Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(3):256-265. doi:10.7150/ijbs.10258 This issue Cite
Research Paper
High-density Lipoprotein Increases the Uptake of Oxidized Low Density Lipoprotein via PPARγ/CD36 Pathway in Inflammatory Adipocytes
Qiaoqing Zhong1, 2, 3, Shuiping Zhao3, Bilian Yu3, Xing Wang4, Robina Matyal5, Yunping Li5, Zhisheng Jiang1 ![]()
1. Post-doctoral Mobile Stations for Basic Medicine, Institute of Cardiovascular Disease and Key Lab for Arteriosclerology of Hunan Province, University of South China, Hengyang, Hunan, 421001, China
2. Department of Cardiology, First People's Hospital of Chenzhou, Chenzhou, 423000, China
3. Department of Cardiology, The second Xiangya Hospital of Central South University, Changsha, 410011, China
4. Department of Cardiology, the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, 510080, China
5. Department of Anesthesia and Critical Care, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USA.
Received 2014-8-4; Accepted 2014-12-18; Published 2015-1-15
Abstract

Aim: Previous studies have demonstrated that the dysregulated-secretion of adipokines by adipocytes may contribute to obesity-associated atherosclerosis (As) and high density lipoprotein (HDL) may protect against atherogenesis through multiple pathways. This study was to explore the effect of HDL on the oxLDL uptake in inflammatory adipocytes stimulated by endotoxin lipopolysaccharide (LPS) and the possible mechanism.
Methods and Results: 3T3-L1 adipocytes were cultured and induced to differentiation and maturation. Acute inflammation in adipocytes was induced by LPS (100 ng/ml) for 6 hours. The adipocytes were pretreated with HDL in various concentrations (10, 50, 100 μg/ml) for 16 hours or with specific PPARγ antagonist (GW9662, 10 μM) or agonist (Rosiglitazone, 10 μM) for 30 min before administration of LPS. The results showed that LPS significantly increased the release of inflammation-related adipokines, such as monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor 1 (PAI-1), tumor necrosis factor-alpha (TNF-α), interleukin (IL)-8 and IL-6, while decreasing the release of leptin and adiponectin. Meanwhile, LPS reduced the uptake and degradation of 125I-oxLDL, and down-regulated the expression of PPARγ and CD36. Pretreatment with HDL dose-dependently affected the release of IL-8 and IL-6 and the reduced uptake and degradation of oxLDL of adipocytes stimulated by LPS, accompanied with marked upregulation of PPARγ and CD36 expression. Pretreatment with GW9662 markedly inhibited the upregulation of CD36 expression mediated by HDL (100 μg/ml), while the effects of Rosiglitazone were opposite to GW9662.
Conclusions: HDL may increase oxLDL uptake of inflammatory adipocytes stimulated by LPS via upregulation of PPARγ/CD36 pathway, which may be a new mechanism of anti-atherosclerosis mediated by HDL.
Keywords: Lipoprotein, Atherosclerosis, Inflammation, Lipopolysaccharide
Introduction
There is growing evidence that adipose tissue is an important endocrine organ that secretes various adipokines. Chronic inflammation associated with obesity has been characterized by increased release of cytokines and chemokines in adipose tissue [1-3] and dysregulated secretion of adipokines by adipocytes may be involved in obese-associated pathologies including type 2 diabetes and atherosclerosis (As) [4]. However, less is known about the secretory function of inflammatory adipocytes.
Oxidized low density lipoprotein (oxLDL) is a key element in the development of AS. Peroxisome proliferator-activated receptor γ (PPARγ) and CD36, a plausible pathway for oxLDL uptake in monocytes, is also highly expressed in adipocytes [5, 6]. PPARγ, an important nuclear transcription factor, is involved in regulating pivotal processes including cell differentiation, lipid metabolism and glucose homeostasis in adipocytes [7-9]. CD36, a member of class B scavenger receptor, mediates the endocytosis of oxLDL in phagocytes and is a high-affinity receptor for oxLDL [10, 11].
Adipose tissue has high cholesterol content and acts to buffer the amount of circulating cholesterol. Scavenger receptor class B, such as scavenger receptor class B type I (SR-BI) and CD36 might play significant roles in adipocytes cholesterol and oxLDL metabolism [12, 13]. According to our previous study, HDL-induced cholesterol efflux was associated with SR-BI expression among different groups of adipocytes, and atorvastatin might promote cholesterol efflux from adipocytes to circulation for catabolism and prevent adipocyte cholesterol from over-accumulation, which is associated with hypercholesterolemia through stimulation of SR-BI expression [12]. Fibrates, and other lipid-lowering drugs, enhanced CD36 mediated endocytic uptake and degradation of oxLDL in adipocytes from hypercholesterolemia rabbits. The mechanism of fenofibrate action may be through regulating PPARγ and CD36 expression [13]. Additionally, PPARγ is thought to directly induce CD36 expression and mediate oxLDL uptake in monocytes to promote foam cell formation [14, 15]. These results suggested that PPARγ and CD36 are related to the oxLDL metabolism, but the detailed molecular mechanism is not fully elucidated. Especially, little is known concerning the ability of adipocytes in the uptake and degradation of oxLDL under inflammatory conditions.
It is well-known that high density lipoprotein (HDL) has significant anti-atherogenic properties. However, the mechanism by which HDL prevents atherogenesis has not been completely elucidated. Recent studies have demonstrated that HDL may diminish lipid accumulation in adipocytes through phosphorylation of PPARγ [16], which possibly could account for the favourable effects of HDL on adipokines secretion. Therefore, we hypothesized that regulation of adipokines secretion and lipid accumulation in adipocytes by HDL contributes to its anti-atherogenic properties. Our data suggested that HDL might increase the uptake of oxLDL in inflammatory adipocytes stimulated by LPS via upregulation of PPARγ/CD36 pathway, which may be a new mechanism of anti-atherosclerosis mediated by HDL.
