Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(3):284-294. doi:10.7150/ijbs.10248 This issue Cite
Research Paper
5-Fluorouracil Chemotherapy of Gastric Cancer Generates Residual Cells with Properties of Cancer Stem Cells
Zhi-Yuan Xu1*, Jia-Ning Tang1*, Hua-Xia Xie1, Yi-An Du2, Ling Huang2, Peng-Fei Yu2, Xiang-Dong Cheng1 ![]()
1. Department of Gastrointestinal Surgery, The First Affiliated Hospital of Zhejiang Chinese Medicine University, Hangzhou, Zhejiang, China
2. Department of Abdominal cancer Surgery, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, China
* These two authors contributed equally to this work.
Received 2014-8-2; Accepted 2014-12-12; Published 2015-1-21
Abstract

Background: 5-Fluorouracil (5Fu) chemotherapy is the first treatment of choice for advanced gastric cancer (GC), but its effectiveness is limited by drug resistance. Emerging evidence suggests that the existence of cancer stem cells (CSCs) contributes to chemoresistance. The aim of the present study was to determine whether 5Fu chemotherapy generates residual cells with CSC-like properties in GC. Methods: Human GC cell lines, SGC7901 and AGS, were exposed to increasing 5Fu concentrations. The residual cells were assessed for both chemosensitivity and CSC-like properties. B lymphoma Mo-MLV insertion region 1 (BMI1), a putative CSC protein, was analyzed by immunohistochemical staining and subjected to pairwise comparison in GC tissues treated with or without neoadjuvant 5Fu-based chemotherapy. The correlation between BMI1 expression and recurrence-free survival in GC patients who received 5Fu-based neoadjuvant chemotherapy was then examined. Results: The residual cells exhibited 5Fu chemoresistance. These 5Fu-resistant cells displayed some CSC features, such as a high percentage of quiescent cells, increased self-renewal ability and tumorigenicity. The 5Fu-resistant cells were also enriched with cells expressing cluster of differentiation (CD)133+, CD326+ and CD44+CD24-. Moreover, the BMI1 gene was overexpressed in 5Fu-resistant cells, and BMI1 knockdown effectively reversed chemoresistance. The BMI1 protein was highly expressed consistently in the remaining GC tissues after 5Fu-based neoadjuvant chemotherapy, and BMI1 levels were correlated positively with recurrence-free survival in GC patients who received 5Fu-based neoadjuvant chemotherapy. Conclusions: Our data provided molecular evidence illustrating that 5Fu chemotherapy in GC resulted in acquisition of CSC-like properties. Moreover, enhanced BMI1 expression contributed to 5Fu resistance and may serve as a potential therapeutic target to reverse chemoresistance in GC patients.
Keywords: Gastric cancer, 5-Fluorouracil (5Fu), Chemoresistance, Cancer stem cells
Introduction
Gastric cancer (GC) is the second leading cause of death from malignant disease worldwide and is a major global public health problem [1]. As many as 42% of total new cases in the world are diagnosed in China [2], most of which are in the advanced stage by the time of diagnosis. Therefore, traditional chemotherapy remains one of the most common treatment modalities in our country. After more than 30 years of clinical research, 5-fluorouracil (5Fu) monotherapy, or its combination with other conventional therapeutics, has become the standard chemotherapy regimen. Unfortunately, the prognosis of advanced GC is grim, with the median overall survival barely exceeding 1 year. Many patients relapse after an initial response to 5Fu-based chemotherapies, and these recurrent cancers exhibit acquired resistance to chemotherapy, resulting in progression and fatality. The phenomenon of GC acquired chemoresistance has been recognized for decades, but the problem remains unsolved. Hence, the mechanisms by which tumor cells survive chemotherapy treatment are an active area of investigation.
Increasing evidence suggests that intratumoral heterogeneity significantly contributes to the phenomenon of acquired chemoresistance. Indeed, intratumoral heterogeneity is now understood to be a defining feature of cancer [3]; in other words, some tumor cells survive chemotherapy exposure and contribute to disease progression, whereas others perish [4]. A diverse range of molecular mechanisms have been implicated in intratumoral heterogeneity, including genetic mutations and interactions with the microenvironment [5]. Recently, emerging studies have highlighted the important contribution of cancer stem cells (CSCs) to intratumoral heterogeneity [6] and acquired resistance. The CSC model proposes that some malignancies comprise a small subset of CSCs that are highly resistant intrinsically to many therapeutic approaches and exhibit tumor-initiating properties in serial transplantation assays. The CSC model comprises an attractive framework to explain acquired resistance, because chemotherapy-resistant CSCs are particularly well suited to initiate progressive disease [4].
