Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(1):99-108. doi:10.7150/ijbs.8621 This issue Cite
Research Paper
CEP2 Attenuates Myoblast Differentiation But Does Not Affect Proliferation
Ming Wu1, Gongshe Yang2, Yaosheng Chen1, Xingyu Zhou1, Hu Chen1, Mingsen Li1, Kaifan Yu1, Xumeng Zhang1, Shuihua Xie1, Ying Zhang1, Guiyan Chu2, Delin Mo1 ![]()
1. State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-Sen University, Guangzhou 510006, China;
2. Laboratory of Animal Fat Deposition and Muscle Development, College of Animal Science and Technology, Northwest A&F University, Yangling, China.
Received 2014-1-1; Accepted 2014-3-11; Published 2015-1-1
Abstract

CEP2 (CDC42EP2) is a member of the CDC42 subfamily that belongs to the Rho family. The Rho family plays an important role in a variety of cellular processes including skeletal myogenesis. Here, we find the expression of CEP2 increased significantly during C2C12 myogenesis. Overexpression of CEP2 could attenuate myoblast differentiation, while knockdown of CEP2 by siRNA results in enhancing myogenesis. Furthermore, we demonstrate for the first time that CEP2 attenuates myoblast differentiation via suppression of muscle regulatory factors (MRFs) rather than influencing myoblast proliferation. These results indicate that CEP2 acts as a repressor during myogenesis, which provides new insights into the role of CEP2 in muscle development.
Keywords: CEP2, myogenesis, skeletal muscle, myoblast proliferation.
Introduction
The formation of mature skeletal muscle is a multistep process wherein pluripotent mesodermal cells commit to myoblasts, and then myoblasts withdraw from the cell cycle and fuse into plurinucleated myotubes [1, 2]. The development of skeletal muscle is mainly orchestrated by a family of muscle-regulatory factors (MRFs) including myoD, myf5, myogenin, and mrf4 [3, 4]. At the onset of myoblast differentiation, proliferating myoblasts express myf5 and myoD. When myoblasts are cultured in differentiation medium, myoD induce termination of myoblast proliferation and the expression of myogenin. Besides, the process that coordinates myoblast proliferation and myoblast differentiation needs down-regulation of cell cycle activators such as cyclins and cdks and up-regulation of cell cycle inhibitors such as Rb, p21, p27 and p57 [5-9]. Indeed, a crosstalk between myoblast proliferation and differentiation is important during myogenesis.
MRFs are targets of various signaling pathways including the Rho family of small GTPases which consists of three subfamilies (Rho, Rac, and Cdc42) [10-16]. During myogenesis, Cdc42 (cell division cycle 42 homolog) regulates myoblast fusion by recruiting F-actin and other molecules to the sites of cell-cell contact in vitro and in vivo [17, 18]. To be competent for fusion, myoblasts extend lamellipodium and filopodia for contacting with neighbors [19], which is necessary to recognize each other. Interestingly, CEP2 involved in the formation of pseudopodia by interacting with Cdc42. In addition, co-expression of CEP2 with dominant negative mutant CDC42 or expressing a Cdc42/Rac interactive binding domain mutant of CEP2 didn't induce long pseudopodia formation in fibroblasts [20]. These results suggest CEP2 has a function in myoblast shape changes, recognition and fusion. Previous study has identified that CEP2 is down-regulated in tongue muscle from myogenin-/- embryos using microarray analysis [21], which implies that CEP2 plays a role in skeletal muscle differentiation. Another study reveals CEP2 mRNA is decreased in miR-29a mutant mouse progenitors using gene array, indicating CEP2 links to miR-29a (a promoter) in myogenesis [22, 23]. Transcriptional profiling shows that CEP2 may play pivotal roles in cytokinesis of human cardiomyocytes [24], which shares some of the common myogenic factors. Besides, CEP2 is also known as a Rho GTPase [20, 25-26], which can be regarded as a molecular switch controlling cell behavior [12]. Therefore, the role of CEP2 in myogenesis attracts our interest.
Although the role of CEP2 in fibroblasts is well elaborated, its biological function in myoblasts remains unknown. The aim of the present study was to characterize the CEP2 in myogenesis, and gain insight into its potential biological role in muscle development by overexpressing and knocking down it in C2C12 myoblast.
Materials and Methods
Plasmids and Generation of Lentivirus Cell Lines
The cDNA of mouse CEP2 were separately cloned into pcDNA3.1 vector and lentivirus packing vector pl411G GLUC (Invitrogen, Shanghai, China). The other lentiviral components including pMD2G and PsPAX2 were extracted using ultrapure endotoxin-free extraction kits (Omega, Guangzhou, China).