Materials and Methods
Chemicals and Biological Reagents
Human pre-adipocytes cells (3T3-L1, ATCC) were obtained from the Shanghai Cytobiology Institute of the Chinese Academy of Sciences. Dulbecco's modified Eagle's medium (DMEM), benzylpenicillin and streptomycin were obtained from Gibco-BRL. Fetal bovine serum (FBS) was obtained from Sijiqing Biological Engineering Materials. Lyophilized HDL powder from human plasma was obtained from Sigma, the contents of HDL powder were analyzed and were the same as HDL extracted from human plasma. GW 9662 was purchased from ALEXIS Biochemicals and Rosiglitazone was obtained from GSK Company. Elisa kits for measurement of IL-8 and IL-6 were obtained from the Shanghai Fanbang Chemical Engineering Technology Limited Corporation. TRIzol reagent was purchased from Invitrogen. A first strand cDNA synthesis kit was obtained from Promega. The primers of PPARγ and GAPDH were synthesized by the Beijing Aoke Biological Technology Limited Corporation (Beijing, China).
Cell Culture and Treatment
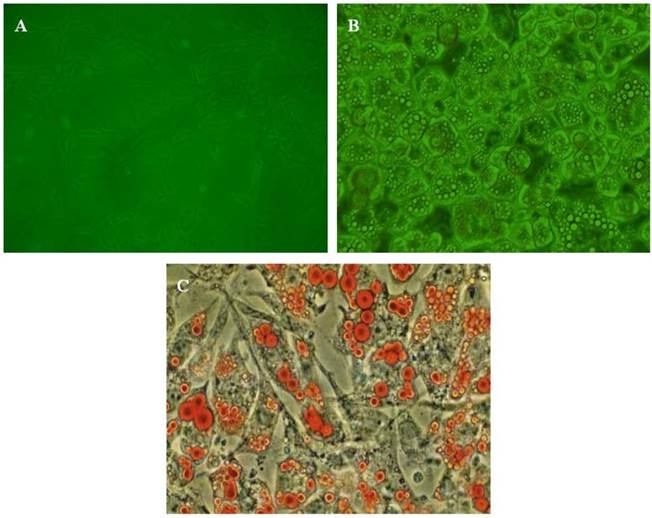
3T3-L1 pre-adipocytes cells were cultured in DMEM according to conventional methods. When the cells reached sub-confluence, cells were put into 6-well or 24-well culture dishes at an optimal density of 2×105 cells/ml. Differentiation was induced by the addition of medium supplemented with 3-isobutyl-1-methylxanthine (0.5 mM), dexamethasone (1 µM) and insulin (10 μg/ml) for 48 hours and then replaced by DMEM containing 10% FBS and insulin (10 μg/ml). The medium was changed every 2 days. Seven days later, differentiated, mature adipose cells were obtained and identified by morphology and Oil red O staining. Matured adipocytes had a round shape and the cytoplasm was completely filled with multiple lipid droplets (Fig. 1). Before our experiment, the conditioned medium was replaced by the medium containing 0.2% BSA for 24 hours. Acute inflammation of adipocytes was induced by treatment with LPS (100 ng/ml) for 6 hours. In order to determine the effect of HDL on the inflammatory adipocytes, cells were treated with HDL in various concentrations (10, 50, 100 μg/ml) for 16 hours, then 100 ng/ml LPS were added for co-incubation for another 6 hours. In order to clarify whether a PPARγ-dependent signal pathway is involved in the effect mediated by HDL, the specific PPARγ antagonist GW 9662 (10 μM) and agonist Rosiglitazone (10 μM) were added 30 min before administration of HDL and LPS.
The identification of adipocytes. A, The morphology of pre-adipocytes; B, The morphology of mature adipocytes; C: Oil red staining of mature adipocytes.
Determination of Inflammation-related Adipokines Release
The levels of inflammation-related adipokines, such as IL-6, IL-8, leptin, adiponectin, MCP-1, PAI-1 or TNF alpha in the conditioned medium were measured using ELISA kits according to the manufacturer's instructions.
Lipoprotein Labeling and Modification
LDL was radio-labeled with 125I according to the previous study [17] before oxidative modification. Then removed free 125I and EDTA by extensive dialysis against PBS and 125I-LDLwas oxidized by incubating with CuSO4 (10 μmol/L) at 37°C for 20 hours. The reaction was terminated by 1.01 mmol EDTA. The degree of oxidation was determined by measuring thiobarbituric reactive substances.
Binding, Cell Association and Degradation of 125I-oxLDL in Adipocytes
Binding, cell association and degradation of 125I-oxLDL was performed as previously described [13, 18, 19]. To determine the specific uptake of 125I-oxLDL in adipocytes, nonspecific uptake was subtracted from the total uptake. Total uptake was determined after incubating with increasing concentrations of 125I-oxLDL (5-75 μg/ml) at 4°C for 2 hours. Nonspecific uptake was measured after incubating adipocytes in the presence of 20-fold excess amounts of unlabeled oxLDL. The cells were washed three times with ice-cold phosphate buffer solution (pH 7.4) containing 0.2% BSA, followed by three washes without albumin. Subsequently, the cells were solubilized in 0.1 mol/L NaOH. Solubilized cells were counted for radioactivity and cellular proteins were measured by the modified Lowry's method. Dissociation constant (KD) and maximal binding (Bmax) were determined from binding studies according to a single-site displacement model. For cell association and degradation assays, cells were incubated with 5-75 μg/mL 125I-oxLDL in the presence or absence of 20-fold excess amounts of unlabeled oxLDL at 37°C for 5 hours. Then the medium was taken and soluble radioactivity in trichloroacetic acid was determined as an index of cellular degradation, and the cell-associated radioactivity and cellular proteins were determined.