Previous studies have shown an association between chemoresistance and acquisition of a CSC-like phenotype in some types of cancers. For example, residual tumor cells found in breast cancer patients that survived conventional therapy have features similar to CSCs [7, 8]. However, it is currently unknown whether 5Fu chemotherapy generates residual cells with CSC-like characteristics in GC. In this study, we assessed 5Fu chemotherapy-induced residual GC cells for CSC-associated biological characteristics to determine whether these cells comprised a CSC-enriched subtype. Then, we confirmed increased expression of B lymphoma Mo-MLV insertion region 1 (BMI1), a key molecule involved in CSC maintenance, in the remaining GC tissue after 5Fu-based chemotherapy. Finally, we discovered a direct relationship between BMI1 expression and disease-free survival in GC patients who underwent surgery after 5FU-based neoadjuvant therapy.
MATERIALS AND METHODS
Cell lines
Both GC cell lines, SGC7901 and AGS, were purchased from the Shanghai Institute of Cell Biology (Shanghai, China) and stored at our institute. The cell lines were maintained under the recommended culture conditions and incubated at 37°C in a humidified environment containing 5% CO2. Prior to 5Fu exposure, 1 × 106 cells were seeded into 10-cm-diameter dishes and cultured for 24 h. The cultures were then maintained in increasing 5Fu concentrations (AGS: 5, 7.5, 10, 20, and 30 μg/mL; SGC790115: 30, 45, and 60 μg/mL; Nantong Jinghua Pharmaceutical Ltd. Co., Jiangsu, China) for 24 h per passage. This process was continued for approximately 3 months.
Patient tissue samples
This study was approved by the Zhejiang Cancer Hospital ethical committee, and written informed consent was obtained prior to use of the resected samples. This retrospective study included 120 consecutive patients who underwent a D2 radical gastrectomy for GC at Zhejiang Cancer Hospital between January 2009 and January 2011. The patient cohort consisted of 74 males and 46 females, and the median age at the time of surgery was 64 years (range: 32 to 73 years). Among the 120 cases of D2 radical gastrectomy, 23 were from the cardia, 32 from the body, and 65 from the antrum. Neoadjuvant chemotherapy was administered to 60 patients before surgery: Folfox (oxaliplatin + 5Fu) in five patients, Xelox (oxaliplatin + capecitabine) in 27 patients, and SOX (oxaliplatin + S1) in 28 patients.
Antibodies and reagents
Monoclonal and polyclonal antibodies targeting cluster of differentiation (CD)324, CD44, CD24, CD117 and CD133 were obtained from Millipore (Melbourne, Australia). Monoclonal antibodies against BMI1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were obtained from Epitomics (San Francisco, CA, USA). Antibodies against tumor necrosis factor alpha (TNF-a), doxorubicin, and cisplatin (DDP) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell proliferation
Cell proliferation was examined using a Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer's instructions. The number of cells seeded into 96-well plates was 5 × 103.
Chemosensitivity assay
Cells were seeded at a concentration of 1,500/well in a 96-well plate. After 24 h, the medium was replaced by fresh medium with or without various concentrations of 5Fu (6.25, 12.5, 25, 50, 100, 200, 400, 800 and 1600 μM). After further incubation for 72 h, we performed a cell viability assay using the CCK-8 Cell Counting Kit (Dojindo Molecular Technologies). Six wells were counted for each drug concentration, and the experiment was replicated three times. The half maximal inhibitory concentration (IC50) value was defined as the concentration that resulted in a 50% reduction in cell growth compared with growth of the control.
Cell cycle and apoptotic rate analyses
Cell cycle and the apoptotic rate were assessed using flow cytometry. For cell cycle analysis, the cells were fixed with ice-cold 75% ethyl alcohol at 4oC overnight and incubated with propidium iodide (BD Biosciences, San Jose, CA, USA) at 4°C in the dark for 30-60 minutes. For apoptotic rate analysis, cells were incubated with Annexin V - fluorescein isothiocyanate (FITC; BD Biosciences) and propidium iodide for 5 minutes at 4°C in the dark. After staining, the cells were analyzed using a flow cytometer (Cytomics FC500; Beckman Coulter, Miami, FL, USA).
Western blot analysis
Proteins were extracted from cell lines using radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China), separated by 8-12% NUPAGE® Bis-Tris gels (Invitrogen, Carlsbad, CA, USA) and transferred onto polyvinylidene difluoride (PVDF) membranes. The following process was finished as standard processes. The rabbit anti-BMI1 monoclonal antibody was diluted 1:2,000.
Single-cell clonogenic assay
A single-cell suspension was prepared by serially diluting the cells to a concentration of 10 cells/mL. The suspension was then seeded into 96-well plates (100 μL/well), and cultured in fetal bovine serum (FBS)-containing medium [9]. The surviving colonies (> 50 cells) were counted after 2 weeks of culture following crystal violet staining. The colony-forming rate was defined as the ratio of the number of colonies formed in culture to the number of cells incubated. This experiment was performed in triplicate.
