Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(9):948-956. doi:10.7150/ijbs.9033 This issue Cite
Short Research Communication
Extraintestinal Helminth Infection Reduces the Development of Colitis-Associated Tumorigenesis
Sonia León-Cabrera1*, Blanca E. Callejas1*, Yadira Ledesma-Soto1, Jossimar Coronel1, Carlos Pérez-Plasencia1, Emma B. Gutiérrez-Cirlos1, Federico Ávila-Moreno1, Miriam Rodríguez-Sosa1, Rogelio Hernández-Pando2, Brenda Marquina-Castillo2, Yolanda I. Chirino1, Luis I. Terrazas1 ![]()
1. Unidad de Biomedicina. Facultad de Estudios Superiores-Iztacala. Universidad Nacional Autónoma de México. Av. de los Barrios 1, Los Reyes Iztacala, Tlalnepantla, Edo. de México. México 54090.
2. Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubiran. Vasco de Quiroga 15, Colonia Sección XVI, Tlalpan, México D.F., México 14000.
* These authors contributed equally in this work.
Received 2014-3-6; Accepted 2014-7-3; Published 2014-8-24
Abstract
Colitis-associated colorectal cancer (CAC) is one of the most common cancers and is closely related to chronic or deregulated inflammation. Helminthic infections can modulate inflammatory responses in some diseases, but their immunomodulatory role during cancer development remains completely unknown. We have analyzed the role of Taenia crassiceps-induced anti-inflammatory response in determining the outcome of CAC. We show that extraintestinal T. crassiceps infection in CAC mice inhibited colonic inflammatory responses and tumor formation and prevented goblet cell loss. There was also increased expression of IL-4 and alternatively activated macrophages markers in colonic tissue and negative immunomodulation of pro-inflammatory cytokine expression. In addition, T. crassiceps infection prevented the upregulation of β-catenin and CXCR2 expression observed in the CAC mice, which are both markers associated with CAC-tumorigenesis, and reduced the numbers of circulating and colonic CD11b+Ly6ChiCCR2+ monocytes. Thus, immunomodulatory activities induced by helminth infections may have a role in the progression of CAC.
Keywords: Colitis-associated colorectal cancer, T. crassiceps
Introduction
Colitis associated cancer (CAC) is one of the most common malignant diseases and is the second leading cause of cancer death in several countries [1]. A relationship between inflammation and colon cancer development has been described worldwide [2]. Patients with inflammatory bowel diseases (IBD), such as Crohn's disease or ulcerative colitis, are more susceptible to developing colorectal cancer [3].
It has been widely reported that some infectious agents, such as viruses and bacteria, can fuel or even be a cause for the development of cancer [4]. The role of parasitic infections in such field has been insufficiently studied, and scarce reports suggest that helminths can increase the risk of cancer [5]. The geographical distribution of gastrointestinal and parasitic infections demonstrated that they are widely distributed in tropical and subtropical areas with greatest numbers occurring in China, Africa, the Americas and East Asia. Concomitantly, epidemiological data provide strong evidence about the incidence of inflammatory bowel disease and colorectal cancer in the past three decades in developed countries [6], suggesting an inverse association between helminth infections and inflammatory bowel disease and colorectal cancer.
Nevertheless, helminth parasites have developed complex strategies to modulate the immune responses of their hosts through versatile immunoregulatory mechanisms. The capability of helminths to modulate an exacerbated inflammatory response has been used advantageously to study their effects on autoimmune diseases [7]; however, the effect of helminth infections on the development of cancer, specifically CAC, remains completely unknown. Here, we address the impact of a helminth infection on tumor development using a well-established chemical colorectal cancer mouse model.
Materials and Methods
Mice
Six to eight week old female BALB/c mice were purchased from Harlan Laboratories (México) and were maintained in a pathogen-free environment at the FES-Iztacala, UNAM animal facilities.
Parasites and infection
Metacestodes of T. crassiceps were harvested from the peritoneal cavity of female BALB/c mice after 2 to 3 months of infection, processed as previously reported [8] and the infection was accomplished by an i.p. injection of 20 small (2mm in diameter) non-budding cysticerci.