Real-time Quantitative PCR (qRT-PCR) Analysis
Total RNA was extracted from cells of 6-well with TRIzol. First strand cDNA was then synthesized from 1 μg total RNA using reverse transcriptase. qRT-PCR, using SYBR Green detection chemistry, was performed on a Roche Light Cycler Run 5.32 Real-time PCR System. The sequences of the qRT-PCR primers are as follows: 5'-TGGAGCCTAAGTTTGAGTTTGC-3' (PPARγ, forward primer), 5'-TGACGATCTGCCTGAGGTCTG-3' (PPARγ, reverse primer); 5'-GCTTGCAACTGTCAGCACAT-3' (CD36, forward primer), 5'-GCCTTGCTGTAGCCAAGAAC-3' (CD36, reverse primer); 5'-TCACTGCCACCCAGAAGAACT-3' (GAPDH, forward primer), and 5'-TGAAGTCGCAGGAGACAACC-3' (GAPDH, reverse primer). Melt curve analyses of all products were performed and shown to produce a single DNA duplex. Quantitative measurements were determined using the △△Ct method and the expression of GAPDH was used as the internal control.
Western Blotting Analysis
For Western blotting, cells were lysed and Western blot analysis was performed according to standard procedures. Protein was loaded on 8% SDS-polyacrylamide electrophoresis gel, electrophoresed for 2 hours at 100V in buffer, and transferred to polyvinylidene fluoride (PVDF) membranes. The primary antibodies were used against CD36, PPARγ and β-actin. Immunoreactivity was detected by ECL test according to the manufacturer's protocol. Protein content was calculated by densitometry using Labwords analysis software.
Small Interference RNA (siRNA) Transfection
Pre-designed siRNA targeted to murine CD36 and an irrelevant 21-nucleotide control siRNA duplex were purchased from Biology Engineering Corporation in Shanghai, China. This mouse CD36 siRNA sequence: Sense, 5'-UAGCUUGGCCAAUAGGACAAAUUCC-3'; Antisense, 5'-GGAAUUUGUCCUAUUGGCCAAGCUA-3'. siRNA were transfected into the cells at a final concentration of 10-30 nM using Lipofectamine RNAiMAX according to the manufacturer's protocol. After 4 hours incubation, the medium was changed to medium, and real-time RT-PCR was performed.
Statistical Analysis
Data are expressed as means±SD and analyzed by ANOVA followed by Newman-Keuls-Student test for multiple comparisons using SPSS 13.0 statistics software. The statistical significance was considered if p<0.05.
Results
Effect of HDL on the Release of Inflammatory-related Adipokines Induced by LPS in Adipocytes
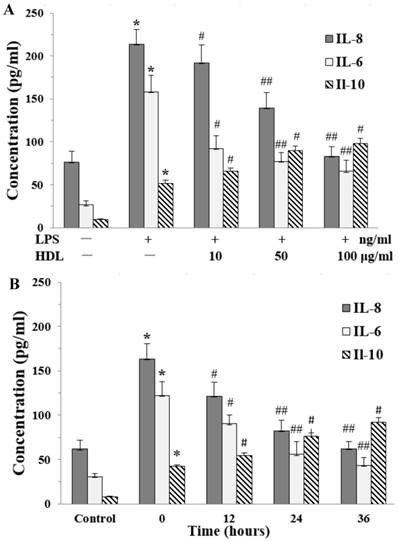
The proinflammatory cytokines interleukin-8 (IL-8) and interleukin-6 (IL-6) play an important role in cell proliferation, survivals, and metabolism. The effects of HDL on release of IL-8 and IL-6 were examined by ELISA. There was a very low level of IL-8 and IL-6 in the base condition. Compared with the control, LPS markedly induced the release of IL-8 and IL-6 in adipocytes, which was attenuated in dose-dependent and time-dependent manners by pretreatment with HDL. We also checked the anti-inflammatory cytokine IL-10, the results showed that LPS increased the level of IL-10. Meanwhile, compared with the cells treated by LPS alone, the level of IL-10 was increased in cells which treated with both HDL and LPS. The effect of HDL was also measured in dose-dependent and time-dependent manners. (Fig. 2A and 2B)
Effects of HDL on the concentration of IL-8 and IL-6 in the medium of inflammatory adipocytes stimulated by LPS. The matured adipocytes were pretreated with HDL (10, 50, 100 μg/ml) for 16 hours or with 100 μg/ml HDL for 12, 24, 36 hours, respectively. Then, cells were stimulated by LPS (100 ng/ml) for 6 hours. Cells were treated with BSA as vehicle control. The concentration of IL-8, IL-6 and IL-10 were measured by ELISA. A, the cells were treated with HDL (10, 50, 100 μg/ml) for 16 hours and LPS for 6 hours, respectively. B, the cells were treated with 100 μg/ml HDL for 12, 24, 36 hours and LPS for 6 hours, respectively. Similar results were obtained in three independent experiments. Data are expressed as means ± S.D. *P<0.01 vs. control; #P<0.05 vs. only treated with LPS or 0 hours; ##P<0.01 vs. only treated with LPS or 0 hours.