Tumorigenicity assays in nude mice
All experimental procedures involving animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals and the institutional ethical guidelines for animal experiments. Female nude mice (4-5 weeks old) were divided randomly into four groups comprised of six mice each: Group 1 was injected with 1 × 106 SGC7901 cells; Group 2 was injected with 1 × 106 SGC7901-FR cells; Group 3 was injected with 5 × 104 SGC7901 cells; and Group 4 was injected with 5 × 104 SGC7901-FR cells. For the injections, tumor cells were suspended in 200 mL phosphate-buffered saline (PBS) and then injected subcutaneously into the anterior flank of the mice. All mice were sacrificed 4-5 weeks after inoculation, and the tumors were harvested and photographed. Tumorigenicity was determined by the tumor incidence (the number of tumors versus the number of injections).
Transfection of BMI1-targeted small interfering RNAs (siRNAs)
The siRNA sequences targeting human BMI1 were as follows: siRNA-1, 5'-CAGATGAAGATAAGAGAAT-3'; siRNA-2, 5'-GAGAAGGAATGGTCCACTT-3'; siRNA-3, 5'-CCAGACCACTACTGAATATAA-3'; A negative control siRNA (NC; 5'-GTGGACTCTTGAAAGTACTAT-3') was also used. These siRNA were designed according to a previous study [10, 11] and produced by Genepharma (Shanghai, China). siRNAs were transfected into cells using Lipofectamine® 2000 (Invitrogen). BMI1 expression was measured by Western blotting to determine the interference effect.
Immunohistochemistry (IHC)
GC tissue samples were fixed in 10% formalin and embedded in paraffin. The slices were soaked in xylene to dewax and hydrated with an ethanol gradient. Then, endogenous peroxidase was deactivated by incubating the slices in 3% H2O2 at room temperature for 5 min, followed by washing with PBS. Next, the antigens were recovered using citrate buffer (0.01 M, pH 6.0), and non-specific antigens were blocked at room temperature for 10 min using 5% normal goat serum. The rabbit anti-human BMI1 monoclonal antibody was added, and the slices were incubated at 4oC overnight and washed with PBS. Then, a biotin-labeled secondary antibody [donkey anti-rabbit immunoglobulin (IgG) polyclonal antibody, biotin conjugated; 1:200 dilution; Abcam, Cambridge, UK] was added, and the slices were incubated at 37oC for 1 h, followed by washing with PBS. Horseradish peroxidase-conjugated streptavidin working solution was added to the slices, followed by washing with PBS. After staining with 3,3'-diaminobenzidine and hematoxylin for 10-15 min, the slices were soaked in an alcohol solution with hydrochloric acid and diluted in ammonia for several minutes. The slices were then dehydrated using an alcohol gradient, cleared with xylene, and mounted in neutral balsam. IHC staining was scored by two pathologists independently. Positive nuclear BMI1 staining was scored and categorized from 0 to 3+ as follows: 0, no staining; 1+, 1-25% of the specimen was stained; 2+, 26-50% of the specimen was stained; and 3+, >50% of the specimen was stained. Scores of 2+ and 3+ were considered to be a positive IHC result.
Statistical analyses
Continuous data were compared between two groups using independent t-tests or paired t-tests, whereas categorical data were analyzed using the chi-square test. Disease-free survival was analyzed using the Kaplan-Meier method. All statistical analyses were performed using SPSS for Windows, v. 16.0 (SPSS, Chicago, IL, USA) and GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA). P < 0.05 was considered to indicate statistical significance.
Results
Residual cells acquired chemoresistance after 5Fu chemotherapy
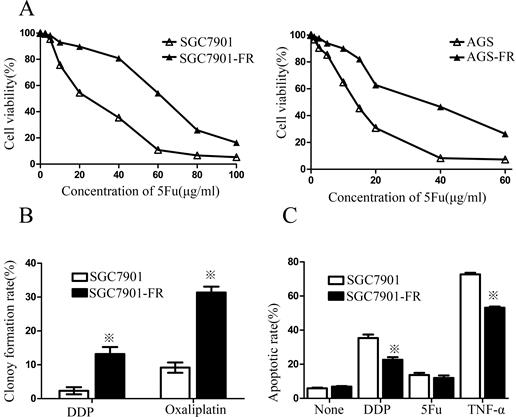
Two cell sublines, SGC7901 5Fu-resistant (SGC7901-FR) and AGS 5Fu-resistant (AGS-FR) cells, were established by repeated subculturing in the presence of stepwise 5Fu concentration increases (AGS-FR: 5, 7.5, 10, 20, and 30 μg/mL; SGC7901-FR: 15, 30, 45, and 60 μg/mL). The IC50 value was considered to be associated with chemoresistance. As shown in Fig. 1A, chemoresistance to 5Fu was observed in both SGC7901-FR and AGS-FR cells. The IC50 values of SGC7901-FR and AGS-FR increased by 2.3- and 2.9-fold, respectively, compared with their parent cell lines. Moreover, we further compared DDP and oxaliplatin sensitivity between SGC7901-FR and SGC7901 cells using a colony formation assay; increased cell survival against both DDP and oxaliplatin was observed in SGC7901-FR cells (Fig. 1B), indicating that these cells acquired multiple drug resistance. In addition, we evaluated the susceptibility of the cells to apoptotic stimuli using flow cytometric analysis. Fig. 1c shows that SGC7901-FR cells exhibited a significantly decreased response to the apoptotic stimulus TNF-α as well as to the cytotoxic agent DDP.