The lentivirus vectors were prepared by transient transfection of 293T cells using the calcium phosphate precipitation method. After incubation for 48, 60 and 72 h, the cell supernatant containing virus-like particles was collected. Subsequently, the viral titers were determined after condensation. When C2C12 cell confluence reached 50-60% in 6-well plates, C2C12 cells were infected with lentivirus-based CEP2 and an empty lentivirus vector (CON) for 12 hours. One or two days later, the GFP expression level was measured by fluorescence microscopy as the transfection efficiency. Cells were then harvested and screened with flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The positive cells obtained were passed, harvested and screened for 6 times. Finally the differentiated cells were used for mRNA and protein level analysis. All transfections were performed in triplicate for each experiment.
Cells Culture and Differentiation Induction
Mouse C2C12 myoblasts were cultured in high-glucose DMEM (Gibco, Guangzhou, China) with 10% (v/v) fetal bovine serum (GM) and were maintained at 37 °C in 5% CO2 incubator. To induce differentiation, culture medium was changed to DMEM with 2% horse serum (DM) (Gibco) when cells reached confluence. The culture medium was replaced at 2-day intervals before the end of the checkpoint.
Cell Transfection
The C2C12 cells were seeded in twelve-well or six-well plates. After 12 hours, when cell confluence reached 50%~60%, the plasmids or siRNA pools were transfected into C2C12 cells using Lipofectamine 2000 transfection reagents (Life Technologies, Shanghai, China) or DharmaFECT siRNA transfection reagents (Thermo Fisher, Guangzhou, China) following the manufacturer's instructions. Nucleic acids and transfection reagents were diluted by Opti-MEM I without Serum Medium (Gibco). The sequences of siRNA pools were listed in Supplementary Material: Table S1.
Real Time RT-PCR (qPCR)
Total RNA was extracted from cells with TRIzol reagent (Life Technologies) and treated with DNase I (Promega, Beijing, China). The concentration and quality of RNA were assessed by NanoDrop ND-1000 (Thermo, Waltham, MA, USA) and denatured gel electrophoresis. Reverse transcription was performed using AMV reverse transcriptase (Promega). The qPCR reaction was carried out in the LightCycler 480 II system (Roche, Basel, Switzerland). The sequences of qPCR primers can be found in Supplementary Material: Table S2. Statistical analysis of mRNA relative expression was performed with Student's t test. The analytic method of 2-ΔΔCt was used, ΔΔCt = [target gene (treatment group) / target gene (control group)] / [house-keeping gene (treatment group) / house-keeping gene (control group)]. *p<0.05; **p<0.01; n ≥ 3 (n: Biological replicates).
Western blot
C2C12 cells were treated with Lysis Buffer (Beyotime Biotechnol, Shanghai, China), which was premixed with PMSF (100:1). The proteins at supernatant were separated by SDS-PAGE, followed by protein transfer to polyvinylidene fluoride membranes using Mini Trans-Blot Cell (Bio-Rad, Hercules, CA, USA). The proteins were detected by primary antibodies specific for Myf5 (#sc-302, Santa Cruz, CA, USA), MyoD (#sc-304, Santa Cruz), MRF4 (#sc-301, Santa Cruz). β-actin (#4970s, CST, Massachusetts, USA ) served as loading controls in all western blot assays. The anti-rabbit secondary antibody (#7074s, CST) was used in this assay. Blots were visualized using a commercial enhanced chemiluminescene (ECL) detection Kit (Thermo Scientific, Beijing, China). All results of western blot were calculated and presented as mean ± S.E.M. *p<0.05; **p<0.01; n=3.
Immunofluorescence
After transfection and differentiation, C2C12 cells were harvested and detected by immunofluorescence following the manufacturer's instructions. Primary anti-myosin (skeletal, fast) antibody (#ab7784, Abcam, Hong Kong, China), anti-CEP2 antibody (#ab173012, Abcam), anti-myogenin antibody (#IMG131, Novus, Littleton, CO) and secondary antibody labeled with Alexa Fluor 555 fluorochrome (red) (Invitrogen) or labeled with FITC 488 fluorochrome (green) (Invitrogen) were used. Images were captured with a fluorescence inverse microscope (ZEISS, Heidenheim, Germany) (5 random fields were captured for each treatment group). Confocal microscopy (Leica Microsystems, Deerfield, IL) was used in the subcellular location of CEP2 protein experiment. n = 3.
Cell Proliferation Analysis
Analysis of Cell Cycle by Flow Cytometry: After transfection for 24 hours, suspension cells were fixed in ice-cold 70% (v/v) ethanol overnight. 50 mg/ml propidium iodide solution (PI) (which contains 100 mg/ml RNase A and 0.2% (v/v) Triton X-100) was added to suspension cells, then those cells were incubated at 4 °C for 30 min. Proliferative index was analyzed by BD FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA) and ModFit software (Verity Software House, Topsham, ME, USA). The percentage of mitotic cells from a total of 10000 cells examined (n = 3) stands for the proliferative index.
