respiratory disorders. Trends Mol Med. 2005;11:23-30
14. Sato-Nishiwaki M, Aida Y, Abe S, Shibata Y, Kimura T. et al. Reduced Number and Morphofunctional Change of Alveolar Macrophages in MafB Gene-Targeted Mice. PLoS One. 2013;8:e73963
15. Kuhn C, Yu SY, Chraplyvy M, Linder HE, Senior RM. The induction of emphysema with elastase. II. Changes in connective tissue. Lab Invest. 1976;34:372-80
16. Groneberg DA, Chung KF. Models of chronic obstructive pulmonary disease. Respir Res. 2004;5:18
17. Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG. et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753-9
18. Ishikawa T, Aoshiba K, Yokohori N, Nagai A. Macrophage colony-stimulating factor aggravates rather than regenerates emphysematous lungs in mice. Respiration. 2006;73:538-45
19. Lucey EC, Keane J, Kuang PP, Snider GL, Goldstein RH. Severity of elastase-induced emphysema is decreased in tumor necrosis factor-alpha and interleukin-1beta receptor-deficient mice. Lab Invest. 2002;82:79-85
20. Tokairin Y, Shibata Y, Sata M, Abe S, Takabatake N. et al. Enhanced immediate inflammatory response to Streptococcus pneumoniae in the lungs of mice with pulmonary emphysema. Respirology. 2008;13:324-32
21. Thurlbeck WM. Measurement of pulmonary emphysema. Am Rev Respir Dis. 1967;95:752-64
22. Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. J Immunol. 2003;171:3128-35
23. Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A. 2004;61:170-77
24. Faner R, Tal-Singer R, Riley JH, Celli B, Vestbo J. et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax. 2014;69:666-72
25. Pinto-Plata V, Casanova C, Mullerova H, de Torres JP, Corado H. et al. Inflammatory and repair serum biomarker pattern: association to clinical outcomes in COPD. Respir Res. 2012;13:71
26. Ruwanpura SM, McLeod L, Miller A, Jones J, Bozinovski S. et al. Interleukin-6 promotes pulmonary emphysema associated with apoptosis in mice. Am J Respir Cell Mol Biol. 2011;45:720-30
27. Tasaka S, Inoue K, Miyamoto K, Nakano Y, Kamata H. et al. Role of interleukin-6 in elastase-induced lung inflammatory changes in mice. Exp Lung Res. 2010;36:362-72
28. Vuillemenot BR, Rodriguez JF, Hoyle GW. Lymphoid tissue and emphysema in the lungs of transgenic mice inducibly expressing tumor necrosis factor-alpha. Am J Respir Cell Mol Biol. 2004;30:438-48
29. Munaut C, Salonurmi T, Kontusaari S, Reponen P, Morita T. et al. Murine matrix metalloproteinase 9 gene. 5'-upstream region contains cis-acting elements for expression in osteoclasts and migrating keratinocytes in transgenic mice. J Biol Chem. 1999;274:5588-96
30. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79-82
31. Wu L, Tanimoto A, Murata Y, Fan J, Sasaguri Y. et al. Induction of human matrix metalloproteinase-12 gene transcriptional activity by GM-CSF requires the AP-1 binding site in human U937 monocytic cells. Biochem Biophys Res Commun. 2001;285:300-7
32. Yoo HG, Shin BA, Park JS, Lee KH, Chay KO. et al. IL-1beta induces MMP-9 via reactive oxygen species and NF-kappaB in murine macrophage RAW 264.7 cells. Biochem Biophys Res Commun. 2002;298:251-6
33. Yu Y, Chiba Y, Sakai H, Misawa M. Possible involvements of nuclear factor-kappa B and activator protein-1 in the tumor necrosis factor-alpha-induced upregulation of matrix metalloproteinase-12 in human alveolar epithelial A549 cell line. J Pharmacol Sci. 2010;112:83-8
34. Grapin-Botton A, Bonnin MA, Sieweke M, Le Douarin NM. Defined concentrations of a posteriorizing signal are critical for MafB/Kreisler segmental expression in the hindbrain. Development. 1998;125:1173-81
35. Artner I, Blanchi B, Raum JC, Guo M, Kaneko T. et al. MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A. 2007;104:3853-8
36. Zhang Y, Ross AC. Retinoic acid and the transcription factor MafB act together and differentially to regulate aggrecan and matrix metalloproteinase gene expression in neonatal chondrocytes. J Cell Biochem. 2012
37. Wu L, Fan J, Matsumoto S, Watanabe T. Induction and regulation of matrix metalloproteinase-12 by cytokines and CD40 signaling in monocyte/macrophages. Biochem Biophys Res Commun. 2000;269:808-15
38. Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805-15
39. Morris L, Graham CF, Gordon S. Macrophages in haemopoietic and other tissues of the developing mouse detected by the monoclonal antibody F4/80. Development. 1991;112:517-26
40. Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM. et al. Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2009;296:L936-46
41. Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP. et al. Identification of myeloid cell subsets in murine lungs using flow cytometry. Am J Respir Cell Mol Biol. 2013;49:180-9
42. Nabarra B, Cavelier JF, Dry M, Dimitriu A. Scanning electron microscopic studies of activated macrophages in the mouse. J Reticuloendothel Soc. 1978;24:489-98
43. Wright JL. Emphysema: concepts under change--a pathologist's perspective. Mod Pathol. 1995;8:873-80
44. Molet S, Belleguic C, Lena H, Germain N, Bertrand CP. et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res. 2005;54:31-6
45. Hautamaki RD. Requirement for Macrophage Elastase for Cigarette Smoke-Induced Emphysema in Mice. Science. 1997;277:2002-4
46. Feinberg MW, Jain MK, Werner F, Sibinga NE, Wiesel P. et al. Transforming growth factor-beta 1 inhibits cytokine-mediated induction of human metalloelastase in macrophages. J Biol Chem. 2000;275:25766-73
47. Suzuki M, Mihara M. Possible roles of adiponectin in inflammatory process of rheumatoid arthritis. Inflammation and Regeneration. 2012;32:193-201
Author contact
![]() Corresponding author: Dr. Yoko Shibata, 2-2-2 Iida-Nishi, Yamagata City, Yamagata 990-9585, Japan. Telephone: +81-23-628-5302 Fax: +81-23-628-5305 Email: shibataid.yamagata-u.ac.jp.
Corresponding author: Dr. Yoko Shibata, 2-2-2 Iida-Nishi, Yamagata City, Yamagata 990-9585, Japan. Telephone: +81-23-628-5302 Fax: +81-23-628-5305 Email: shibataid.yamagata-u.ac.jp.