5Fu-induced residual cells acquired chemoresistance (A) 5Fu sensitivity of parental SGC7901-FR and AGS-FR cells or their residual cells, generated by re-exposure to escalating concentrations of 5Fu, was determined by CCK-8 assay. (B) Colony formation rate after DDP or oxaliplatin treatment in SGC7901 and SGC7901-FR cells. (C) Apoptotic rate of SGC7901 and SGC7901-FR cells, treated with DDP (1μg/mL) for 24 h, 5Fu(20ug/ml) for 24h or TNF-a (10 ng/mL) for 12h, was determined by flow cytometry analysis (※P<0.05).
5Fu-resistant cells contained a higher percentage of quiescent cells
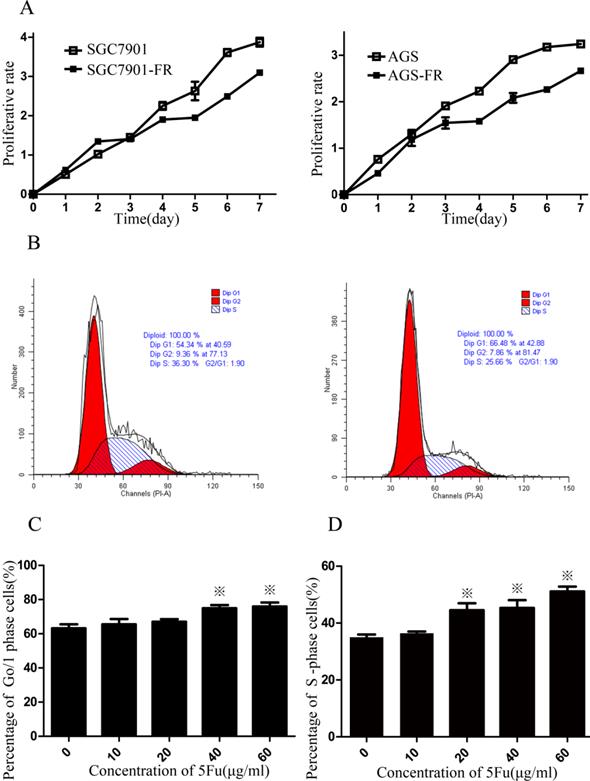
CSC quiescence may account for a possible chemoresistance mechanism. Therefore, we employed the CCK-8 assay to measure cellular viability within 7 days. Both SGC7901-FR and AGS-FR cells showed a significantly slower proliferation rate compared with their parent cells (Fig. 2A). Flow cytometry analysis was then used to assess cell cycle distribution and revealed that 5Fu-resistant cells accumulated in the G0/1 phase (Fig. 2B). Furthermore, we examined SGC7901-FR and SGC7901 cellular responses to 5Fu treatment (10-60 µg/mL concentrations). 5Fu exposure increased the accumulation of SGC7901-FR cells in the G0/1 phase of the cell cycle (63.2% ± 2.3% in control cells vs. 65.6 ± 3.0%, 67.0 ± 1.5%, 74.9% ± 1.9% and 75.9% ± 2.4% in SGC7901-FR cells exposed to 10, 20, 40 and 60 μg/mL 5Fu, respectively; Fig. 2C). In contrast, 5Fu treatment of SGC7901 parental cells induced typical cell accumulation in the S-phase (Fig. 2D).
5Fu-resistance cells contained a high percentage of quiescent cells (A) Proliferative potentials of 5Fu-resistant and their parental cells were assessed using a CCK-8 assay. (B) The cell cycles of SGC7901 (left) and SGC7901-FR (right) cells were compared using flow cytometer. (C) Percentage of cells arrested at the quiescent G0/1 state in SGC7901-FR cells after exposure to 10~60ug/ml 5Fu. (D) Percentage of cells arrested at the S phase in SGC7901 cells after exposure to 10~60ug/ml 5Fu. (※P<0.05)
5Fu-resistant cells were associated with increased self-renewal and tumorigenic ability
Self-renewal is a key characteristic of CSCs. Thus, we examined colony formation of chemoresistant GC cells in culture medium contain containing 1% or 0.1% FBS. In the medium supplemented with 1% FBS, the SGC7901-FR cell colony-forming rate was 12.2% ± 1.2 compared with 3.4% ± 0.72 in SGC7901 cells. However, this difference was not observed in AGS cells, in which both the untreated and 5Fu-resistant cells formed very few colonies. When FBS was reduced to 0.1%, SGC7901-FR cells were still able to form colonies at a rate of 0.56%, whereas control cells did not form colonies (Fig. 3A).
