Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(3):246-255. doi:10.7150/ijbs.11138 This issue Cite
Research Paper
MicroRNA-1 Participates in Nitric Oxide-Induced Apoptotic Insults to MC3T3-E1 Cells by Targeting Heat-Shock Protein-70
Yong-Eng Lee1,2, Chung-Ye Hong3, Yi-Ling Lin2, Ruei-Ming Chen2,4,5, ![]()
1. Department of Orthopedic Surgery, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan
2. Cell Biology and Molecular Image Research Center, Taipei Medical University-Wan Fang Hospital, Taipei, Taiwan
3. Department of Internal Medicine, Taipei Medical University-Wan Fang Hospital, Taipei, Taiwan
4. Graduate Institute of Medical Sciences, College of Medicine, Taipei Medical University, Taipei, Taiwan.
5. Anesthetics and Toxicology Research Center, Taipei Medical University Hospital, Taipei, Taiwan.
Received 2014-11-22; Accepted 2015-1-6; Published 2015-1-15
Abstract

Our previous studies showed that nitric oxide (NO) could induce osteoblast apoptosis. MicroRNA-1 (miR-1), a skeletal- and cardiac muscle-specific small non-coding RNA, contributes to the regulation of multiple cell activities. In this study, we evaluated the roles of miR-1 in NO-induced insults to osteoblasts and the possible mechanisms. Exposure of mouse MC3T3-E1 cells to sodium nitroprusside (SNP) increased amounts of cellular NO and intracellular reactive oxygen species. Sequentially, SNP decreased cell survival but induced caspase-3 activation, DNA fragmentation, and cell apoptosis. In parallel, treatment with SNP induced miR-1 expression in a time-dependent manner. Application of miR-1 antisense inhibitors to osteoblasts caused significant inhibition of SNP-induced miR-1 expression. Knocking down miR-1 concurrently attenuated SNP-induced alterations in cell morphology and survival. Consecutively, SNP time-dependently inhibited heat-shock protein (HSP)-70 messenger (m)RNA and protein expressions. A bioinformatic search predicted the existence of miR-1-specific binding elements in the 3'-untranslational region of HSP-70 mRNA. Downregulation of miR-1 expression simultaneously lessened SNP-induced inhibition of HSP-70 mRNA and protein expressions. Consequently, SNP-induced modifications in the mitochondrial membrane potential, caspase-3 activation, DNA fragmentation, and apoptotic insults were significantly alleviated by miR-1 antisense inhibitors. Therefore, this study showed that miR-1 participates in NO-induced apoptotic insults through targeting HSP-70 gene expression.
Keywords: Nitric oxide, MicroRNA-1, Apoptosis, HSP-70, Mitochondria
Introduction
Osteoblasts play a critical role in bone formation (1). During bone remodeling, old bone is resorbed by osteoclasts, and then new bone is formed by osteoblasts, which are differentiated from mesenchymal stem cells (2). However, a variety of systemic and local factors contributes to regulation of osteoblast activities (3, 4). Nitric oxide (NO), a reactive oxygen species (ROS), was reported to regulate osteoblast proliferation, differentiation, and mineralization (4, 5). In addition, our previous study showed that overproduction of NO elevated cellular ROS levels and concurrently induced apoptotic insults to osteoblasts via an intrinsic mitochondrion-dependent mechanism (6-8). In comparison, pretreatment with low concentrations of NO can protect osteoblasts against oxidative stress-induced injury through an antiapoptotic pathway (3, 9, 10). As a result, NO can function like a double-edged sword to control osteoblast activities by regulating cell survival or death. However, insults to osteoblasts may cause an imbalance in bone remodeling and severe perturbations in skeletal structure and function, ultimately leading to bone diseases such as osteoporosis, osteosclerosis, and osteopetrosis.
Micro (mi)RNAs that contain about 22 nucleotides belong to endogenously expressed small non-coding RNAs (11). miRNAs take part in crucial roles in adjusting biological processes, including cell differentiation, proliferation, and apoptosis (12). miRNAs can posttranscriptionally regulate specific gene expressions by base pairing with target messenger (m)RNAs, leading to mRNA cleavage or translational silencing (11, 12). For example, our previous study showed that miR-210 can mediate hypoxia-induced apoptosis of neuroblastoma cells by targeting expression of the antiapoptotic B-cell lymphoma (Bcl)-2 gene (13). In comparison, miR-1 was implicated as skeletal- and cardiac muscle-specific miRNA that plays pathological and biological roles in regulating cell apoptosis, ischemic myocardial injuries, cardiogenesis, conduction, myogenesis, and hypertrophy (14, 15). A previous study reported that reducing miR-1 expression protected cardiomyocytes against ischemic injury (16). Recently, miR-1 was also shown to participate in controlling brain development and neuronal activity (17, 18). miR-1 was more highly expressed in osteosarcoma cells compared to normal osteoblasts (19, 20). Furthermore, such induction of miR-1 gene expression is directly associated with controlling cell proliferation and cell cycle. However, the roles of miR-1 in NO-induced alterations in osteoblast activities are little known.