EdU assay: After cells were transfected for 24 hours, EdU reagents (RIBOBIO, Guangzhou, China) (final concentration, 10 mM) were added for 4 hours. 4% paraformaldehyde was used for cell fixation, the proportion of EdU-positive cells were detected by fluorescence inverse microscope (5 random fields were captured for each treatment group, n = 3).
AlamlaBlue assay: 6 hours post-transfection, alamlaBlue reagents (Invitrogen) (final concentration, 10% v/v) were added into fresh complete medium (n = 6). The fluorescence value was detected by a microplate reader (BioTek, Vermont, USA) at 6 or 12-hour intervals.
Statistical analysis
All results are presented as mean ± standard errors of the means (SEM) based on at least three independent experiments. Unless additional notion, the differences between groups were analyzed with a Student's two-tailed t test when only two groups were compared or ANOVA when more than two groups were compared. *p<0.05; **p<0.01; n ≥ 3.
Results
CEP2 is increased during C2C12 myoblast differentiation
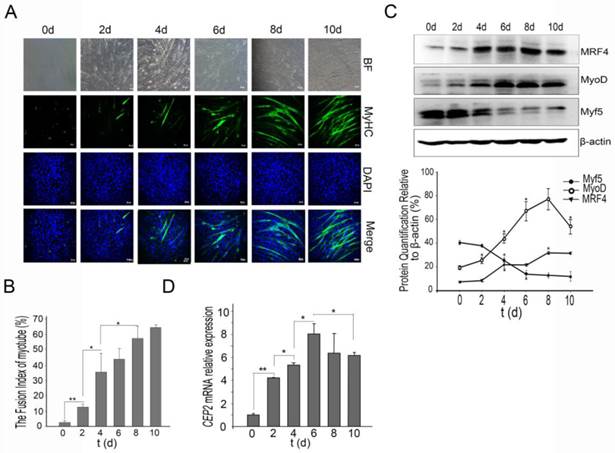
The first issue that we sought to address was whether CEP2 participates in C2C12 myoblast differentiation. For this purpose, we established a C2C12 induction model that was confirmed by immunofluorescence of MyHC (a muscle-specific protein) (Figure 1A). The fusion index of myotube increased regularly (Figure 1B). During differentiation, Myf5 protein exerted high expression at the initiation of C2C12 differentiation, and declined rapidly after 2 days. Unlike Myf5, MyoD protein gradually was up-regulated, but declined after 8 days. MRF4 protein exhibited continuous increase (Figure 1C). CEP2 expression was continuous increased significantly, and then gradually decreased at the end of C2C12 differentiation (Figure 1D).
CEP2 was upregulated during C2C12 myoblast differentiation. (A) A normal model of C2C12 cells differentiation was established using DMEM with 2% horse serum (DM). Every two days, MyHC protein was detected by anti-myosin (skeletal, fast) antibody (green). The nuclei were stained with DAPI (blue). BF: bright field. Scale bar = 50μm. (B) The fusion index (percentage of the number of nuclei residing in the MyHC-positive cells divided by the total number of the nuclei) was calculated during differentiation. *p<0.05; **p<0.01, n = 3. (C) Western blotting analysis of myogenic factors during differentiation. *p<0.05; n = 3. (D) CEP2 expression in C2C12 cells during differentiation. The level of mRNA was in relation to that on 0 days. *p<0.05; **p<0.01, n = 3 (three independent replicates per group).
Up-regulation of CEP2 attenuates myoblast differentiation
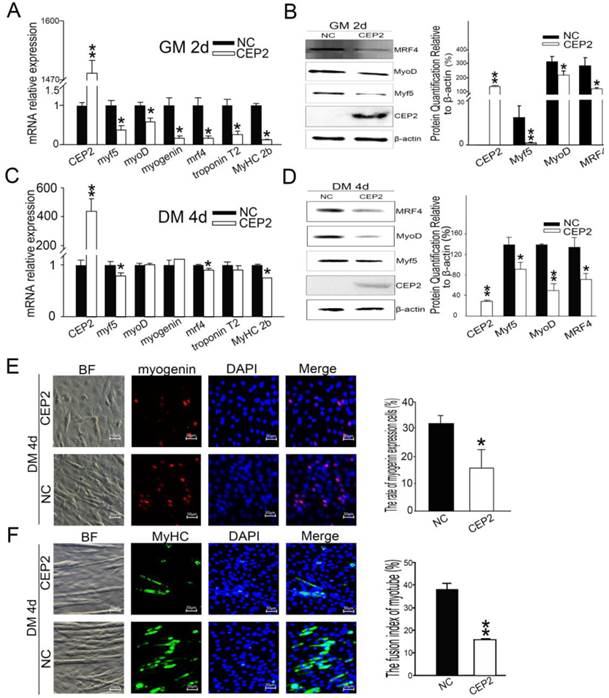
To address whether CEP2 gene influences myoblast differentiation, we detected muscle-specific gene expression after transfection with pcDNA3.1-CEP2. CEP2 was up-regulated significantly in C2C12 cells (Figure 2A). As a result, the mRNA level of myf5, myoD, myogenin, mrf4, troponin T2 and MyHC 2b were decreased significantly (Figure 2A). Myf5, MyoD, MRF4 expression were decreased as well (Figure 2B). When the cells cultured in DM for 4 days, the mRNA level of myf5, mrf4, and MyHC 2b were down-regulated significantly (Figure 2C). Myf5, MyoD and MRF4 expression were all decreased (Figure 2D). These results suggested that overexpression of CEP2 can inhibit the expression of muscle-specific gene. Immunofluorescence assay showed that myogenin and MyHC expression was decreased in DM on the 4th d, which also proved the inhibitory effect of CEP2 (Figure 2E and F). Furthermore, we found that CEP2 was distributed in the nucleus and cytoplasmic of myoblast and myotube (Supplementary Material: Figure S1). Besides, CEP2 tends to express in the nucleus of myotube. It indicated that CEP2 influencing myogenesis might be due to position effect. In summary, overexpression of CEP2 could inhibit myoblast differentiation.