5Fu-resistant cells were associated with increased self-renewal and tumorigenic ability (A) Colony formation rate of SGC7901 and SGC7901-FR cells in 1% FBS medium or 0.1% FBS medium. (B) Comparison of the colony formation rate between SGC7901 and SGC7901-FR cells using single-cell clonogenic assay. (C) Comparison of the tumorigenic ability between SGC7901 and SGC7901-FR cells using tumorigenicity assays on injection of 5×104 cells. (※P<0.05)
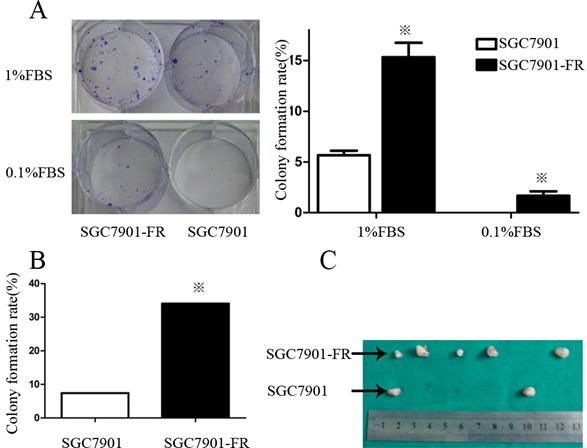
We further compared the self-renewal ability between SGC7901-FR and SGC7901 cells using a single-cell clonogenic assay. Fourteen clones formed from 41 (34.1%) SGC7901-FR single cells after 14 days in culture. However, only four clones formed from 54 (7.4%) single SGC7901 cells. The single-cell clonogenicity capacity of SGC7901-FR cells was significantly higher than that of the SGC7901 cells (Fig. 3B).
We then evaluated the tumorigenic ability of SGC7901-FR and SGC7901 cells via xenotransplant growth in vivo. Tumor incidence was not significantly different between SGC7901-FR and SGC7901 cells following the injection of 1 × 106 cells. However, following injection of 5×104 cells, SGC7901-FR cells formed five tumors out of six injections, whereas SGC7901 cells formed two tumors out of six injections (Fig. 3C). These lines of evidence demonstrated that these chemoresistant GC cells displayed some cancer stem cell features.
5Fu-resistant cells had a diverse stem cell marker expression pattern
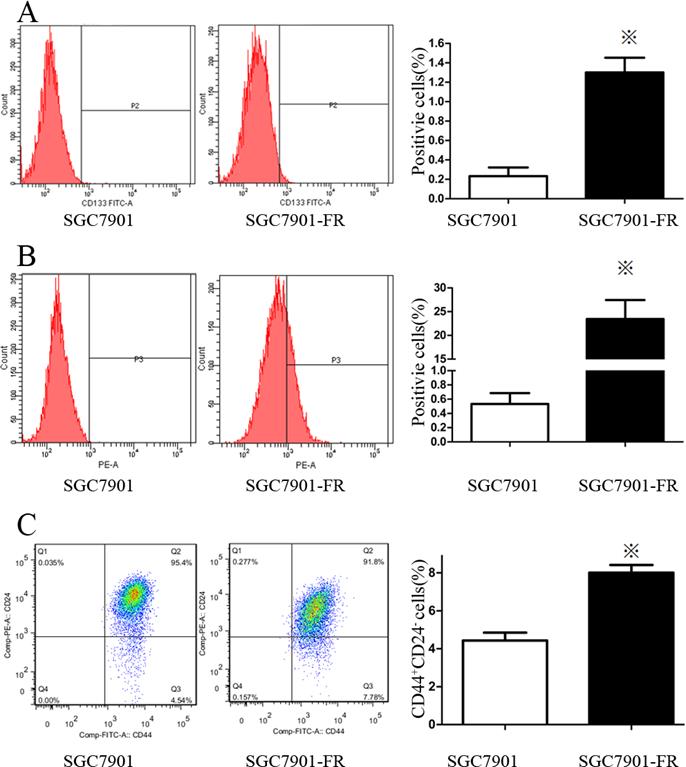
We used flow cytometry to analyze the expression of cell surface stem cell markers, including CD133, CD25, CD24, CD117, CD44 and CD326. SGC7901-FR cells highly expressed a couple of these markers, including CD133 (Fig. 4A) and CD326 (Fig. 4B). No changes were observed in CD25, CD117 or CD44 expression, while CD24 expression was significantly lower in SGC7901-FR cells than in SGC7901 cells. Moreover, the SGC7901-FR cells contained a larger CD44+CD24- subpopulation (Fig. 4C), which appears to be relatively more resistant to chemotherapy [12].