Apoptosis of osteoblasts can determine the bone cell population in skeletal tissues and consequently plays a critical role in physiologically regulating bone turnover and regeneration (21). In the clinic, early osteoblast apoptosis can lead to impaired osteogenesis and the pathogenesis of post-transplant osteoporosis (22). A mixture of intrinsic and extrinsic factors contributes to the regulation of cell apoptosis (23). Our previous studies showed that NO-induced oxidative stress can stimulate translocation of the proapoptotic Bax protein from the cytoplasm to the outer membranes of mitochondria, which subsequently induces osteoblast apoptosis via an intrinsic mitochondrion-dependent mechanism (6, 7). Heat shock protein (HSP)-70 are a family of highly conserved proteins whose expressions are either constitutive or inducible in response to different physiological and environmental conditions (24). Being a survival gene, HSP-70 can be rapidly expressed to build up cytoprotection against various types of cellular stress, including elevated temperatures, mechanical damage, hypoxia, lowered pH, and ROS generation (25). Kumei et al. (2003) reported that microgravity inhibited the production of HSP-70 in osteoblasts, which may cause osteoblast damage (26). In H9c2 rat ventricular cells, miR-1 was overproduced in response to oxidative stress and afterward triggered cell apoptosis by targeting HSP-60 and HSP-70 (14). However, whether or not miR-1 is involved in NO-induced insults to osteoblasts has not previously been appraised. Therefore, in this study, we evaluated the roles of miR-1 in NO-induced osteoblast injury and the possible mechanisms.
Materials and Methods
Cell culture and drug treatment
Mouse MC3T3-E1 cells purchased from American Type Culture Collection (Rockville, MD, USA) were seeded in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, L-glutamine, penicillin (100 IU/ml), and streptomycin (100 μg/ml) in 75-cm2 flasks at 37 °C in a humidified atmosphere of 5% CO2. Sodium nitroprusside (SNP) purchased from Sigma (St. Louis, MO, USA) was freshly dissolved in phosphate-buffered saline (PBS) (0.14 M NaCl, 2.6 mM KCl, 8 mM Na2HPO4, and 1.5 mM KH2PO4) in the absence of light. Our previous studies showed that SNP at 2 mM can induce insults to osteoblasts (3, 6-8). In this study, MC3T3-E1 cells were treated with 2 mM SNP for various time intervals. Control cells received PBS only.
Quantification of cellular NO and intracellular ROS
Levels of cellular NO were determined by the Griess reaction (7). After drug treatment, the culture medium was collected and centrifuged. Following a reaction of the supernatant with sulfanilamide and N-1-napthylethylenediamine, a colorimetric azo compound was formed and quantified using a microplate photometer (Anthos Labtec Instruments, Lagerhausstrasse, Wales, Austria). Amounts of intracellular ROS were quantified following a previously described method (8). Briefly, 5 x 105 osteoblasts were cotreated with SNP and 2',7'-dichlorofluorescin diacetate, an ROS-sensitive dye. After drug treatment, osteoblasts were suspended in PBS buffer. Relative fluorescent intensities were quantified using a flow cytometer (Becton Dickinson, San Jose, CA, USA).
Assays of cell morphologies and cell survival
Cell survival was determined by a trypan blue exclusion assay (18). Briefly, MC3T3-E1 cells (2 x 104 cells) were seeded in 24-well tissue culture plates. After drug treatment, cells were trypsinized with 0.1% trypsin-EDTA (Gibco-BRL). Following centrifugation and washing, MC3T3-E1 cells were suspended in PBS and stained with trypan blue dye (Sigma). Fractions of dead cells with a blue signal were visualized and counted using a reverse-phase microscope (Nikon, Tokyo, Japan). Cell morphologies were observed and photographed using a microscope (Nikon).
Quantification of the mitochondrial membrane potential (MMP)
The MMP was determined using flow cytometry (27). Briefly, MC3T3-E1 cells (5 × 105 cells) were cultured in 12-well tissue culture plates overnight. After drug treatment, cells were harvested and incubated with 3,3'-dihexyloxacarbocyanine iodide at 37 °C for 30 min in a humidified atmosphere of 5% CO2. After washing and centrifugation, cell pellets were suspended in PBS buffer. Intracellular fluorescent intensities were analyzed using a flow cytometer (EPICS XL, Beckman Coulter, Fullerton, CA, USA).
Assay of caspase-3 activity
Caspase-3 activity was determined by a fluorogenic substrate assay (28). Briefly, caspase-3 activity was analyzed by incubating cell lysates with the fluorogenic substrate, Asp-Glu-Val-Asp (DEVD), in a cell-free buffer system consisting of 10 mM Hepes (pH 7.4), 220 mM mannitol, 68 mM sucrose, 2 mM NaCl, 2.5 mM KH2PO4, 0.5 mM EGTA, 2 mM MgCl2, 5 mM pyruvate, 0.1 mM PMSF, and 1 mM dithiothreitol. Intensities of the fluorescent products in cells were measured with a LS55 luminescence spectrometer (Perkin Elmer, Norwalk, CT, USA).
Quantification of DNA fragmentation
DNA fragmentation was quantified using a cellular DNA fragmentation enzyme-linked immunosorbent assay (ELISA) kit (Boehringer Mannheim, Indianapolis, IN, USA) (8). Briefly, MC3T3-E1 cells (2 x 105) were subcultured in 24-well tissue culture plates and labeled with 5-bromo-2'-deoxyuridine (BrdU) at 100 µM overnight. After being harvested, 100 μl of a cell suspension was added to 96-well tissue culture plates. Following drug treatment, amounts of BrdU-labeled DNA in the cytoplasm were quantified using an Anthos 2010 microplate photometer (Anthos Labtec Instruments, Lagerhausstrasse, Wals/Salzburg, Austria) at a wavelength of 450 nm.
Analysis of apoptotic cells
Cell apoptosis was quantified using propidium iodide (PI) according to a previously described method (3). After drug treatment, MC3T3-E1 cells and mouse MC3T3-E1 cells were harvested and fixed in cold 80% ethanol. Following centrifugation and washing, fixed cells were stained with PI and analyzed by a flow cytometer (Beckman Coulter).