Additionally, we measured the inflammatory-related adipokines release in adipocytes, such as leptin, adiponectin, monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor 1 (PAI-1) and tumor necrosis factor-alpha (TNF-α) by ELISA. The leptin and adiponectin levels in medium decreased after LPS treatment, compared with the non-treated control, while the levels of MCP-1, PAI-1 and TNF-α increased after LPS treatment. Meanwhile, in comparison with cells treated with LPS alone, cells treated with both LPS and HDL showed increased leptin and adiponectin levels while decreased levels of MCP-1, PAI-1 and TNF-α. The effect was dose-dependent as shown in Table 1 by pretreatment with HDL.
Effect of HDL on Inflammatory-related Adipokines Release induced by LPS in the Medium of Adipocytes
| Control | LPS | LPS and 10 μg/ml HDL | LPS and 50 μg/ml HDL | LPS and 100 μg/ml HDL | |
|---|---|---|---|---|---|
| Leptin | 205.7±22.3 | 107.6±11.6* | 120.6±10.7# | 142.5±13.4# | 159.4±13.2# |
| Adiponectin | 443.8±35.7 | 211.6±25.7* | 257.3±24.6# | 303.7±23.9# | 363.2±29.8# |
| MCP-1 | 638.3±78.5 | 933.6±87.4* | 875.1±69.5# | 821.0±77.4# | 766.2±90.7# |
| PAI-1 | 776.7±89.3 | 1132.2±90.1* | 1020.0±98.6# | 890.7±78.5# | 810.5±80.5# |
| TNF-alpha | 180.7±20.6 | 560.6±60.3* | 476.3±45.1# | 395.8±28.2# | 329.7±29.4# |
MCP-1, monocyte chemoattractant protein-1; PAI-1, plasminogen activator inhibitor 1; TNF-alpha, Tumor necrosis factor-alpha. The matured adipocytes were pretreated with HDL (10, 50, 100 μg/ml) for 16 hours. Then, cells were stimulated by LPS (100 ng/ml) for 6 hours. Cells were treated with BSA as vehicle control. The concentration of IL-8 and IL-6 were measured by ELISA. Similar results were obtained in three independent experiments. Data are expressed as means ± S.D. *P<0.05 vs. control; #P<0.05 vs. only treated with LPS.
HDL Attenuated the Inhibition of oxLDL Uptake and Degradation Induced by LPS in Adipocytes
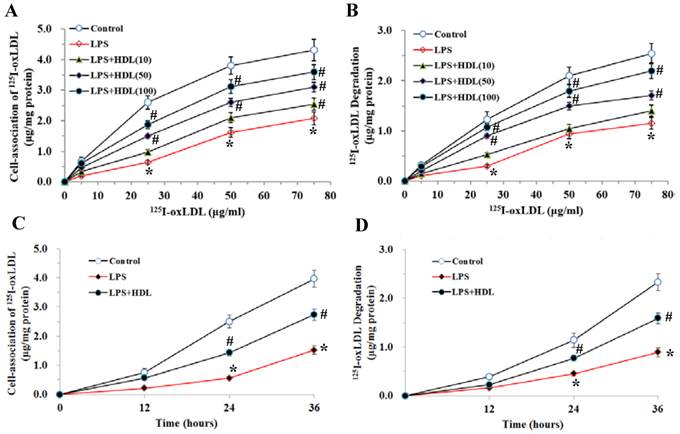
The specific uptake of 125I-oxLDL in adipocytes exhibited a dose-dependent saturation pattern. Stimulation with LPS (100 ng/ml, 6 hours) markedly inhibited the endocytic uptake and degradation of 125I-oxLDL in adipocytes (P<0.01). Pretreatment with HDL (10, 50, 100 μg/ml) significantly attenuated the inhibition of 125I-oxLDL uptake and degradation by LPS in concentration-dependent manners (Fig. 3A and 3B). We also used 100μg/ml HDL to treat the adipocytes for 12, 24 and 36 hours and tested the uptake and degradation of oxLDL. As demonstrated, with the extension of time, HDL gradually attenuated the inhibition of 125I-oxLDL uptake and degradation by LPS (Fig. 3C and 3D).
Effects of HDL on cell-association of 125I-ox-LDL and its degradation in adipocytes treated with LPS. The matured adipocytes were pretreated with HDL (10, 50, 100 μg/ml) for 16 hours or with 100 μg/ml HDL for 12, 24, 36 hours, respectively. Then, cells were stimulated by LPS (100 ng/ml) for 6 hours. Cells were treated with BSA as vehicle control. For cell association and degradation assays, cells were incubated with 5-75 μg/mL 125I-ox-LDL in the presence or absence of 20-fold excess amounts of unlabeled ox-LDL at 37°C for 5 hours. The cell-association radioactivity (A and C) and trichloroacetic acid-soluble radioactivitly in the medium (B and D) were determined as described previously. Similar results were obtained in three independent experiments. Data are expressed as means ± S.D. *P<0.05 vs. control; #P<0.05 vs. treated with LPS-treated groups.