Expression pattern of stem cell markers in SGC7901 and SGC7901-FR cells (A) CD133 expression in SGC7901 and SGC7901-FR cells. (B) CD326 expression in SGC7901 and SGC7901-FR cells. (C) CD44+CD24- subpopulation in SGC7901 and SGC7901-FR cells. (※P<0.05)
BMI1 protein was highly expressed in 5Fu-resistant GC cells
In addition to cell surface markers in 5Fu-resistant cells, we evaluated BMI1 gene expression, which is related to stemness putatively. Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) revealed increased BMI1 expression in both SGC7901-FR and AGS-FR cells compared with their parental cells. Increased BMI1 expression was also observed in other chemoresistant cancer cells, such as SGC7901-OXA cells, which were derived from SGC7901 cells exposed to oxaliplatin, and Bel7402-DR cells, which were derived from Bel7402 hepatocellular cancer cells exposed to DDP (Fig. 5A). These data suggest that enhanced BMI1 expression is common in different types of chemoresistant cells and is not related to specific drug resistance. Western blotting was used to assess BMI1 protein expression and showed elevated BMI1 protein expression in both SGC7901-FR and AGS-FR cells (Fig. 5B).
High expression of BMI1 was associated with chemoresistance (A) Expression levels of BMI1 in untreated or chemoresistant cells were measured by qRT-PCR. (B) Expression of BMI1 and was measured in AGS, AGS-FR, SGC7901 and SGC7901-FR cells by western-blot. (C) Knockdown of BMI1 mRNA expression in SGC7901-FR cells through siRNA transfection was confirmed by qRT-PCR. (D) SGC7901-FR cells transfected with NC or siRNA for 48 h were treated with DDP or 5Fu at indicated concentrations for 24 h, and cell viability was then examined with CCK-8 assay. (※P<0.05)
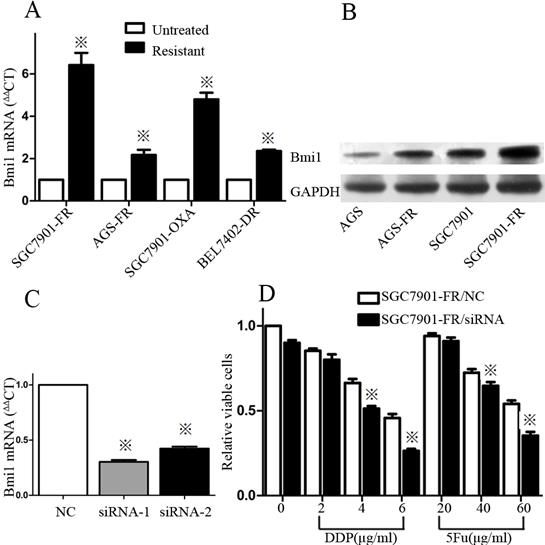
To determine the role of BMI1 in regulating chemosensitivity, we performed siRNA knockdown in SGC7901-FR cells. BMI1 expression was effectively knocked down by siRNA1 and siRNA2 (Fig. 5C). The CCK-8 assay showed that BMI1 knockdown resulted in a significantly increased response of SGC7901-FR cells to the chemotherapeutic drugs DDP and 5Fu (Fig. 5D).
BMI1 protein expression was high in GC tissues after 5Fu-based neoadjuvant chemotherapy and correlated with cancer relapse
Since BMI1 was highly expressed in chemoresistant cells, we expected to see enriched BMI1 expression in residual GC tissues after 5Fu-based neoadjuvant chemotherapy. We evaluated this concept in 60 patient pairs with or without neoadjuvant chemotherapy (Fig. 6). Our results showed that most clinicopathologic features were comparable between two groups (Table 1), however, the BMI1 positive expression (2+ to 3+) rate in GC tissues following neoadjuvant chemotherapy was 58.3% (35 / 60), which was significantly higher than that observed in GC tissues without neoadjuvant chemotherapy (21 / 60, 35%; p=0.01). This indicates that chemotherapy may preselect for GC stem cells.