Development of colitis-associated colorectal cancer (CAC)
An extensively used CAC model was developed as previously described [9]. Briefly, T. crassiceps-infected mice (6 weeks post-infection) or age-matched uninfected mice were i.p. injected with Azoxymethane (AOM) at 12.5 mg/kg (Sigma, USA). Five days later, dextran sulfate sodium (DSS, MW: 35 000-50 000, MP Biomedicals. Solon, OH, USA) dissolved at 2% in drinking water was administered ad libitum for 7 consecutive days. Afterwards, the mice were maintained with regular water for 14 days and were subjected to two more DSS cycles. The mice were sacrificed on day 68; the colon was removed, weighed and submitted for macroscopic inspection, histopathological examination and mRNA expression assays. The mouse infection scheme and treatment are showed in Fig. 1A.
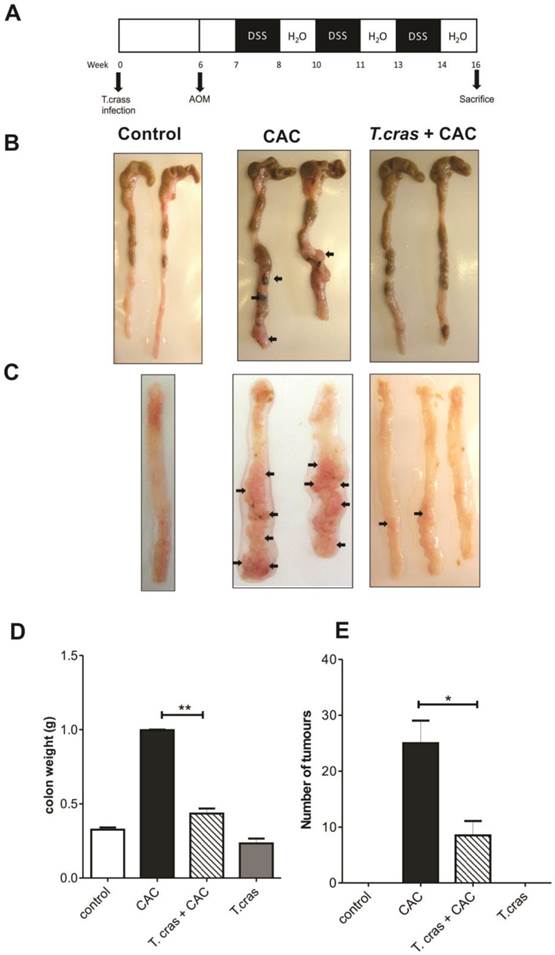
T. crassiceps infection significantly decreases colitis associated tumorigenesis. (A). Protocol for helminth infection and CAC induction. (B) Excised colon from the cecum to the rectum of the control mice, CAC mice and T. crassiceps+CAC mice. (C) Opened and washed colon from the proximal to the distal section of the control mice, CAC mice and T. crassiceps+CAC mice. (D) Colon weight after removing and flushing the cecum and rectum. (E) Tumor number count. The data are presented as the mean ± SE. Arrows show tumors. *P < 0.05, **P < 0.003, n= 5 mice per group. All data are representative of two independent experiments.
Histology
For histological analysis, longitudinal sections from the large intestine were immediately fixed by immersion in 10% formaldehyde dissolved in PBS, after one day the tissue was dehydrated in gradient alcohol concentrations and embedded in paraffin. Sections (5µm) were stained with hematoxilin/eosin to visualize neutrophils or with alcian blue stain to visualize the goblet cells, using an optical microscopy (Axio Vert.A1, Carl Zeiss). These cells were quantified (percentage) from at least 20 crypts per region in five fields in four different slides per animal at 400X magnification. For Immunohistochemistry staining of β-catenin and F4/80, the sections were incubated with optimal dilutions of anti-mouse beta-catenin (Gene Tex, Inc. USA) and anti-mouse F4/80 (e-Bioscience, USA) overnight at 4°C, and then developed following the conventional technique.
Colon Culture
Sections of 0.5 cm of the proximal colon were cut and washed three times with PBS. The colon fragments were placed into 24-well plate's culture in DMEM medium with penicillin and streptomycin and stimulated or not with LPS (1µg/mL) and cultured at 37°C with 5% CO2. Supernatants were harvested 24h later and the concentration of TNF-α was determined by ELISA (Biolegend, San Diego, CA. USA).