Overexpression of CEP2 mediated by plasmids inhibited myoblast differentiation. RNA (A) and protein (B) of CEP2 and myogenic factors were analyzed in GM after 2 days post-transfection. Results were presented as mean ± S.E.M. After differentiation for 4 days, the mRNA (C) and protein (D) of myogenic factors were analyzed again. Myogenin (red) (E) and MyHC (green) (F) were detected using immunofluorescence in DM. The rate of myogenin expression cells was calculated. Scale bar = 50μm. NC: native control (pcDNA3.1); CEP2 (pcDNA3.1-CEP2). *p<0.05; **p<0.01; n ≥ 3.
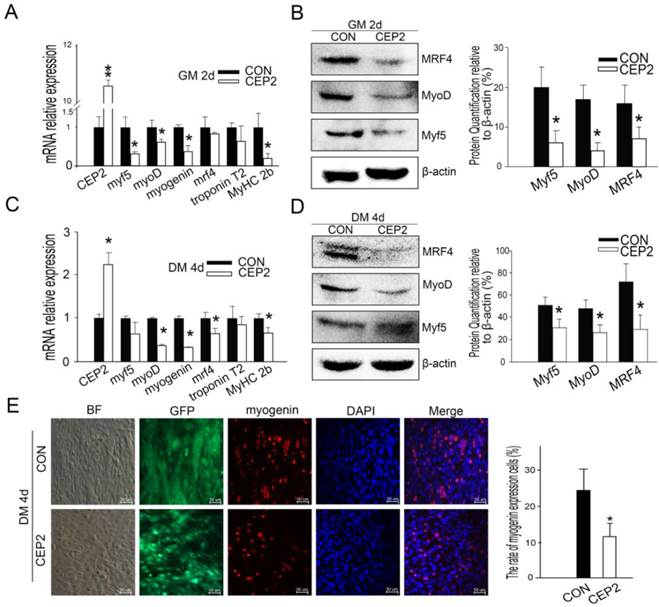
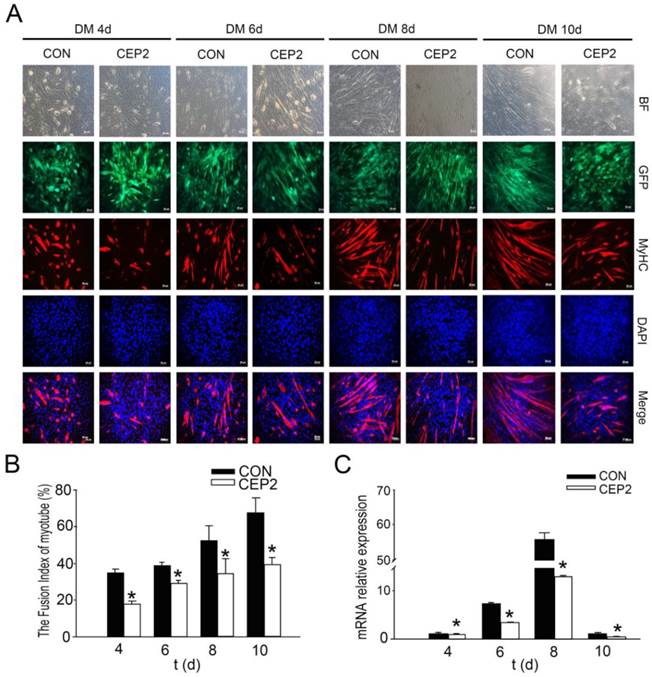
Compared with the plasmid, lentivirus-mediated transfection presented more stable performance and was therefore employed to study the inhibitory effect of CEP2 on differentiation. Consequently, CEP2 showed significant up-regulation both in GM on the 2nd d and in DM on the 10th d (Supplementary Material: Figure S2 A and B). These results presented more stable expression of CEP2. In GM on the 2nd d, the mRNA level of myf5, myoD, myogenin and MyHC 2b were decreased significantly (Figure 3A). Myf5, MyoD and MRF4 were down-regulated significantly (Figure 3B). After differentiation for 4 days, the mRNA level of myoD, myogenin, mrf4 and MyHC 2b were decreased significantly (Figure 3C). Myf5, MyoD and MRF4 were all reduced (Figure 3D). The rate of myogenin expression cells decreased, demonstrating that the protein level of myogenin declined (Figure 3E). To confirm the inhibitory role of CEP2, MyHC proteins were detected on the 4th, 6th, 8th and 10th day of post-transfection. As a result, the expression of MyHC declined at four stages mentioned above, demonstrating that myoblast differentiation was attenuated (Figure 4). Furthermore, this kind of inhibition mainly occurred in earlier period of differentiation (Figure 4A). After differentiation for 10 days, MyoD protein increased significantly (Supplementary Material: Figure S2 E and F), although the expression of myf5, mrf4 and MyHC 2b decreased significantly (Supplementary Material: Figure S2 D). It suggested that the expression of CEP2 can result in differentiation delay of myoblasts after 10 days but does not inhibit myoblast differentiation completely. In conclusion, up-regulation of CEP2 could attenuate myoblast differentiation.