The clinicopathological feature of GC patients with or without neoadjuvant chemotherapy
| Clinicopatholocical variables | Number of each group | neoadjuvant chemotherapy | X2 | P value | |
|---|---|---|---|---|---|
| no | yes | ||||
| All case | 120 | 60 | 60 | ||
| Age(years) | 1.222 | 0.269 | |||
| ≤50 | 52 | 23 | 29 | ||
| >50 | 68 | 37 | 31 | ||
| Gender | 0.926 | 0.336 | |||
| Male | 79 | 37 | 42 | ||
| female | 41 | 23 | 18 | ||
| HP | 1.477 | 0.224 | |||
| positive | 86 | 40 | 46 | ||
| negative | 34 | 20 | 14 | ||
| Size of tumor, (cm) | 2.155 | 0.142 | |||
| <5(small) | 66 | 37 | 29 | ||
| ≥5(large) | 54 | 23 | 31 | ||
| Location of tumor | 1.095 | 0.578 | |||
| Cardia | 16 | 7 | 9 | ||
| Body | 35 | 20 | 15 | ||
| antrum | 69 | 33 | 36 | ||
| Depth of tumor invasion | 5.735 | 0.125 | |||
| T1 | 9 | 4 | 5 | ||
| T2 | 13 | 9 | 4 | ||
| T3 | 25 | 16 | 9 | ||
| T4 | 73 | 31 | 42 | ||
| Lymph node metastasis | 1.443 | 0.23 | |||
| present | 99 | 52 | 47 | ||
| Absent | 21 | 8 | 13 | ||
| Stage | 0.745 | 0.388 | |||
| I, II | 28 | 16 | 12 | ||
| III, IV | 92 | 44 | 48 | ||
| Grade of differentiation | 1.137 | 0.286 | |||
| well and moderate | 29 | 17 | 12 | ||
| Poor and not | 91 | 43 | 48 | ||
| BMI1 expression | 6.562 | 0.01 | |||
| Positive (2+ ~ 3+) | 56 | 21 | 35 | ||
| Negative (0 ~ 1+) | 64 | 39 | 25 | ||
| Serum CEA value(ug/L) | 0.862 | 0.353 | |||
| <25 | 71 | 38 | 33 | ||
| ≥25 | 49 | 22 | 27 | ||
| Serum CA199 value(ug/L) | 0.154 | 0.695 | |||
| <25 | 82 | 40 | 42 | ||
| ≥25 | 38 | 20 | 18 | ||
Immunohistochemical staining for BMI1 in GC tissues with or without 5Fu-based neoadjuvant chemotherapy (A) No staining in GC tissues without neoadjuvant chemotherapy. (B) 1+ staining in GC tissues without neoadjuvant chemotherapy. (C) 2+ staining in GC tissues received neoadjuvant chemotherapy. (D) 4+ staining in GC tissues received neoadjuvant chemotherapy. Magnification ×400.

To examine the predictive role of BMI1 on survival, we analyzed the BMI1 level in a cohort of 60 GC patients who received surgery after neoadjuvant chemotherapy. Univariate analysis indicated that the 3-year cumulative recurrence-free survival in GC patients with negative BMI1 expression was significantly better than in those with positive BMI1 expression (HR = 2.455, 95% confidence interval, CI: 1.108~5.440), suggesting that BMI1 could be a novel independent prognostic factor for recurrence-free survival after surgery for patients who received neoadjuvant chemotherapy.
Discussion
Over 50 years have passed since 5Fu was developed, but it still plays a key role in chemotherapeutic regimens for GC. The oral 5Fu derivatives capecitabine and S-1 were developed in recent years, and these drugs have been widely used instead of 5Fu due to their greater efficacy and lower toxicity. The antitumor effects of 5Fu can be further enhanced when used in combination with DDP (FP therapy), which has become a standard treatment and has been used as a reference arm in various clinical trials [13-15]. Other 5Fu-containing doublet therapy (i.e., either using different oral fluorouracil prodrug formulations or in combination with a second platinum or non-platinum active drug) or triplet therapy (i.e., in combination with a third chemotherapeutic drug) has also improved the therapeutic outcomes of GC. Despite these impressive accomplishments, virtually all GCs eventually become resistant to 5Fu, with a median time to progression of 0-8 months. Therefore, the mechanisms underlying acquired resistance to 5Fu in GC are an active area of investigation.
To date, several lines of evidence suggest that the existence of CSCs contributes to the acquisition of chemotherapy resistance. Tumors are heterogeneous, comprised of a population of CSCs that are resistant to cytotoxic drugs and responsible for tumor recurrence [6]. Recent clinical studies have begun to monitor the prevalence of CSCs during chemotherapy and provide evidence implicating CSCs in acquired resistance. For example, in a large cohort of acute myeloid leukemia (AML) patient samples, high CSC frequency at diagnosis is correlated with high residual disease frequency after chemotherapy and poor prognosis [16]. In addition, AML minimal residual disease detected during complete remission was enriched in CD34+CD38-cells, the abundance of which after therapy was associated with subsequent relapse [17].