RNA isolation and real-time PCR
RNA was isolated from colon tissues using a TRIzol extraction. Tissues were first disrupted in a tissue homogenizer (Bullet Blender®; Next Advance). Semi-quantitative RT-PCR was performed to assess the expression of mRNAs for YM-1, arginase, FIZZ1, iNOS, and CXCR2. cDNA was synthesized from the isolated RNA using a RevertAid H Minus First Strand cDNA synthesis Kit (Thermo Scientific). mRNA expression for β-catenin, IFN-γ, TNF-α, TGF-β and IL-4 was performed in a LightCycler 2.0 system (Roche Applied Science) using a KAPA SYBR® FAST One-Step qRT-PCR Roche LightCycler® (KapaBiosystems), according to the manufacturer's instructions. The oligonucleotide primer sequences are described in Table 1 and 2. All quantifications were normalized to GAPDH levels. The relative expression of the target genes was analyzed by the ΔΔCt method. Control group was used as “comparator samples” for quantification of those corresponding to test samples.
Genes and their respective sequences used to determine alternative activation of macrophages by semi-quantitative RT-PCR.
| Gene | Sequence | Melting Temperature |
|---|---|---|
| GAPDH | F-TCGGTGTGAACGGATTTGGC R-CTCTTGCTCAGTGTCCTTGC | 56° |
| Arg-1 | F-CAG AAG AAT GGA AGA GTC AG R-CAG ATA TGC AGG GAG TCA CC | 54° |
| Ym1 | F-TCACAGGTCTGGCAATTCTTCTG R-TTTGTCCTTAGGAGGGCTTCCTC | 56° |
| Fizz1 | F-GGTCCCAGTGCATATGGATGAGACCATAG R-CACCTCTTCACTCGAGGGACAGTTGGCAGC | 65° |
| iNOS | F-GCCACCAACAATGGCAACAT R-AAGACCAGAGGCAGCACATC | 60° |
| CXCR2 | F-CACCGATGTATACCTGCTGA R-ACGCAGTACGACCCTCAAAC | 63° |
Primer sequences for quantitative RT-PCR.
| Gene | Sequence | Melting Temperature |
|---|---|---|
| β-Catenin | F-GGCCTCTGATAAAGGCAACTG R-CCGAGCAAGGATGTGGAGAG | 57° |
| IFN-γ | F-AGCGGCTGACTGAACTCAGATTGTAG R-GTCACAGTTTTCAGCTGTATAGGG | 57° |
| TNF-α | F-GGCAGGTCTACTTTGGAGTCATTGC R-ACATTCGAGGCTCCAGTGAATTCG | 59° |
| TGF-β | F-GCCCTTCCTGCTCCTCAT R-TTGGCATGGTAGCCCTTG | 63° |
| IL-4 | F-CGAAGAACACCACAGAGAGTGAGCT R-GACTCATTCATGGTGCAGCCTTATCG | 58° |
Cell Isolation and Flow Cytometry Analysis
For the isolation of the colonic lamina propria cells a standard procedure previously reported was used [10]. For flow cytometry, single cell suspensions of the lamina propria and the circulation obtained during the sacrifice were stained with anti-CD11b, anti-Ly6C (Biolegend, San Diego, CA), and anti-CCR2 (R&D Systems, USA) antibodies for 30 min at 4° C. The cells were washed twice and analyzed using the FACSCalibur system and Cell Quest software (Becton Dickinson, USA).
Statistical Analysis
The data were analyzed by either a one-way ANOVA followed by Tukey´s Multiple comparisons test or by an unpaired two-tailed t test with GraphPad Prism 4 (San Diego, CA).