HDL Up-regulated the Expression of PPARγ and CD36 in Adipose cells Treated with LPS
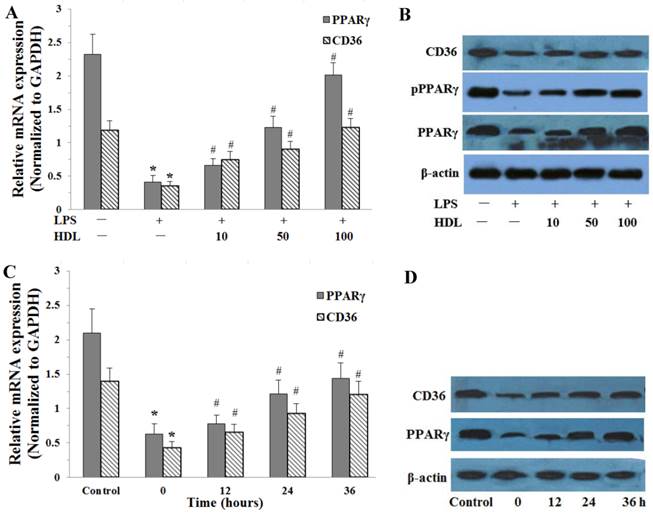
The effects of HDL on the PPARγ and CD36 expressions were examined by real-time quantitative PCR and Western blotting analysis in adipose cells. Compared with the control, LPS (100 ng/ml, 6 hours) significantly down-regulated the expression of PPARγ and CD36 expression in adipocytes (P<0.01). After cells were incubated with different concentrations of HDL (10, 50, 100 μg/ml) or 100 μg/ml HDL treated different times (12, 24, 36 hours), the down-regulation of PPARγ and CD36 expressions induced by LPS were significantly attenuated. The effects of HDL were shown in concentration-dependent and time-dependent manner (Fig.4).
Effects of HDL on the expression of PPARγ, phosphorylation PPARγ (pPPARγ) and CD36 in adipose cells treated with LPS. The matured adipocytes were pretreated with HDL (10, 50, 100 μg/ml) for 16 hours or with 100 μg/ml HDL for 12, 24, 36 hours, respectively. Then, cells were stimulated by LPS (100 ng/ml) for 6 hours. Cells were treated with BSA as vehicle control. A and C, PPARγ, CD36 mRNA expression were measured by real-time PCR. B and D, PPARγ, pPPARγ and CD36 protein expression were measured by Western immunoblotting assays. Similar results were obtained in three independent experiments. Data are expressed as means ± S.D. *P<0.05 vs. control; #P<0.05 vs. only treated with LPS or 0 hours.
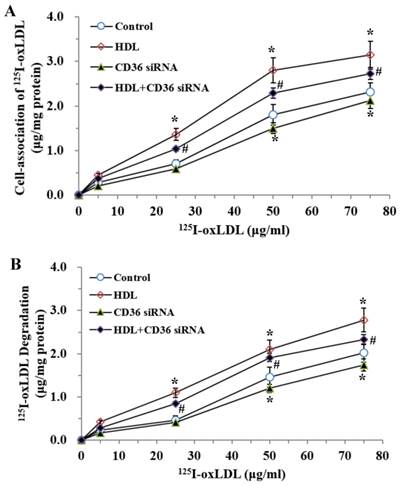
Down-regulation of CD36 Expression Attenuated the Inhibition of oxLDL Uptake and Degradation Induced by LPS in Adipocytes
CD36, a member of class B scavenger receptors, mediates the endocytosis of oxLDL in phagocytes and is a high-affinity receptor for oxLDL. To confirm the effects of CD36 on oxLDL uptake and degradation induced by LPS, we treated the cells with CD36 siRNA and (or) HDL, respectively. As demonstrated (Fig.5), co-treatment with CD36 siRNA and HDL decreased the oxLDL uptake and degradation compared with those treated by HDL only.
Effects of CD36 siRNA on cell-association of 125I-ox-LDL and its degradation in adipocytes treated with LPS. The matured adipocytes were transfected with CD36 siRNA and (or) pretreated with HDL (100 μg/ml) for 16 hours. Then, cells were stimulated by LPS (100 ng/ml) for 6 hours. Cells were treated with LPS as vehicle control. For cell association and degradation assays, cells were incubated with 5-75 μg/mL 125I-ox-LDL in the presence or absence of 20-fold excess amounts of unlabeled ox-LDL at 37°C for 5 hours. The cell-association radioactivity (A) and trichloroacetic acid-soluble radioactivitly in the medium (B) were determined as described previously. Similar results were obtained in three independent experiments. Data are expressed as means ± S.D. *P<0.05 vs. control; #P<0.05 vs. treated with HDL treated groups.
PPARγ was Involved in the Uptake and Degradation of oxLDL Induced by HDL in Adipocytes
To further study whether the effect of HDL is mediated by the PPARγ/CD36 pathway, fully differentiated 3T3-L1 adipocytes were pretreated with 10μmol/L PPARγ antagonist (GW9662) for 30 min before administration of HDL and LPS. The CD36 expressions were examined by real-time quantitative PCR and Western blotting analysis. The results showed that GW9662 significantly attenuated the effect of HDL on the expression of CD36 stimulated by LPS (Fig. 6A and 6B). At the same time, oxLDL uptake and degradation in cells treated with GW9662 and HDL was significantly decreased as compared with those treated by HDL only (Fig. 6C and 6D).
PPARγ is involved in HDL-induced uptake of oxLDL in adipocytes treated with LPS. The matured adipocytes were transfected with PPARγ antagonist (GW9662, 10 μMol/L) and/or PPARγ agonist (Rosiglitazone, 10 μMol/L), and/or pretreated with HDL (100 μg/ml) for 16 hours. Then, cells were stimulated by LPS (100 ng/ml) for 6 hours. Cells were treated with LPS as vehicle control. A and E, CD36 mRNA expression were measured by real-time PCR. B and F, CD36 protein expression was measured by Western immunoblotting assays. For cell association and degradation assays, cells were incubated with 5-75 μg/mL 125I-ox-LDL in the presence or absence of 20-fold excess amounts of unlabeled ox-LDL at 37°C for 5 hours. The cell-association radioactivity (C and G) and trichloroacetic acid-soluble radioactivitly in the medium (D and H) were determined as described previously. Similar results were obtained in three independent experiments. All data are expressed as means ± S.D. *P<0.05 vs. control; #P<0.05 vs. treated with HDL.