Up-regulation of CEP2 mediated by lentivirus inhibited myoblast differentiation. qPCR (A) and (B) western blotting for myogenic factors of CEP2-expressing cells and control cells were detected in GM on the 2nd d. After differentiation for 4 days, the mRNA (C) and protein (D) of myogenic factors were analyzed again. (E) The rate of myogenin expression cells was analyzed with immunofluorescence. Scale bar = 50μm. CON: native control (an empty lentivirus vector); CEP2 (lentivirus-based CEP2). GFP: represents lentivirus infection efficiency.*p<0.05; **p<0.01; n ≥ 3.
CEP2 inhibited the expression of MyHC during differentiation. (A) MyHC protein was probed by anti-myosin antibody (red) in DM on the 4th, 6th, 8th and 10th d, respectively. GFP: represents lentivirus infection efficiency. Scale bar = 50μm. (B) The fusion index of myotubes was calculated at four stages. (C) RNA analysis of MyHC 2b at four stages.*p<0.05; **p<0.01, n ≥ 3.
Knockdown of CEP2 enhances myogenesis
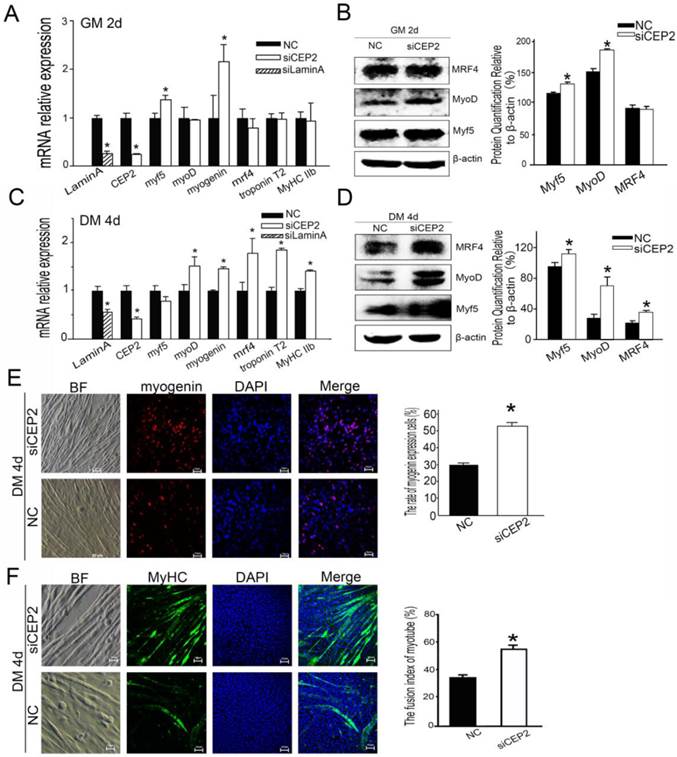
To further confirm the role of CEP2 in attenuating myoblast differentiation, CEP2 knockdown cells were generated using siRNA. As a result, the CEP2 mRNA level was 75% lower than the control at 24 h post-transfection (Figure 5A). In this assay, the mRNA level of myf5 and myogenin were increased significantly (Figure 5A), and the protein level Myf5 and MyoD was increased as well (Figure 5B). After differentiation for 4 days, the CEP2 mRNA level was also significantly lower than the control (Figure 5C). And the RNA level and protein level of myogenic factors showed a significantly increase except for myf5 (Figure 5C and D). The expression of myogenin and MyHC protein were also increased in CEP2 knockdown cells (Figure 5E and F). These results indicated that CEP2 knockdown could enhance myogenesis.