RNA isolation and quantitative polymerase chain reaction (qPCR) for miR-1 expression
Total RNA was isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and the expression of miR-1 by MC3T3-E1 cells was evaluated using a TaqMan miRNA assay kit (Applied Biosystems, Foster City, CA, USA) (13). Single-stranded complementary (c)DNA was synthesized from 50 ng of total RNA using looped primers of the TaqMan miRNA reverse- transcription kit (Applied Biosystems). miR-1 cDNA was amplified by the qPCR sequence- specific primers from the TaqMan miRNA assay in the ABI PRISM 7700 sequence detection system (Applied Biosystems). miRNA expression was normalized to that of the small nuclear RNA, RNU6B.
Suppression of miR-1 expression
Expression of miR-1 by MC3T3-E1 cells was suppressed using an Anti-miR™ miRNA inhibitor (hsa-miR-1; Applied Biosystems) (18). The Anti-miR™ miRNA Inhibitor Negative Control #1 (Applied Biosystems) served as a negative control in the experiments. Hsa-miR-1 and the anti-miRNA inhibitor negative control were transfected into MC3T3-E1 cells using the siPORT NeoFX transfection agent (Applied Biosystems) according to the manufacturer's instructions.
Immunoblotting analyses of HSP-70 and β-actin
Protein analyses were carried out according to a previously described method (29). After drug treatment, cell lysates were prepared in ice-cold radioimmunoprecipitation assay buffer (25 mM Tris-HCl (pH 7.2), 0.1% sodium dodecylsulfate (SDS), 1% Triton X-100, 1% sodium deoxycholate, 0.15 M NaCl, and 1 mM EDTA). Protein concentrations were quantified using a bicinchonic acid protein assay kit (Pierce, Rockford, IL, USA). Proteins (50 μg/well) were subjected to SDS-polyacrylamide gel electrophoresis (PAGE), and transferred to nitrocellulose membranes. A rabbit polyclonal antibody against human HSP-70 purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA, dilution 1:500) was used to detect HSP-70. Cellular β-actin protein was immunodetected using a mouse monoclonal antibody against mouse β-actin (Sigma, dilution 1:2500) as the internal standard. The secondary antibodies used were horseradish peroxidase (HRP) anti-rabbit (Santa Cruz Biotechnology, dilution 1:2500) and HRP anti-mouse (Sigma, dilution 1:2500). After adding enhanced chemiluminescence substrates to react with these secondary antibodies according to the instruction of an enhanced chemiluminescence detection system of the Western Lightning Plus-ECL (Perkin Elmer), these protein bands were observed and quantified using a digital imaging system (UVtec, Cambridge, UK).
qPCR analyses of HSP-70 and β-actin
mRNA from MC3T3-E1 cells was prepared for the qPCR analyses of HSP-70 and β-actin mRNA as described previously (30). Oligonucleotides for the PCR analyses of HSP-70 and β- actin mRNA were designed and synthesized by Clontech Laboratories (Palo Alto, CA, USA). The oligonucleotide sequences of the respective upstream and downstream primers for these three mRNA analyses were 5'-CCGCCTACTTCAACGACTC-3' and 5'- TCTTGAACTCCTCCACGAAG-3' for HSP-70 and 5'-GTGGGCCGCTCTAGGCACCAA-3' and 5'-CTCTTTGATGTCACGCACGATTTC-3' for β-actin. A qPCR analysis was carried out using iQSYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and the MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad).
Statistical analysis
Statistical differences between the control and drug-treated groups were considered significant when the p value of Duncan's multiple-range test was < 0.05. Statistical analysis between drug-treated groups was carried out using a two-way analysis of variance (ANOVA).Results
SNP increased levels of cellular NO and ROS and induced apoptotic insults to osteoblasts
NO and intracellular ROS were quantified to determine levels of SNP-induced oxidative stress to osteoblasts (Fig. 1A, B). Exposure of MC3T3-E1 cells to 2 mM SNP for 1 h caused a significant 67% elevation in cellular NO levels (Fig. 1A). After exposure for 3, 6, 12, and 24 h, SNP increased cellular NO by 3-, 6-, 8-, and 13-fold, respectively. In comparison, treatment of MC3T3-E1 cells with SNP for 1 h increased levels of intracellular ROS by 75% (Fig. 1B). When exposed to SNP for 3, 6, 12, and 24 h, amounts of intracellular ROS were augmented 4-, 5-, 7-, and 8-fold compared to the control group, respectively.
Effects of sodium nitroprusside (SNP) on cellular nitrite and intracellular reactive oxygen species (ROS) levels and osteoblast insults. MC3T3-E1 cells were exposed to 2 mM SNP for 1, 3, 6, 12, and 24 h. Amounts of cellular nitrite (A) and intracellular ROS (B) were quantified by the Griess reaction method and flow cytometry, respectively. Cell survival was determined using a trypan blue exclusion method (C). Caspase-3 activity was assayed by a fluorogenic substrate assay (D). DNA fragmentation (E) and apoptotic cells (F) were analyzed using an ELISA method and flow cytometry. Each value represents the mean ± SEM, n = 6. * Values significantly differ from the respective control, p < 0.05.