Results and Discussion
Helminth infection reduces CAC development
Because chronic or deregulated inflammation is a well-recognized risk factor for colorectal carcinoma development and because helminth infections are largely known to modulate inflammatory responses, this study was conducted to determine whether a helminth infection could impact the progression of CAC. During the course of the experiments, the uninfected mice (CAC group) exhibited several symptoms, such as piloerection and bloody diarrhea, whereas the T. crassiceps+CAC mice did not show any symptoms (data no shown). All the animals from the CAC group had diverse numbers and sizes of polypoid tumors, whereas 20% of the T. crassiceps+CAC mice were free from these types of lesions. CAC mice had numerous reddish polypoid tumors in the whole large intestine; macroscopic damage and pathological alterations were clearly visible. In particular, the middle area of the transverse and descending colon had numerous lesions (Fig. 1B, 1C). Indeed, after the colons were opened and washed out, multiple tumors were found in the middle to distal colon of the CAC mice (Fig. 1C). In contrast, the T. crassiceps+CAC mice had more normal shape colons and a significant and visible reduction in the number of tumors (Fig. 1C). The weight of the excised colons from the cecum to the rectum of the CAC mice showed a 2.5-fold of increase, whereas the T. crassiceps infection prevented this increase (Fig. 1D), most likely due to the lower number of tumors, as the T. crassiceps+CAC mice had a 64% decrease in the expected tumor formation (Fig. 1E). These data indicate that the presence of the extraintestinal larval stage of T. crassiceps can modulate CAC progression.
T. crassiceps infection reduces CAC-induced pathologic alterations
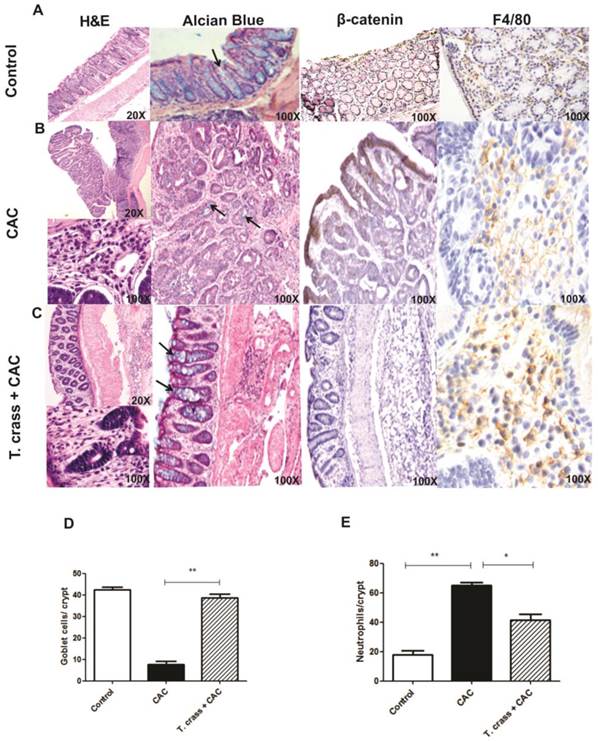
The histological study of the large intestine in the CAC mice without T. crassiceps infection revealed extensive chronic inflammation localized in the lamina propria, with numerous and large polypoid tumors constituted by well-formed glands revisted by atypical epithelial cells with large and dysplastic nucleus and numerous mitotic figures that corresponded to well differentiated adenocarcinoma (Fig 2B). Numerous neoplastic glands showed intraluminal abscess constituted by cellular debris and numerous neutrophils, a remarkable decrease of goblet cells (stained with Alcian blue, arrows) in these polypoid lesions was also observed (Fig 2B, D, E). These polypoid lesions also showed strong β-catenin immune-staining, which is a marker for abnormal cell proliferation and for tumorigenesis (Fig 2B). In contrast, T. crassiceps+CAC displayed lesser inflammatory infiltrate and small polypoid lesions constituted by hyperplastic epithelium with mild dysplasia and normal appearance and numbers of goblet cells, similar to the control mice (Fig 2C, D). Moreover, the colon tissue from these animals was negative for β-catenin immune-staining (Fig 2C) suggesting that these mice did not develop tumorigenesis. Immune-staining for F4/80, a macrophage marker, in colon tissue was performed. Both groups of mice were positive for this marker indicating macrophage recruitment, however, the intensity of this mark was higher in T. crassiceps+CAC group (Fig 2A-C), suggesting an increased recruitment of macrophages. Conversely, neutrophil recruitment was higher in the CAC group as observed by H&E histology and as is evidenced in the higher magnification photo and counting of these cells in the crypts (Fig 2B, E).