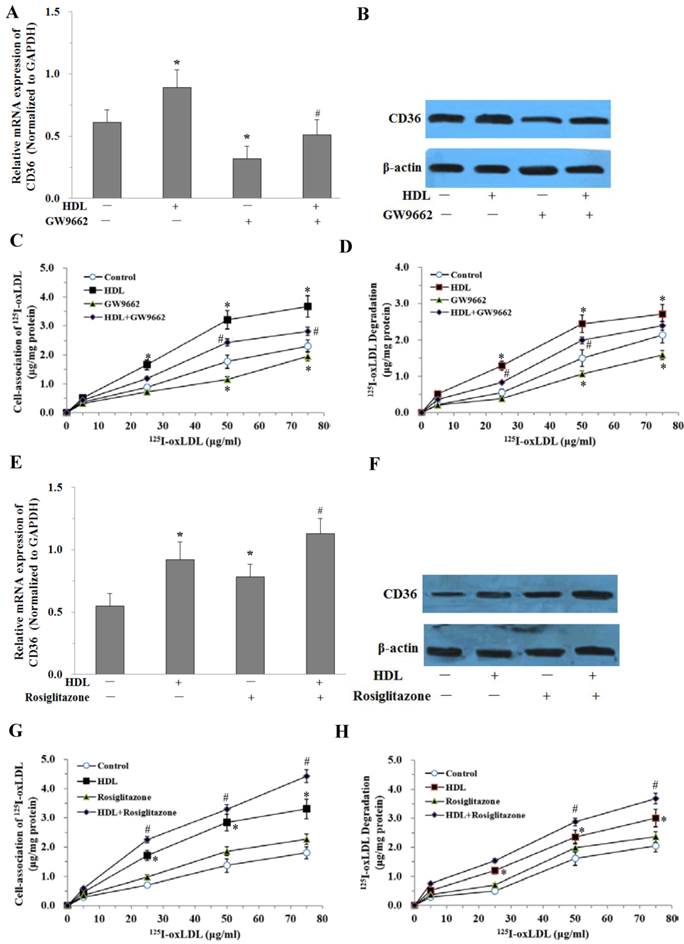
We further investigated the effects of PPARγ agonist (Rosiglitazone) on the uptake and degradation of oxLDL by HDL in adipocytes. Treatment with Rosiglitazone (10μmol/L) and HDL up-regulated CD36 mRNA and protein expressions compared with those treated by HDL only (Fig. 6E and 6F). At the same time, oxLDL uptake and degradation in cells treated with Rosiglitazone and HDL was significantly increased as compared with those treated by HDL only (Fig. 6G and 6H).
Discussion
High-density lipoproteins (HDL) protect against the development of atherosclerotic coronary heart disease. In part, this reflects the ability of HDL to promote the efflux of cholesterol from macrophages in the artery wall but given that atherosclerosis is an inflammatory disorder, it may also reflect anti-inflammatory properties of HDL [20]. The present study demonstrated that HDL treatment decreased the release of inflammation-related adipokines and attenuated the inhibition of oxLDL uptake and degradation stimulated by LPS, accompanied with upregulation of the expression of PPARγ and CD36. Pretreatment with PPARγ antagonist GW9662 significantly inhibited the favorable effect of HDL on the uptake of oxLDL and CD36 expression.
Many studies have demonstrated that adipose tissue is not only an energy-regulating organ, but also an important endocrine organ that can secrete various adipokines and chemokines. Interestingly, recent studies have demonstrated that culture of newly differentiated adipocytes can be efficiently and rapidly converted into macrophages and robustly secrete cytokines/chemokines [21], suggesting that adipocytes may play a potential role in the inflammatory process of obesity-associated diseases. IL-8, one of the super families of chemokines, is secreted by a series of proatherogenic cells including adipocytes and plays an important role in the inflammatory process. IL-6 is an interleukin that acts as both a pro-inflammatory and anti-inflammatory cytokine and its circulating levels were associated with obesity. It was previously reported that the inflammatory mediators LPS and TNF-alpha (TNF-α) had major stimulatory effects on the expression and secretion of IL-6, MCP-1 and TNF-α in canine adipocytes [22]. Other research showed that LPS also induces IL-6 and IL-8 but not TNF-α in human adipocytes [23]. We also used LPS to induce inflammatory reactions of adipocytes and showed that LPS had a similar effect on the release of inflammation-related adipokines such as IL-6 and IL-8.
The beneficial effects of HDL in protecting against the development of As is thought to be mediated predominately through increasing cellular cholesterol efflux, inhibiting oxidative modification of LDL, favourable adipokines secretion, antioxidant and anti-inflammatory profile. However, the studies that focus on the relationship between HDL and adipocytes are inconclusive. Our previous studies showed that HDL induced cholesterol efflux in adipocytes isolated from hypercholesterolemic rabbits [12] and others reported that HDL could inhibit the uptake of oxidized lipids in macrophage and cellular lipid accumulation in adipocytes [24]. The present study further observed the effect of HDL on the adipokines and oxLDL uptake in inflammatory adipocytes stimulated by LPS. The data showed that HDL treatment attenuated the increased release of MCP-1, PAI-1, TNF-α, IL-8 and IL-6, and attenuated the decreased release of leptin and adiponectin in a dose-dependent manner.