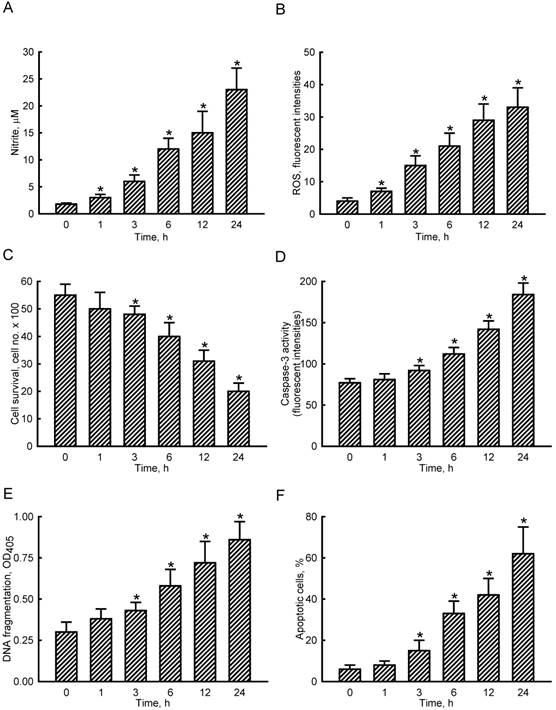
To evaluate NO-induced insults to osteoblasts, cell survival, caspase-3 activity, DNA fragmentation, and apoptotic cells were assayed (Fig. 1C-F). Exposure of MC3T3-E1 cells to SNP for 1 h did not affect cell survival (Fig. 1C). However, after treatment with SNP for 3, 6, 12, and 24 h, cell survival decreased by 13%, 28%, 44%, and 64%, respectively. In parallel, SNP caused respective 19%, 45%, 84%, and 139% increases in caspase-3 activities following SNP administration for 3, 6, 12, and 24 h (Fig. 1D). Exposure of MC3T3-E1 cells to SNP for 3, 6, 12, and 24 h led to respective 43%, 93%, 140%, and 187% induction of DNA fragmentation (Fig. 1E). Consequently, analysis of the cell cycle revealed that when treated with SNP for 3, 6, 12, and 24 h, the proportions of MC3T3-E1 cells undergoing apoptosis increased by 15%, 33%, 42%, and 62%, respectively (Fig. 1F).
NO induced miR-1 expression in osteoblasts
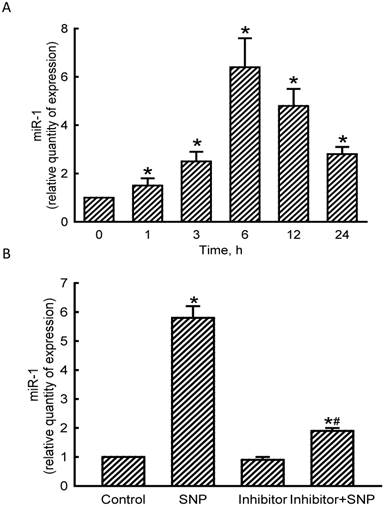
Levels of miR-1 in MC3T3-E1 cells were quantified to evaluate the effects of NO on the expression of this small non-coding RNA (Fig. 2). Exposure of MC3T3-E1 cells to SNP for 1 h led to a significant 50% induction in miR-1 expression (Fig. 2A). At 3, 6, 12, and 24 h after exposure to SNP, amounts of miR-1 were augmented by 2.5-, 6.4-, 4.8-, and 2.8-fold, respectively. Application of miR-1 antisense inhibitors to MC3T3-E1 cells slightly decreased basal levels of miR-1 (Fig. 2B). However, SNP-induced miR-1 expression was significantly reduced by 67% following pretreatment with miR-1 antisense inhibitors (Fig. 2B).
Effects of sodium nitroprusside (SNP) on microRNA-1 (miR-1) expression. MC3T3- E1 cells were exposed to 2 mM SNP for 1, 3, 6, 12, and 24 h. Levels of miR-1 were quantified using a quantitative PCR analysis (A). MC3T3-E1 cells were treated with SNP, hsa-miR-1 (a miR-1 inhibitor), and their combination for 6 h. The Anti-miR™ miRNA Inhibitor Negative Control #1 (Control) was transfected into MC3T3-E1 cells as a negative control. Amounts of miR-1 were quantified using a quantitative PCR analysis (B). Each value represents the mean ± SEM, n = 6. * and # Values significantly (p < 0.05) differ from the respective control and SNP-treated groups, respectively.
miR-2 participated in NO-induced osteoblast death
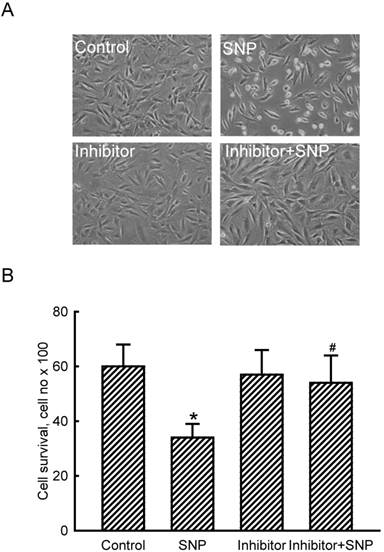
The role of miR-1 in NO-induced insults was further determined (Fig. 3). Exposure of MC3T3-E1 cells to SNP decreased the cell number and concurrently induced a shrunken morphology (Fig. 3A). Application of miR-1 antisense inhibitors did not affect the cell number or cell shape but caused significant alleviation of SNP-induced cell insults (Fig. 3A). Treatment with SNP caused a significant 47% decrease in cell survival (Fig. 3B). In comparison, knocking down miR-1 significantly lowered SNP-induced death of MC3T3-E1 cells.
Roles of micro RNA-1 (miR-1) in sodium nitroprusside (SNP)-induced osteoblast injuries. MC3T3-E1 cells were treated with SNP, hsa-miR-1 (an miR-1 inhibitor), and their combination for 12 h. The Anti-miR™ miRNA Inhibitor Negative Control #1 (Control) was transfected into MC3T3-E1 cells as a negative control. Cell morphology was observed with a microscope (A). Cell survival was determined using a trypan blue exclusion method (B). Each value represents the mean ± SEM, n = 6. * and # Values significantly (p < 0.05) differ from the respective control and SNP-treated groups, respectively.