T. crassiceps infection greatly inhibits the pathology of colitis associated tumorigenesis. Representative histological features from the colon (A) control, (B) CAC and (C) T. crassiceps+CAC mice taken at day 68 after CAC induction and stained with Hematoxilin and Eosin (H&E), Alcian Blue for visualizing goblet cells, and immunohistochemistry stain for β-catenin and F4/80. (D) Number of goblet cells and (E) number of neutrophils located in distal colons. These cells were quantified from at least 20 crypts per region in five fields in four different slides per animal. Alcian blue stained goblet cells (arrows). Data are expressed as means ± SEM. * P< 0.05 ** P< 0.01.
Helminth infection induces differential AAMs and inflammatory marker expression during the course of CAC
We next determined the expression levels of typical molecular markers associated with both CAC development and helminth infections. We found that T. crassiceps infection markedly increased the mRNA expression of Ym-1, Arg-1, and Fizz-1 in the colonic tissue from the CAC-induced mice, all of these markers are characteristic of alternative activated macrophages (AAMs) (Fig. 3A-C); concomitantly, iNOS expression was significantly reduced in the T. crassiceps+CAC mice compared with the CAC mice (Fig. 3D). These data together with the higher number of F4/80+ cells as revealed by immune-staining in the T. crassiceps+CAC mice suggest a preferential recruitment of AAMs induced by the presence of this helminth. We next evaluated the mRNA expression profile of IFN-γ, TNF-α, TGF-β and IL-4 in the colon. The expression of these genes was significantly enhanced, particularly TNF-α (Fig. 3F) and, to a lesser degree, IFN-γ (Fig. 3E) and TGF-β (Fig. 3G) but not IL-4 (Fig. 3H), in the CAC mice. However, in the T. crassiceps+CAC mice the expression of TNF-α, IFN-γ and TGF-β, was reduced, but IL-4 expression was remarkably enhanced in the colonic tissue compared with the CAC animals (Fig. 3E-H). Thus, infection with T. crassiceps remarkably reduced the inflammatory infiltrate and number and size of polypoid tumors in the colon and inhibited the development of hyperplasia. This observation was correlated with attenuated production of potent inflammatory mediators, such as TNF-α and IFN-γ, both of which are known to orchestrate the development of CAC.
T. crassiceps infection induces a differential expression of AAM and pro-inflammatory markers during CAC. A semi-quantitative RT-PCR or quantitative RT-PCR analysis was performed on the total RNAs extracted from the colons and was normalized to GAPDH. mRNA expression of (A) Ym-1, (B) Arginase 1, (C) FIZZ1, (D) iNOS, (E) IFN-γ, (F) TNF-α, (G) TGF-β and (H) IL-4. (I) TNF-α production in ex-vivo tissue colon cultures of CAC and T. crassiceps+CAC mice stimulated with LPS were measured by ELISA. (J) β-catenin and (K) CXCR2 relative expression. The ratios of the mRNA levels were expressed relative to those of the mRNA levels in the untreated control group. Values are mean ± SD (n=5 mice/group) *P<0.05, **P<0.03.