Knockdown of CEP2 enhanced myogenesis. RNA (A) and protein (B) analysis of myogenic factors after transfection of siCEP2 to C2C12 in GM on the 2nd d. siLamin A: positive control (siRNA pool targeted to Lamin A). After differentiation for 4 days, the mRNA (C) and protein (D) of myogenic factors was analyzed. The rate of myogenin expression cells (E) and MyHC (F) were detected with immunofluorescence in DM on the 4th d. Scale bar = 50μm. siCEP2: siRNA pool targeted to CEP2, NC: native control (No target siRNA pool). *p<0.05; n ≥ 3.
CEP2 cannot influence cell proliferation
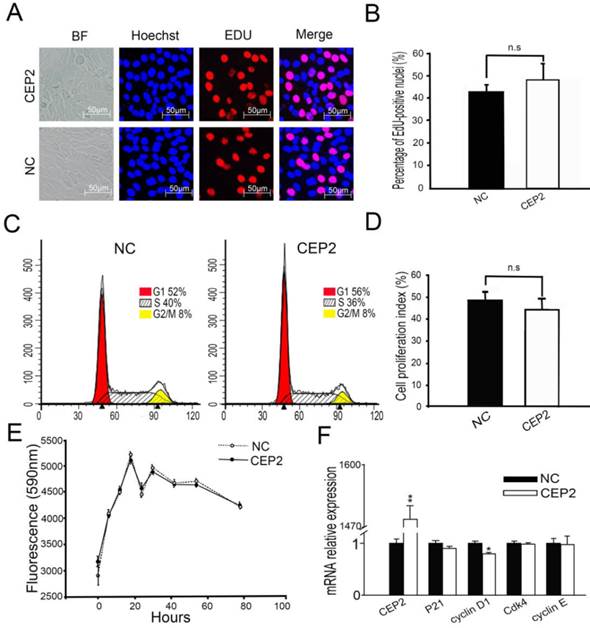
Before cell cycle arrest and fusion into myotubes, the cell cycle of myoblasts undergoes a phase of active proliferation [5, 6]. The molecular events maintain the balance between proliferation and differentiation of skeletal myoblasts [27], by shedding light on the crosstalk mechanisms taking place between cell cycle regulators and myogenic factors [28-31]. In order to address whether CEP2 gene influences cell proliferation, EdU experiment assay and flow cytometry analysis were performed at 24 h post-transfection (Figure 6A, B, C and D). It showed that overexpression of CEP2 did not change cell proliferation significantly, which was confirmed by alamlaBlue assay. This assay was performed after transfection with pcDNA3.1-CEP2 for 6 h and the dynamic curve of fluorescence values was plotted during cell growth (Figure 6E). This finding was similar to the result of experiment in which CEP2 gene was knocked down by siRNA (Supplementary Material: Figure S3). In summary, CEP2 could not influence cell proliferation significantly.
Overexpression of CEP2 did not influence cell proliferation. (A) The cells were fixed for EDU (red) immunostaining at 24 h post-transfection. Scale bar = 50μm. (B) The proportion of proliferation cells in EDU assay was presented as mean ± S.E.M (n=3). (C) C2C12 cells were collected for PI staining and FACSCalibur cell cycle assay. (D) The proliferative index in FACSCalibur assay was presented as mean ± S.E.M (n=3) (E) The fluorescence value was measured every 6, 12 or 24 hours using alamlaBlue assay after 6 hours post-transfection (n = 6). (F) RNA analysis of cell cycle-specific genes at 24 h post-transfection. CEP2: pcDNA3.1-CEP2; NC, negative control (pcDNA3.1). *p<0.05; n ≥ 3.
Discussion
In this study, the effects of CEP2 on myogenesis were investigated. Our results show that the expression of CEP2 is increased during myogenic differentiation. Further, CEP2 overexpression led to attenuating myogenic differentiation, while knockdown of CEP2 resulted in enhancing myogenesis. For the first time, we report here that CEP2 attenuates the myoblast differentiation by suppressing MRFs.
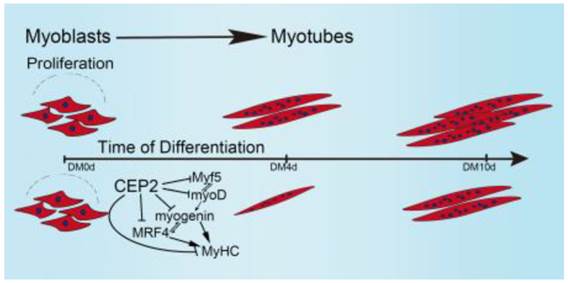
Taken together, these results allow us to propose a model demonstrating the biological role of CEP2 in myogenesis (Figure 7). In our model, when GM was changed to DM, myoblasts express myf5 and myoD which resulted in termination of myoblast proliferation and the expression of myogenin, MRF4, and MyHC. Up-regulation of CEP2 inhibits the expression of MRFs and MyHC, ultimately attenuating myoblast differentiation. On the other hand, knockdown of CEP2 promotes the expression of MRFs and MyHC, and thus stimulating myoblast differentiation.