NO inhibited HSP-70 mRNA and protein expressions in osteoblasts
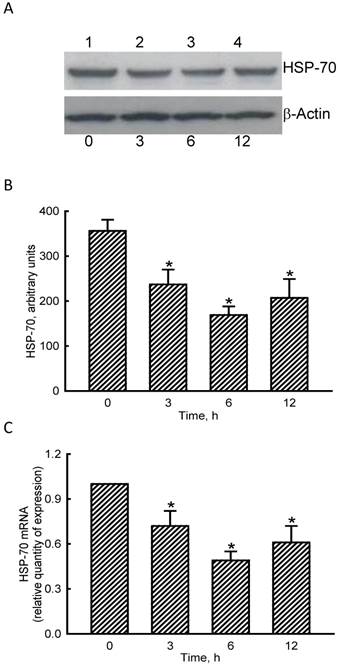
RNA and protein analyses were carried out to evaluate the effects of NO on HSP-70 expression (Fig. 4). Exposure of MC3T3-E1 cells to SNP for 3 h diminished levels of HSP-70 (Fig. 4A, top panel, lane 2). Amounts of HSP-70 were more obviously reduced after exposure to SNP for 6 and 12 h (lanes 3 and 4). β-Actin was immunodetected as the internal standard (bottom panel). These protein bands were quantified and statistically analyzed (Fig. 4B). Treatment of MC3T3-E1 cells with SNP for 3, 6, and 12 h caused significant 33%, 53%, and 42% decreases in levels of HSP-70. Real-time PCR analyses showed that treatment of MC3T3-E1 cells with SNP for 3, 6, and 12 h inhibited HSP-70 mRNA expression by 35%, 56%, and 44%, respectively (Fig. 4C).
Effects of sodium nitroprusside (SNP) on HSP-70 mRNA and protein expressions. MC3T3-E1 cells were exposed to 2 mM SNP for 3, 6, and 12 h. Amounts of HSP-70 were immunodetected (A, top panel). β-Actin was analyzed as the internal control (bottom panel). These immunorelated protein bands were quantified and statistically analyzed (B). Levels of HSP-70 mRNA were quantified using a quantitative PCR (C). Each value represents the mean ± SEM, n = 6. * Values significantly differ from the respective control, p < 0.05.
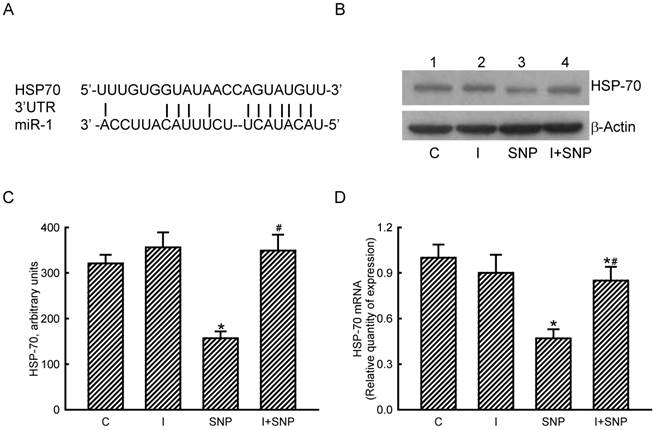
miR-1 targeted HSP-70 translation in osteoblasts
The roles of miR-1 in regulating HSP-70 gene expression were validated (Fig. 5). Results of a bioinformatic search revealed that a predicted miR-1-specific binding element exists in the 3'-untranslated region (UTR) of HSP-70 mRNA (Fig. 5A). Treatment of MC3T3-E1 cells with miR-1 antisense inhibitors did not lead to apparent changes in basal levels of HSP-70 (Fig. 5B, top panel, lane 2). However, SNP caused a decline in the amount of HSP-70 in MC3T3-E1 cells (lane 3). In comparison, pretreatment with miR-1 antisense inhibitors obviously lowered the SNP-induced reduction in HSP-70 production (lane 4). Levels of β-actin were immunodetected as the internal standard (bottom panel). These protein bands were quantified and statistically analyzed (Fig. 5C). Treatment of MC3T3-E1 cells with SNP caused a 51% decrease in the amount of HSP-70. Meanwhile, application of miR-1 antisense inhibitors significantly attenuated the SNP-induced decrease in cellular HSP-70 levels (Fig. 5C). Real-time PCR analyses further revealed that the SNP-induced inhibition of HSP-70 mRNA expression was lessened by 45% after knocking down miR-1 expression (Fig. 5D).
Roles of microRNA-1 (miR-1) in sodium nitroprusside (SNP)-induced inhibition of heat shock protein (HSP)-70 mRNA and protein expressions. A bioinformatic approach was conducted to predict the existence of miR-1-specific binding elements in the 3'-UTR of HSP-70 mRNA (A). MC3T3-E1 cells were treated with SNP, hsa-miR-1 (an miR-1 inhibitor), and their combination for 12 h. The Anti-miR™ miRNA Inhibitor Negative Control #1 (Control) was transfected into MC3T3-E1 cells as a negative control. Amounts of HSP-70 were immunodetected (B, top panel). β-Actin was analyzed as the internal control (bottom panel). These immunorelated protein bands were quantified and statistically analyzed (C). Levels of HSP-70 mRNA were quantified using a quantitative PCR (D). Each value represents the mean ± SEM, n = 6. * and # Values significantly (p < 0.05) differ from the respective control and SNP- treated groups, respectively.