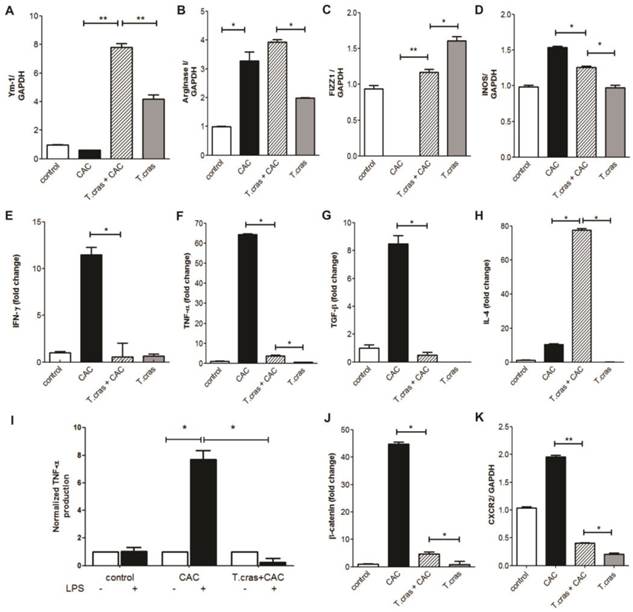
A feature of CAC is the chronic intestinal inflammation that mediates the breakdown of the protective intestinal barriers causing increased accessibility of the microbiota to the inflamed epithelium. This in turn may activate immune or epithelial cells through TLR-pathways, thereby amplifying the inflammatory response and generating tissue damage in the colon [11]. Excessive TLR-signaling can itself drive strong and sustained pro-inflammatory responses that fuel tumorigenesis. In this regard, T. crassiceps infection is able to block TLR signaling in macrophages, inhibiting IL-12, IL-23, TNF-α, and NO production, all of which are importantly involved in CAC development [12]. In order to explore whether this possibility was happening in our model we performed ex-vivo tissue colon cultures to evaluate the response to LPS. In line with this we detected an increase in TNF-α production in whole colon tissue cultures of CAC mice in response to LPS as compared with similar colon tissue cultures from T. crassiceps+CAC mice (Fig. 3I). These data may indicate that chronic T. crassiceps infection down-regulate the inflammatory response induced by LPS in vivo, suggesting that the immunomodulatory effects of this intraperitoneal infection could induce a refractory state to LPS in tissue colon during CAC development. Moreover, DCs exposed to helminth-derived products became unresponsive to TLR-stimulation in vitro [13, 14]. Indeed, T. crassiceps excreted/secreted products impair the pro-inflammatory response of DCs exposed to different TLR-ligands by blocking the activation of NFκB [8], an important regulator of tumor initiation and progression.
Conversely, IL-4 expression was enhanced locally in the colon. This cytokine may also down-regulate macrophage pro-inflammatory activation and promote AAMs. It has been well established that in addition to anti-inflammatory activity, AAMs also have the ability to repair tissue. Thus, it may be that robust early recruitment of AAMs during CAC development helps to resolve tissue damage and avoid the concomitant inflammation generated by DSS exposure thereby preventing the development of CAC. In this system, arginase 1 expression induced by the presence of T. crassiceps may control the harmful inflammation that occurs during CAC, consequently contributing to tissue repair in the colon and to the maintenance of the mucosal barrier. Moreover, the number and size of the goblet cells was preserved in T. crassiceps+CAC mice, suggesting that T. crassiceps, like other helminths, could increase the number of these cells [15, 16]. Goblet cells are involved in regulating both the mucosal barrier and the relative composition of the luminal microbiota by mucin production [17]. Therefore, the high expression of IL-4 in the colon of the T. crassiceps+CAC mice could promote the production of Th2 cytokines favoring goblet cell hyperplasia. The mucus production by goblet cells in turn may limit bacterial access to the epithelial cells and prevent chronic inflammation during CAC. Indeed, mucin 2-deficient mice develop spontaneous colitis and colonic cancers as a result of the commensal bacteria that are in direct contact with the epithelial cells [18].
We also looked for a classical molecular marker of tumorigenesis, such as β-catenin. Remarkably, we found that the CAC mice had enhanced mRNA expression of β-catenin in the colonic tissue, and importantly, the infection attenuated this increase (Fig. 3J). This is an interesting finding, as β -catenin plays an important role in cell proliferation, and its enhanced expression has been associated with colorectal carcinogenesis [19].
Given that a large number of neutrophils were observed in the colonic infiltrate in the CAC mice, we evaluated the mRNA expression of the chemokine receptor CXCR2, which is mainly expressed in these cells. CXCR2 expression was significantly increased in the CAC mice, but T. crassiceps infection down-regulated the expression of this chemokine neutrophil receptor in the colonic tissue (Fig. 3K). The chemokine receptor CXCR2 is a key mediator of neutrophil migration and is also involved in tumor development [20], CXCR2 is significantly up-regulated in various stages of CAC, and polymorphisms in the CXCR2 gene are associated with colon cancer progression and recurrence [21], our data here are suggestive that this helminthic infection could have potential to modulate neutrophil tumor-promoting functions.