An effective inflammatory response requires the balance between appropriate activation of inflammatory mechanisms and effective resolution of the inflammatory state. This balance is orchestrated, at least in part, by the balance of pro- and anti-inflammatory cytokines secreted. IL-10 is a major anti-inflammatory cytokine and LPS can induce IL-10 secretion [25, 26]. Previous studies show that IL-10 is a critical component of the host's natural defense against the development of pathologic responses to LPS although it is not responsible for LPS-induced tolerance [27]. In this study, our results show that HDL decreased IL-6 and IL-8 levels and increased IL-10 level in adipocytes. This is consistent with the idea that HDL can inhibit LPS-induced inflammatory response by decreasing the pro-inflammatory cytokines (IL-6, IL-8) levels but enhancing anti-inflammatory cytokine (IL-10) expression. A previous study also found that cholesterol has similar affection in inflammation modulation in macrophage [28]. Meanwhile, we also found that HDL attenuated the reduced uptake and degradation of oxLDL by LPS from adipocytes. These results suggested that HDL mediated a protective effect on adipocytes in the inflammatory state.
However, the molecular mechanisms responsible for the favorable effect of HDL on inflammatory adipocytes are not completely elucidated. Our present study found that HDL markedly increased the oxLDL uptake of adipocytes, accompanied with upregulation of the mRNA expression of PPARγ and CD36. PPARγ is a central regulator of adipocyte differentiation and lipid accumulation through transcriptional regulation of genes encoding proteins involved in lipid metabolism and the inflammatory reaction. Several PPARγ-responsive genes have been identified such as CD36 are highly expressed in adipocytes [29]. CD36 is a major regulator of long-chain FA transport across the cell membrane and acts as a scavenger receptor for oxLDL in macrophages. It was reported that PPARγ activation promotes oxLDL uptake in monocytes via induction of the scavenger receptor CD36 [15]. Furthermore, CD36 has been previously demonstrated to internalize oxLDL in adipocytes, suggesting that CD36 might participate in the clearance of circulating oxLDL like macrophages and directly mediate oxLDL uptake and degradation in 3T3-L1 adipocytes cells [30]. There was a conflicting report that oxLDL induced CD36 overexpression by up-regulating nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2), independent of PPARγ [31]. Our previous study has shown that GW9662 significantly abolished the niacin-induced increase in PPARγ, pPPARγ expression and cholesterol efflux to ApoA-I. It suggest that the PPARγ pathway may be involved in the cholesterol efflux in adipocytes [32]. In the present study, the selective PPARγ antagonist GW9662 was used to elucidate whether the PPARγ pathway is involved in the favorable effect of HDL on adipocytes. The results have demonstrated that pretreatment with GW9662 markedly inhibited the upregulated expression of CD36 mediated by HDL. Thus, we concluded that HDL may inhibit adipokines release and promote oxLDL uptake and degradation in adipocytes stimulated by LPS via PPARγ-CD36 pathway.
In conclusion, HDL reduced the release of adipokines and increased oxLDL uptake of inflammatory adipocytes stimulated by LPS, which may be explained by activation of the PPARγ/CD36 signaling pathway. These finding are likely to further enrich the pathophysiology of adipocytes and explain the favorable effect of HDL on inflammatory diseases like As.
Acknowledgements
This work was supported by Arteriosclerology Hunan Science and Technology Innovation Team, the Research Fund of Hunan Provincial Department of Public Health (B2010-013), the Research Fund of the Chenzhou Scientific and Technological Administration and the Research Found of First People's Hospital of Chenzhou City (N2012-001).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Samad F, Ruf W. Inflammation, obesity, and thrombosis. Blood. 2013;122:3415-22
2. Lee J. Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Archives of pharmacal research. 2013;36:208-22
3. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. The Journal of clinical investigation. 2003;112:1785-8
4. Bondia-Pons I, Ryan L, Martinez JA. Oxidative stress and inflammation interactions in human obesity. Journal of physiology and biochemistry. 2012;68:701-11
5. Mottillo EP, Bloch AE, Leff T, Granneman JG. Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) alpha and delta in brown adipocytes to match fatty acid oxidation with supply. The Journal of biological chemistry. 2012;287:25038-48
6. Townsend KL, An D, Lynes MD, Huang TL, Zhang H, Goodyear LJ. et al. Increased mitochondrial activity in BMP7-treated brown adipocytes, due to increased CPT1- and CD36-mediated fatty acid uptake. Antioxidants & redox signaling. 2013;19:243-57
7. Kawahara A, Haraguchi N, Tsuchiya H, Ikeda Y, Hama S, Kogure K. Peroxisome proliferator-activated receptor gamma (PPARgamma)-independent specific cytotoxicity against immature adipocytes induced by PPARgamma antagonist T0070907. Biological & pharmaceutical bulletin. 2013;36:1428-34
8. Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors: shifting endocrinology into reverse. Science. 1999;284:757-60
9. Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS. et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Molecular cell. 1999;4:611-7