miR-1 mediated NO-induced insults to osteoblasts via an apoptotic mechanism
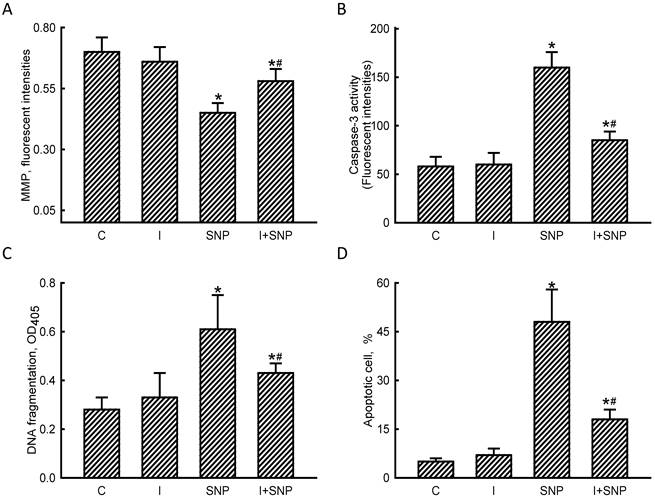
The MMP, caspase-3 activity, DNA fragmentation, and apoptotic cells were assayed to evaluate the mechanisms of miR-1-involved osteoblast insults (Fig. 6). Exposure of MC3T3-E1 cells to SNP decreased the MMP by 36% (Fig. 6A). Application of miR-1 antisense inhibitors did not affect the MMP but caused a significant 53% alleviation in the SNP-induced reduction in the MMP. In comparison, SNP increased caspase-3 activity by 2.8-fold, but pretreatment with miR-1 antisense inhibitors lowered such augmentation by 47% (Fig. 6B). The SNP-induced DNA fragmentation of MC3T3-E1 cells was suppressed by 35% following treatment with miR-1 inhibitors (Fig. 6C). Consequently, reducing miR-1 expression simultaneously diminished 58% of SNP-induced osteoblast apoptosis (Fig. 6D).
Roles of microRNA-1 (miR-1) in sodium nitroprusside (SNP)-induced osteoblast apoptosis. MC3T3-E1 cells were treated with sodium nitroprusside (SNP), hsa-miR-1 (a miR-1 inhibitor), and their combination for 12 h. The Anti-miR™ miRNA Inhibitor Negative Control #1 (Control) was transfected into MC3T3-E1 cells as a negative control. The membrane potential of mitochondria was quantified using flow cytometry (A). Caspase-3 activity was assayed by a fluorogenic substrate assay (B). DNA fragmentation (C) and apoptotic cells (D) were analyzed using an ELISA method and flow cytometry, respectively. Each value represents the mean ± SEM, n = 6. * and # Values significantly (p < 0.05) differ from the control and SNP-treated groups, respectively.
Discussion
This study showed that miR-1 participates in NO-induced insults to osteoblasts. After exposure to SNP, the levels of cellular NO in MC3T3-E1 cells time-dependently increased. SNP can be decomposed into NO in the presence of a biological system, reducing agents, and visible light (31). In parallel, NO released by SNP elevated oxidative stress and decreased cell survival. Interestingly, treatment of MC3T3-E1 cells with SNP induced miR-1 expression in a time-dependent manner. Li et al. (2014) used Northern blot analyses to identify the specific expression of miR-1 in growth plate cartilage and muscle tissue (32). The present study further demonstrated that miR-1 can be detected in osteoblasts. We also showed induction of miR-1 in MC3T3-E1 cells following SNP treatment. In comparison, treatment of MC3T3-E1 cells with miR-1 antisense inhibitors caused significant inhibition of SNP-induced expression of this small non-coding RNA. At the same time, knocking down miR-1 expression alleviated SNP-induced insults to osteoblasts. Osteoblasts contribute to bone formation (1). However, plenty of endogenous and exogenous factors can damage osteoblasts (4). During bone inflammation, NO is usually overproduced (4, 5). This study provides in vitro evidence to show the roles of miR-1 in NO-induced osteoblast injury. Therefore, miR-1 may be clinically applied as an effective biomarker for the diagnosis and prognosis of inflammation-related bone disorders such as bone trauma, bone fracture, and osteoporosis.
miR-1 takes part in NO-induced damage to osteoblasts through an apoptotic mechanism. Exposure of MC3T3-E1 cells to SNP induced shrunken morphologies, caspase-3 activation, DNA fragmentation, and cell cycle arrest at the sub-G1 phase. Morphological shrinkage, caspase-3 activation, DNA fragmentation, and cell cycle arrest are typical features of cells undergoing apoptosis (3, 7). There are two typical apoptotic mechanisms: intrinsic mitochondrion-dependent and extrinsic mitochondria-independent pathways (23). Our previous studies showed that overproduction of NO in osteoblasts can stimulate translocation of Bax, a proapoptotic protein, from the cytoplasm to the outer membranes of mitochondria (6, 7, 9). In this study, we demonstrated that after exposure to SNP, the MMP of MC3T3-E1 cells decreased. Alterations in the membrane potential imply the involvement of mitochondria in NO-induced apoptotic insults to osteoblasts (33). Thus, SNP may induce apoptosis of MC3T3-E1 cells through activation of a mitochondrion-dependent pathway. In comparison, knocking down miR-1 expression using its specific antisenses inhibited SNP-induced miR-1 synthesis and simultaneously attenuated NO donor-triggered changes in the MMP, caspase-3 activation, DNA fragmentation, and cell cycle arrest. Therefore, miR-1 transduces NO-induced apoptotic insults to MC3T3-E1 cells via an intrinsic mitochondrion-dependent mechanism. A previous study reported that under hypoxic conditions, miR-1 was induced and transduced a mitochondrion-dependent apoptotic signals in neuronal cells (18). This study further showed the molecular mechanisms of miR-1-involved regulation of NO-induced osteoblast apoptosis.