Helminth infection prevents the increase of circulating CD11b+Ly6ChiCCR2+ and its recruitment into the colonic lamina propria
We analyzed the presence of inflammatory Ly6Chi monocytes in the peripheral circulation and colonic lamina propria. Ly6C monocytes differ in their expression of a major chemokine receptor CCR2. CCR2 is responsible for the recruitment of Ly6Chi monocytes to peripheral sites of inflammation. CCR2 monocyte depletion has been associated with control of inflammatory lesions, prevention of persistent inflammation and achieves control repair mechanisms [22]. CD11b+Ly6ChiCCR2+ monocytes were detected at a high frequency in the circulation of the CAC animals (Fig. 4A-B). Conversely, CD11b+Ly6CloCCR2- cells were significantly increased in the T. crassiceps+CAC mouse group (Fig. 4C). Mice in the CAC group had an increase in CD11b+Ly6ChiCCR2+ cells in the lamina propria of the colonic tissue that was blocked in the T. crassiceps+CAC mice (Fig. 4D). Indeed, the CAC group recruited lower percentages of CD11b+Ly6CloCCR2- cells into the colonic lamina propria (Fig. 4E).
T. crassiceps infection reduces the number of inflammatory monocytes during CAC. (A) Representative flow cytometry plots from control mice, CAC mice and T. crassiceps+CAC mice gated on CD11b+ living cells isolated from the circulation. Quantification of (B) CD11b+Ly6ChiCCR2+ cells (region A) and (C) CD11b+Ly6CloCCR2- (region B) cells from the circulation. Percentage of (D) CD11b+Ly6ChiCCR2+ and (E) CD11b+Ly6CloCCR2- in colonic lamina propria isolated cells. Data are representative of 2 independent experiments. Values are mean ± SD (n=4 mice/group) *P < 0.05, **P<0.03.
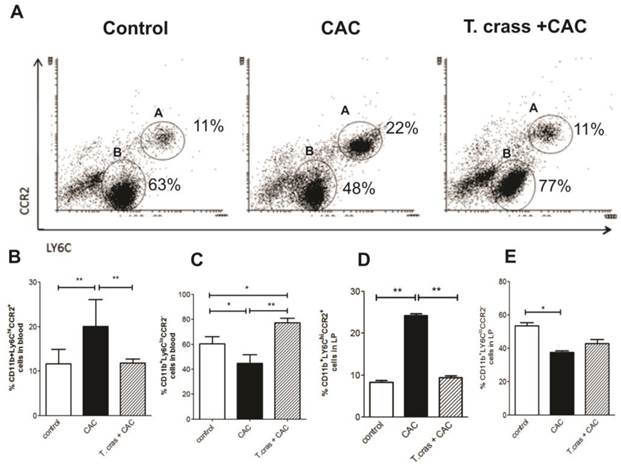
Ly6Chi monocytes are recruited to the inflamed colon in a CCR2 dependent fashion and give rise to pro-inflammatory effector cells that sense bacterial products and critically promote inflammation through the production of pro-inflammatory mediators [23]. The fact that T. crassiceps infection reduced the number of inflammatory monocytes in both the circulation and the lamina propria of the colon implies a strong regulatory activity triggered by this helminth, which could also contribute to a reduction in tumor development induced by CAC.
In sum, we demonstrate here for the first time that a helminth infection inhibits the number of colon tumors and, consequently, the development of CAC. This is most likely due to the capacity of T. crassiceps infection to down-modulate inflammatory responses. Several potential mechanisms may be triggered by T. crassiceps to inhibit tumorigenesis in this CAC model, which would be worthwhile analyzing in more detailed studies and may include: a) the recruitment of AAMs with tissue repair ability; b) lack of a response to TLR-driven inflammation; c) increase or maintenance of goblet cells and mucus production; d) blocking the recruitment of inflammatory monocytes; e) blocking of the intracellular signaling pathways associated with inflammation; and f) lowering the expression of β-catenin (proto-oncogenes). Thus, the immunoregulatory activities of helminths may be useful in modulating the outcome of inflammatory-associated cancers in addition to their well-known role in modulating autoimmunity.