A model illustrating the biological role of CEP2 in myogenesis. DM: differentiation medium.
Critical for skeletal muscle differentiation are the down-regulation of cell cycle activators such as cyclins and cdks [9], and the up-regulation of cell cycle inhibitors such as p21 and p57 [5, 9, 27]. Crosstalk between cell cycle regulators and myogenic factors is important during myoblast differentiation [5, 6]. Cyclin E-Cdk2 phosphorylates the Ser200 of MyoD, which in turn causes degradation of MyoD and promotion of late G1-Phase [29]. Cyclin D1 can regulate MyoD expression and the onset of myogenesis by controlling the subcellular location of Cdk4 protein [27]. We investigated whether the role of CEP2 in myogenic differentiation was caused by crosstalk between cell cycle regulators and MRFs. Unexpectedly, neither overexpression nor knockdown of CEP2 significantly influenced cell cycle progression or cell proliferation. Even though cyclin D1 and p21 expression levels had changed, they didn't significantly influence cell cycle events in our investigation. It suggested that CEP2 attenuating myoblast differentiation does not mainly via regulatory mechanisms between cell cycle regulators and myogenic factors.
The effect of CEP2 (a Rho GTPase) on myogenic differentiation is mediated by MRFs in our research. CEP2 overexpression significantly inhibited MRFs expression while CEP2 knockdown markedly up-regulated myogenic factor such as MyoD. This study demonstrated that CEP2 is a repressor of myogenesis. CEP2 is a member of the CEP proteins family, most of which act as Cdc42 effector proteins via a CRBI domain, which binds Cdc42 [20]. Cdc42 is essential for differentiation and myoblast fusion in vivo and vitro [17-19, 32]. Cdc42 inhibits the expression of myogenin, troponin T and MyHC, which ultimately blocks myogenesis via activation of the C-jun N-terminal kinase (JNK) pathway [32, 33]. However, it has been suggested that Cdc42 exerts distinct functions under different conditions: For example, Cdc42 has a dual function in myogenesis due to its activation of the stress-activated protein kinase (SAPK) as well as p38, which both are necessary for myogenesis [13, 32].
Pseudopodia formation cannot be induced with a Cdc42/Rac interactive binding domain mutant of CEP2 or a dominant negative Cdc42 with CEP2 in fibroblasts [20]. These results suggest that CEP2 regulates myogenic differentiation through acting as a downstream factor of Cdc42. It showed that CEP2 attenuating myogenesis could be mediated by JNK pathway, which has a crosstalk with MRFs and finally attenuates myogenesis. Additionally, a series of other mechanisms are involved in CEP2-attenuated myogenic differentiation. It has been shown that expression of CEP2 reduces levels of E-cadherin at adherens junctions and alters the organization of F-actin and Vinculin [12, 20]. This way, cytoskeletal proteins and the extracellular matrix are also involved in myoblast differentiation [34-36].
Supplementary Material
Figures S1-S3, Tables S1-S2.
Acknowledgements
This research was supported by the Natural Science Foundation of China (U1201213, 31272417) and the Fundamental Research Funds for the Central Universities (12lgpy44).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rudnicki MA, Jaenisch R. The MyoD family of transcription factors and skeletal myogenesis. Bioessays. 1995;17:203-209
2. Perry RL, Rudnicki MA. Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci. 2000;5:750-767
3. Yokoyama S, Asahara H. The myogenic transcriptional network. Cell Mol Life Sci. 2011;68:1843-1849
4. Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol. 2005;16:585-595
5. Kitzmann M, Fernandez A. Crosstalk between cell cycle regulators and the myogenic factor MyoD in skeletal myoblasts. Cell Mol Life Sci. 2001;58:571-579
6. Lassar AB, Skapek SX, Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr opin cell biol. 1994;6:788-794
7. Zabludoff SD, Csete M, Wagner R. et al. p27Kip1 is expressed transiently in developing myotomes and enhances myogenesis. Cell Growth Differ. 1998;9:1-11
8. Zhang P, Wong C, Liu D. et al. p21CIP1 and p57KIP2 control muscle differentiation at the myogenin step. Genes Dev. 1999;13:213-224
9. De Falco M, De Luca A. Involvement of cdks and cyclins in muscle differentiation. Eur J Histochem. 2006;50:19-23
10. M.Hindi S, Tajrishi MM, Kumar A. Signaling mechanisms in mammalian myoblast fusion. Sci Signal. 2013;6:1-9
11. Terada K, Misao S, Katase N. et al. Interaction of Wnt signaling with BMP/Smad signaling during the transition from cell proliferation to myogenic differentiation in mouse myoblast-derived cells. Int J Cell Biol. 2013;2013:1-11
12. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629-635
13. Bryan BA, Li D, Wu X. et al. The Rho family of small GTPases: crucial regulators of skeletal myogenesis. Cell Mol Life Sci. 2005;62:1547-1555
14. Doherty JT, Lenhart KC, Cameron MV. et al. Skeletal muscle differentiation and fusion are regulated by the BAR-containing Rho-GTPase-activating protein (Rho-GAP), GRAF1. J Biol Chem. 2011;286:25903-25921
15. Charrasse S, Causeret M, Comunale F. et al. Rho GTPases and cadherin-based cell adhesion in skeletal muscle development. J Muscle Res Cell Moti. 2003;24:309-313
16. Takano H, Komuro I, Oka T. et al. The Rho family G proteins play a critical role in muscle differentiation. Mol Cell Biol. 1998;18:1580-1589
17. Heller H, Gredinger E, Bengal E. Rac1 inhibits myogenic differentiation by preventing the complete withdrawal of myoblasts from the cell cycle. J Biol Chem. 2001;276:37307-316
18. Abmayr SM, Pavlath GK. Myoblast fusion: lessons from flies and mice. Development. 2012;139:641-656
19. Abramovici H, Gee SH. Morphological changes and spatial regulation of diacylglycerol kinase-ζ, syntrophins, and Rac1 during myoblast fusion. Cell moti cytoskeleton. 2007;64:549-567
20. Hirsch DS, Pirone DM, Burbelo PD. A new family of Cdc42 effector proteins, CEPs, function in fibroblast and epithelial cell shape changes. J Biol Chem. 2001;276:875-883
21. Davie JK, Cho JH, Meadows E. et al. Target gene selectivity of the myogenic basic helix-loop-helix transcription factor myogenin in embryonic muscle. Dev Biol. 2007;311:650-664
22. Han YC, Park CY, Bhagat G. et al. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J Exp Med. 2010;207(3):475-489
23. Zhou L, Wang L, Lu L. et al. A novel target of microRNA-29, Ring1 and YY1-binding protein (Rybp), negatively regulates skeletal myogenesis. J Biol Chem. 2012;287:25255-25265
24. Laframboise W, Petrosko P, Krill-Burger J. et al. Proteins secreted by embryonic stem cells activate cardiomyocytes through ligand binding pathways. J Proteomics. 2010;73(5):992-1003
25. Joberty G, Perlungher RR, Macara IG. The Borgs, a new family of Cdc42 and TC10 GTPase-interacting proteins. Mol Cell Biol. 1999;19(10):6585-6597
26. Joberty G, Perlungher RR, Sheffield PJ. et al. Borg proteins control septin organization and are negatively regulated by Cdc42. Nat Cell Biol. 2001;3(10):861-866
27. Zhang JM, Qin W, Zhao XH. et al. Coupling of the cell cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO J. 1999;18:926-933
28. Guo K, Walsh K. Inhibition of myogenesis by multiple Cyclin-Cdk complexes: coordinate regulation of myogenesis and cell cycle activity at the level of E2F. J Biol Chem. 1997;272:791-797
29. Tintignac LA, Leibovitch MP, Kitzmann M. et al. Cyclin E-cdk2 phosphorylation promotes late G1-phase degradation of MyoD in muscle cells. Exp Cell Res. 2000;259:300-307
30. Puri PL, Balsano C, Burgio VL. et al. MyoD prevents cyclinA/cdk2 containing E2F complexes formation in terminally differentiated myocytes. Oncogene. 1997;14:1171-1184
31. Zhang JM, Zhao XH, Qin W. et al. Direct inhibition of G1 cdk kinase activity by MyoD promotes myoblast cell cycle withdrawal and terminal differentiation. EMBO J. 1999;18:6983-6993
32. Meriane M, Roux P, Primig M. et al. Critical activities of Rac1 and Cdc42Hs in skeletal myogenesis: antagonistic effects of JNK and p38 pathways. Mol Biol cell. 2000;11:2513-2528
33. Vasyutina E, Martarelli B, Brakebusch C. et al. The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc Natl Acad Sci U S A. 2009;106:8935-8940
34. Velleman SG, Shin J, Li XH, et al Review. The skeletal muscle extracellular matrix: possible roles in the regulation of muscle development and growth. Can J Anim Sci. 2012;92:1-10
35. Sequeira V, L.A.M Nijenkamp L, A.Regan J. et al. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta. 2014;1838:700-722
36. Nayak RC, Chang KH, Vaitinadin NS. et al. Rho GTPases control specific cytoskeleton-dependent functions of hematopoietic stem cells. Immunol Rev. 2013;256:255-268
Author contact
![]() Corresponding author: Delin Mo. Address: State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-Sen University, Guangzhou 510006, China. Email: modelinsysu.edu.cn; Tel: (86) 20-39332991; Fax: (86) 20-39332940.
Corresponding author: Delin Mo. Address: State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-Sen University, Guangzhou 510006, China. Email: modelinsysu.edu.cn; Tel: (86) 20-39332991; Fax: (86) 20-39332940.