HSP-70 contributes to NO-induced osteoblast apoptosis. Treatment of MC3T3-E1 cells with SNP decreased levels of cellular HSP-70 in a time-dependent manner. In parallel, expression of HSP-70 mRNA by MC3T3-E1 cells was inhibited by SNP. Thus, the SNP-induced suppression of HSP-70 expression occurs at least at a pretranslational level. Under exposure to various types of stress, HSP-70 can be rapidly and massively induced (24). Functionally, HSP-70 was implicated as a survival gene because this early expressed protein is able to build up cytoprotection against temperature alterations, mechanical damage, hypoxia, lowered pH, and ROS generation (25). Our previous study showed that when HSP-70 expression was knocked down, hypoxic stress could provoke more apoptosis of neuronal cells (18). In osteoblasts, HSP-70 expression was inhibited by microgravity (26). Consequently, such a change in HSP-70 production would lead to osteoblast damage. The present study showed that administration of SNP to MC3T3-E1 cells inhibited HSP-70 mRNA and protein production and concurrently induced cell apoptosis. Therefore, HSP-70 is involved in NO-induced apoptosis of osteoblasts.
miR-1 can target HSP-70. The present data reveal that treatment of MC3T3-E1 cells with miR-1 antisense inhibitors caused significant attenuation of SNP-induced changes in miR-1 expression and HSP-70 mRNA and protein syntheses. Our results of a bioinformatic search further predicted the existence of miR-1-specific binding elements in the 3'-UTR of HSP-70 mRNA. In rat ventricular H9c2 cells, miR-1 was shown to be overproduced in response to oxidative stress and afterward triggered cell apoptosis by targeting HSP-60 and HSP-70 (14). In our lab, we also showed that miR-1 can target HSP-70 and mediate hypoxia-induced apoptotic insults to mouse neuro-2a cells (18). Preconditioning of the heart was developed as a powerful experimental tool to discover novel signaling mechanisms in cardioprotection. Salloum et al. (2010) showed that miR-1 is one of multiple miRNAs the gene expressions of which can be regulated in cardiac preconditioning (34). Additionally, miR-1 was reported to drive the synthesis of the cardioprotective protein, HSP-70. Knockdown of HSP-70 may induce loss of synaptic mitochondria and subsequent cell death in a Drosophila Parkinson's disease model (35). As a result, miR-1 can target HSP-70 and then transduces NO-induced apoptotic insults to osteoblasts.
In summary, this study showed that exposure of MC3T3-E1 cells to SNP time-dependently elevated cellular NO and ROS levels. In parallel, SNP decreased cell survival and induced DNA fragmentation and cell apoptosis. Expression of miR-1 by MC3T3-E1 cells was induced by SNP. Knocking down miR-1 expression using its specific antisense inhibitors simultaneously attenuated SNP-induced osteoblast death. Treatment of MC3T3-E1 cells with SNP augmented HSP-70 mRNA and protein expressions. A bioinformatic search revealed the existence of miR-1-specific binding elements in the 3'-UTR of HSP-70. Reducing miR-1 expression could lead to alleviation of SNP-induced inhibition of HSP-70 mRNA production. Subsequently, SNP-induced alterations in the MMP, caspase-3 activation, DNA fragmentation, and cell apoptosis were significantly lowered by miR-1 antisense inhibitors. Therefore, miR-1 is involved in NO-induced apoptosis of osteoblasts via an intrinsic mitochondrion-dependent mechanism that targets HSP-70 expression.
Abbreviations
ANOVA, analysis of variance; Bcl-2, B-cell lymphoma-2; BrdU, 5-bromo-2'-deoxyuridine; cDNA, complementary DNA; DEVD, Asp-Glu-Val-Asp; DMEM, Dulbecco's modified Eagle's medium; ELISA, enzyme-linked immunosorbent assay; HSP, heat-shock protein; miR, microRNA; MMP, mitochondrial membrane potential; mRNA, messenger RNA; NO, nitric oxide; PAGE, polyacrylamide gel electrophoresis; PBS, phosphate-buffered saline; PI, propidium iodide; qPCR, quantitative polymerase chain reaction; ROS, reactive oxygen species; SNP, sodium nitroprusside; SDS, sodium dodecylsulfate.
Acknowledgements
This study was supported by Shin Kong Wu Ho-Su Memorial Hospital (SKH-TMU-101-04), Wan-Fang Hospital (101-wf-eva-08), and the National Science Council (NSC95-2314-B-038-029-MY3 and NSC101-2314-B-038-003-MY3), Taipei, Taiwan.
Competing interests
The authors declare that they have no competing interests.