Acknowledgements
Sonia León-Cabrera was supported by a postdoctoral fellowship from Dirección General de Asuntos del Personal Académico, UNAM. This work is part of the requirements to obtain the PhD degree in Programa de Doctorado en Ciencias Biomedicas, UNAM (FES-Iztacala) for Blanca E. Callejas, who is supported by a fellowship from CONACYT (México). This work was supported by Grants 167799 CONACYT, PAPCA-2013 FES-Iztacala 16, 19, 28. The authors thank Dr. Jorge Barrios-Payán for his help on tissue processing and histology.
Conflict of interest
The authors declare not conflict of interest.
References
1. Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E. et al. Cancer statistics, 2004. CA: a cancer journal for clinicians. 2004;54:8-29
2. Kraus S, Arber N. Inflammation and colorectal cancer. Current opinion in pharmacology. 2009;9:405-10
3. Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World journal of gastroenterology: WJG. 2008;14:3937-47
4. Piazuelo MB, Epplein M, Correa P. Gastric cancer: an infectious disease. Infectious disease clinics of North America. 2010;24:853-69 vii
5. Sripa B, Brindley PJ, Mulvenna J, Laha T, Smout MJ, Mairiang E. et al. The tumorigenic liver fluke Opisthorchis viverrini--multiple pathways to cancer. Trends in parasitology. 2012;28:395-407
6. http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx
7. McSorley HJ, Maizels RM. Helminth infections and host immune regulation. Clinical microbiology reviews. 2012;25:585-608
8. Terrazas CA, Alcantara-Hernandez M, Bonifaz L, Terrazas LI, Satoskar AR. Helminth-excreted/secreted products are recognized by multiple receptors on DCs to block the TLR response and bias Th2 polarization in a cRAF dependent pathway. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:4547-60
9. Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta pharmacologica Sinica. 2007;28:1450-9
10. Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. 2007;2:2307-11
11. Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229-44
12. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011;70(Suppl 1):i104-8
13. van Liempt E, van Vliet SJ, Engering A, Garcia Vallejo JJ, Bank CM, Sanchez-Hernandez M. et al. Schistosoma mansoni soluble egg antigens are internalized by human dendritic cells through multiple C-type lectins and suppress TLR-induced dendritic cell activation. Mol Immunol. 2007;44:2605-15
14. Segura M, Su Z, Piccirillo C, Stevenson MM. Impairment of dendritic cell function by excretory-secretory products: a potential mechanism for nematode-induced immunosuppression. European journal of immunology. 2007;37:1887-904
15. Koyasu S, Moro K. Type 2 innate immune responses and the natural helper cell. Immunology. 2011;132:475-81
16. Leon-Cabrera S, Cruz-Rivera M, Mendlovic F, Romero-Valdovinos M, Vaughan G, Salazar AM. et al. Immunological mechanisms involved in the protection against intestinal taeniosis elicited by oral immunization with Taenia solium calreticulin. Experimental parasitology. 2012;132:334-40
17. Allen JE, Wynn TA. Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathog. 2011;7:e1002003
18. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB. et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117-29
19. Cooper HS, Murthy S, Kido K, Yoshitake H, Flanigan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis. 2000;21:757-68
20. Wang B, Hendricks DT, Wamunyokoli F, Parker MI. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer research. 2006;66:3071-7
21. Zhang W, Stoehlmacher J, Park DJ, Yang D, Borchard E, Gil J. et al. Gene polymorphisms of epidermal growth factor receptor and its downstream effector, interleukin-8, predict oxaliplatin efficacy in patients with advanced colorectal cancer. Clinical colorectal cancer. 2005;5:124-31
22. Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139-55
23. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G. et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37:1076-90
Author contact
![]() Corresponding author: Luis I. Terrazas, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. De los Barrios 1, Los Reyes Iztacala, Tlalnepantla, Edo. de Mexico. Mexico 54090. Phone: +52 55 5623 1333 ext 39773. e-mail: literrazasiztacala.unam.mx
Corresponding author: Luis I. Terrazas, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. De los Barrios 1, Los Reyes Iztacala, Tlalnepantla, Edo. de Mexico. Mexico 54090. Phone: +52 55 5623 1333 ext 39773. e-mail: literrazasiztacala.unam.mx