10. Sundaresan S, Shahid R, Riehl TE, Chandra R, Nassir F, Stenson WF. et al. CD36-dependent signaling mediates fatty acid-induced gut release of secretin and cholecystokinin. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:1191-202
11. Takai M, Tsuzuki S, Matsuno Y, Kozai Y, Eguchi A, Matsumura S. et al. Assessment of key amino-acid residues of CD36 in specific binding interaction with an oxidized low-density lipoprotein. Bioscience, biotechnology, and biochemistry. 2013;77:1134-7
12. Zhao SP, Wu ZH, Hong SC, Ye HJ, Wu J. Effect of atorvastatin on SR-BI expression and HDL-induced cholesterol efflux in adipocytes of hypercholesterolemic rabbits. Clinica chimica acta; international journal of clinical chemistry. 2006;365:119-24
13. Zhao SP, Wu J, Zhang DQ, Ye HJ, Liu L, Li JQ. Fenofibrate enhances CD36 mediated endocytic uptake and degradation of oxidized low density lipoprotein in adipocytes from hypercholesterolemia rabbit. Atherosclerosis. 2004;177:255-62
14. Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. The Journal of biological chemistry. 1993;268:11811-6
15. Han S, Sidell N. Peroxisome-proliferator-activated-receptor gamma (PPARgamma) independent induction of CD36 in THP-1 monocytes by retinoic acid. Immunology. 2002;106:53-9
16. Zhao SP, Yang J, Li J, Dong SZ, Wu ZH. Effect of niacin on LXRalpha and PPARgamma expression and HDL-induced cholesterol efflux in adipocytes of hypercholesterolemic rabbits. International journal of cardiology. 2008;124:172-8
17. Salacinski PR, McLean C, Sykes JE, Clement-Jones VV, Lowry PJ. Iodination of proteins, glycoproteins, and peptides using a solid-phase oxidizing agent, 1,3,4,6-tetrachloro-3 alpha,6 alpha-diphenyl glycoluril (Iodogen). Analytical biochemistry. 1981;117:136-46
18. Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S. et al. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. The Journal of biological chemistry. 2001;276:3195-202
19. Sakai M, Kobori S, Matsumura T, Biwa T, Sato Y, Takemura T. et al. HMG-CoA reductase inhibitors suppress macrophage growth induced by oxidized low density lipoprotein. Atherosclerosis. 1997;133:51-9
20. Barter P. Effects of inflammation on high-density lipoproteins. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:1062-3
21. Charriere G, Cousin B, Arnaud E, Andre M, Bacou F, Penicaud L. et al. Preadipocyte conversion to macrophage. Evidence of plasticity. The Journal of biological chemistry. 2003;278:9850-5
22. Ryan VH, German AJ, Wood IS, Hunter L, Morris P, Trayhurn P. Adipokine expression and secretion by canine adipocytes: stimulation of inflammatory adipokine production by LPS and TNFalpha. Pflugers Archiv: European journal of physiology. 2010;460:603-16
23. Hoch M, Eberle AN, Peterli R, Peters T, Seboek D, Keller U. et al. LPS induces interleukin-6 and interleukin-8 but not tumor necrosis factor-alpha in human adipocytes. Cytokine. 2008;41:29-37
24. Han J, Hajjar DP, Zhou X, Gotto AMJr, Nicholson AC. Regulation of peroxisome proliferator-activated receptor-gamma-mediated gene expression. A new mechanism of action for high density lipoprotein. The Journal of biological chemistry. 2002;277:23582-6
25. Barsig J, Kusters S, Vogt K, Volk HD, Tiegs G, Wendel A. Lipopolysaccharide-induced interleukin-10 in mice: role of endogenous tumor necrosis factor-alpha. European journal of immunology. 1995;25:2888-93
26. Torres MB, Vega VL, Bedri M, Saad D, Trentzsch H, Reeves RH. et al. IL-10 plasma levels are elevated after LPS injection in splenectomized A/J mice. The Journal of surgical research. 2005;129:101-6
27. Berg DJ, Kuhn R, Rajewsky K, Muller W, Menon S, Davidson N. et al. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. The Journal of clinical investigation. 1995;96:2339-47
28. Ma L, Dong F, Zaid M, Kumar A, Zha X. ABCA1 protein enhances Toll-like receptor 4 (TLR4)-stimulated interleukin-10 (IL-10) secretion through protein kinase A (PKA) activation. The Journal of biological chemistry. 2012;287:40502-12
29. Zhao SP, Zhang DQ. Atorvastatin enhances cellular uptake of oxidized LDL in adipocytes from hypercholesterolemic rabbits. Clinica chimica acta; international journal of clinical chemistry. 2004;339:189-94
30. Kuniyasu A, Hayashi S, Nakayama H. Adipocytes recognize and degrade oxidized low density lipoprotein through CD36. Biochemical and biophysical research communications. 2002;295:319-23
31. D'Archivio M, Scazzocchio B, Filesi C, Vari R, Maggiorella MT, Sernicola L. et al. Oxidised LDL up-regulate CD36 expression by the Nrf2 pathway in 3T3-L1 preadipocytes. FEBS letters. 2008;582:2291-8
32. Wu ZH, Zhao SP. Niacin promotes cholesterol efflux through stimulation of the PPARgamma-LXRalpha-ABCA1 pathway in 3T3-L1 adipocytes. Pharmacology. 2009;84:282-7
Author contact
![]() Corresponding author: Dr. Zhisheng Jiang. Post-doctoral Mobile Stations for Basic Medicine, Institute of Cardiovascular Disease and Key Lab for Arteriosclerology of Hunan Province, University of South China, Hengyang, Hunan, 421001, China. E-mail: zsjiang2005com; Tel.: +86-734-8281279
Corresponding author: Dr. Zhisheng Jiang. Post-doctoral Mobile Stations for Basic Medicine, Institute of Cardiovascular Disease and Key Lab for Arteriosclerology of Hunan Province, University of South China, Hengyang, Hunan, 421001, China. E-mail: zsjiang2005com; Tel.: +86-734-8281279