References
1. Takeda S, Karsenty G. Central control of bone formation. Journal of Bone and Mineral Metabolism. 2001;19:195-8
2. Partridge NC, Alcorn D, Michelangeli VP. et al. Functional properties of hormonally responsive cultured normal and malignant rat osteoblastic cells. Endocrinology. 1981;108:213-9
3. Chen RM, Lin YL, Chou CW. GATA-3 transduces survival signals in osteoblasts through upregulation of bcl-xL gene expression. Journal of Bone and Mineral Research. 2010;25:2193-204
4. Ikeda K, Takeshita S. Factors and mechanisms involved in the coupling from bone resorption to formation: how osteoclasts talk to osteoblasts. Journal of Bone Metabolism. 2014;21:163-7
5. Evans DM, Ralston SH. Nitric oxide and bone. Journal of Bone and Mineral Research. 1996;11:300-5
6. Chen RM, Liu HC, Lin YL. et al. Nitric oxide induces osteoblast apoptosis through the de novo synthesis of Bax protein. Journal of Orthopaedic Research. 2002;20:295-302
7. Chen RM, Chen TL, Chiu WT. et al. Molecular mechanism of nitric oxide-induced osteoblast apoptosis. Journal of Orthopaedic Research. 2005;23:462-8
8. Wei JD, Lin YL, Tsai CH. et al. SATB2 participates in regulation of menadione-induced apoptotic insults to osteoblasts. Journal of Orthopaedic Research. 2012;30:1058-66
9. Chang CC, Liao YS, Lin YL. et al. Nitric oxide protects osteoblasts from oxidative stress- induced apoptotic insults via a mitochondria-dependent mechanism. Journal of Orthopaedic Research. 2006;24:1917-25
10. Tai YT, Cherng YG, Chang CC. et al. Pretreatment with low nitric oxide protects osteoblasts from high nitric oxide-induced apoptotic insults through regulation of c-Jun N-terminal kinase/c-Jun-mediated Bcl-2 gene expression and protein translocation. Journal of Orthopaedic Research. 2007;25:625-35
11. Pallante P, Battista S, Pierantoni GM. et al. Deregulation of microRNA expression in thyroid neoplasias. Nature Review Endocrinology. 2014;10:88-101
12. Negrini M, Nicoloso MS, Calin GA. MicroRNAs and cancer--new paradigms in molecular oncology. Current Opinion in Cell Biology. 2009;21:470-9
13. Chio CC, Lin JW, Cheng HA. et al. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Archives of Toxicology. 2013;87:458-68
14. Xu C, Lu Y, Pan Z. et al. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. Journal of Cell Science. 2007;120:3045-52
15. Townley-Tilson WH, Callis TE, Wang D. MicroRNAs 1, 133, and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. International Journal of Biochemistry and Cell Biology. 2010;42:1252-5
16. Zhang Y, Zhang L, Chu W. et al. Tanshinone IIA inhibits miR-1 expression through p38 MAPK signal pathway in post-infarction rat cardiomyocytes. Cell Physiology and Biochemistry. 2010;26:991-998
17. Varendi K, Kumar A, Härma MA. et al. miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cellular and Molecular Life Sciences. 2014;71:443-56
18. Chang CY, Lui TN, Lin JW. et al. Roles of microRNA-1 in hypoxia-induced apoptotic insults to neural cells. Archives of Toxicology. 2014 DOI: 10.1007/s00204-014-1364-x
19. Namlos HM, Meza-Zepeda LA, Baroy T. Modulation of the osteosarcoma expression phenotype by microRNAs. PLoS One. 2012;7:e48086
20. Novello C, Pazzaglia L, Cingolani C. et al. miRNA expression profile in human osteosarcoma: role of miR-1 and miR-133b in proliferation and cell cycle control. International Journal of Oncology. 2013;42:667-75
21. Hock JM, Krishnan V, Onyia JE. et al. Osteoblast apoptosis and bone turnover. Journal of Bone and Mineral Research. 2001;16:975-84
22. Rojas E, Carlini RG, Clesca P. et al. The pathogenesis of osteodystrophy after renal transplantation as detected by early alterations in bone remodeling. Kidney International. 2003;63:1915-23
23. Goyal L. Cell death inhibition: keeping caspases in check. Cell. 2001;104:805-8
24. Young JC, Agashe VR, Siegers K. et al. Pathways of chaperone-mediated protein folding in the cytosol. Nature Reviews Molecular Cell Biology. 2004;5:781-91
25. Patterson AJ, Zhang L. Hypoxia and fetal heart development. Current Molecular Medicine. 2010;10:653-66
26. Kumei Y, Morita S, Shimokawa H. et al. Inhibition of HSP70 and a collagen-specific molecular chaperone (HSP47) expression in rat osteoblasts by microgravity. Annals of the New York Academy of Sciences. 2003;1010:476-80
27. Chang HC, Chen TG, Tai YT. et al. Resveratrol attenuates oxidized LDL-evoked Lox-1 signaling and consequently protects against apoptotic insults to cerebrovascular endothelial cells. Journal of Cerebral and Blood Flow Metabolism. 2011;31:842-54
28. Lin JW, Chen JT, Hong CY. et al. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic Bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro-Oncology. 2012;14:302-14
29. Ho MH, Liao MH, Lin YL. et al. Improving effects of chitosan nanofiber scaffolds on osteoblast proliferation and maturation. International Journal of Nanomedicine. 2014;9:4293-304
30. Wu TT, Tai YT, Cherng YG. et al. GATA-2 transduces LPS-induced il-1β gene expression in macrophages via a toll-like receptor 4/MD88/MAPK-dependent mechanism. PLoS One. 2013;8:e72404
31. Bates JN, Baker MT, Guerra R Jr. et al. Harrison DG: Nitric oxide generation from nitroprusside by vascular tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochemical Pharmacology. 1991;42:s157-65
32. Li P, Wei X, Guan Y. et al. MicroRNA-1 regulates chondrocyte phenotype by repressing histone deacetylase 4 during growth plate development. FASEB Journal. 2014 pii:fj.13-249318
33. Ho WP, Chan WP, Hsieh MS. et al. Runx2-mediated Bcl-2 gene expression contributes to nitric oxide protection against oxidative stress-induced osteoblast apoptosis. Journal of Cellular Biochemistry. 2009;108:1084-93
34. Salloum FN, Yin C, Kukreja RC. Role of microRNAs in cardiac preconditioning. Journal Cardiovascular Pharmacology. 2010;56:581-8
35. Zhu JY, Vereshchagina N, Sreekumar V. et al. Knockdown of Hsc70-5/mortalin induces loss of synaptic mitochondria in a Drosophila Parkinson's disease model. PLoS One. 2013;8:e83714
Author contact
![]() Corresponding author: Ruei-Ming Chen, PhD., Graduate Institute of Medical Sciences College of Medicine, Taipei Medical University, 250 Wu-Hsing St., Taipei 110, Taiwan. Tel: 886-2-27361661 ext. 3222; Fax: 886-2-86621119; E-mail: rmchenedu.tw
Corresponding author: Ruei-Ming Chen, PhD., Graduate Institute of Medical Sciences College of Medicine, Taipei Medical University, 250 Wu-Hsing St., Taipei 110, Taiwan. Tel: 886-2-27361661 ext. 3222; Fax: 886-2-86621119; E-mail: rmchenedu.tw