Similarly, CSC enrichment after chemotherapy has been documented in epithelial malignancies. For instance, studies using clinical breast cancer samples support the hypothesis that residual disease after neoadjuvant chemotherapy is enriched in CSCs [18]. Li et al. [12] evaluated breast cancer treated with neoadjuvant chemotherapy and found that cells absent in CSC marker expression were killed by chemotherapy, while cells expressing CSC markers appeared to be resistant. In murine colon cancer cell xenograft models, the treatment of mice with chemotherapeutic agents enriched the tumor xenografts in epithelial surface antigen (ESA)+CD44+ and ESA+CD44+CD166+ CSCs [19]. Moreover, CSCs were analyzed for chemoresistance ex vivo. For example, in human colon cancer, CD133+ CSCs were highly resistant to 5Fu and oxaliplatin [20]. CD44highCD24low breast cancer CSCs also appear to be intrinsically resistant to conventional chemotherapy [12] and ionizing radiation [21].
Consistent with the above observations, when the GC cell lines SGC7901 and AGS were treated with 5Fu in the present study, the emergent resistant subpopulations were enriched in cells with CSC-like characteristics, including increased in vitro self-renewal ability, resistance to multiple chemotherapeutic drugs, enhanced tumorigenic potential, and elevated expression of known cancer stem cell markers, such as CD133 and EpCAM. Collectively, these data suggest that 5Fu-resistant GC cells were enriched in CSCs.
A wealth of experimental data suggest that quiescence is a vital feature of CSCs [22], and quiescence is considered to be a resistance mechanism in response to some chemotherapeutics [23]. In chronic myeloid leukemia (CML), exposure of patient-derived cells to imatinib resulted in the enrichment of a quiescent CD34+ population [24]; similar results were observed in an independent study by Jorgensen et al. [25]. Moreover, quiescent patient-derived CD34+ CML cells were sensitized to imatinib in vitro by cell cycle induction using granulocyte colony stimulating factor [26]. In colon cancer, 5Fu-resistant cells express a typical CSC-like phenotype and enter into a reversible quiescent G0 state on re-exposure to higher 5Fu concentrations. Consistent with these findings, we found that residual GC cells contained a larger fraction of slowly growing cells that accumulated in the G0 phase of the cell cycle after 5Fu chemotherapy. The growth curve also revealed that the 5Fu-resistant cells proliferated at a slower rate than their parental counterparts. In addition, the percentage of quiescent SGC7901-FR cells increased gradually following 5Fu re-exposure. Because 5Fu activity is dependent on cell cycle progression [27], remaining in a quiescent state may be a possible mechanism of 5Fu resistance in GC cells.
Emerging evidence suggests that CSC chemoresistance is due in part to the activation of different molecules, including BMI1, a polycomb group family member [28]. First, many CSC populations have been reported to aberrantly express BMI1. For example, head and neck squamous cell carcinoma CSCs express BMI1 differentially between the CD44+ and CD44- populations. BMI1 is reportedly expressed highly in CD133+ murine liver CSCs and plays a role in the maintenance of hepatic stem / progenitor cells [29]. Secondly, BMI1 is required for the self-renewal and maintenance of CSCs. Previous reports suggested that BMI1 overexpression controls self-renewal and cell cycle by regulating the expression of proteins with cellular anti-proliferative properties, such as p16INK4a and p14ARF [30, 31]. Lentiviral knockdown of BMI1 considerably decreased the number of hepatocellular carcinoma CSCs [32]. In mice, BMI1 is essential for the self-renewal of hematopoietic and neural stem cells [33]. Third, BMI1 is reportedly associated with the protection of tumor cells from chemotherapy. High BMI1 levels in ovarian CSCs increase resistance to cisplatin and paclitaxel [34]. Qin et al. [35] observed that BMI1 silencing enhanced 5Fu antitumor activity in nasopharyngeal carcinoma. Crea et al. [36] also reported that BMI1 silencing enhanced docetaxel activity and impaired antioxidant responses in prostate cancer.
Given its prominent role in chemoresistance, we sought to investigate the role of BMI1 in the GC response to 5Fu in this study. We found that intermittent exposure to 5Fu significantly increased BMI1 levels in GC cells. Simultaneously, 5Fu-based neoadjuvant chemotherapy in advanced GC patients also induced increased the levels of BMI1, thus providing one explanation for the more aggressive and greater metastatic potential of relapsed drug-resistant tumors. Furthermore, we found that elevated BMI1 levels were correlated with reduced disease-free survival in patients with GC who had received adjuvant chemotherapy. Although the role of BMI1 in predicting overall survival was not evaluated in this study due to the insufficient follow-up period, our data support the potential of BMI1 as a marker to predict tumor relapse.
In conclusion, our data demonstrated that 5Fu chemotherapy in GC cell lines preselects distinct drug-resistant clonal populations enriched in cells with CSC-like phenotypes. These 5Fu-resistant cells displayed enhanced BMI1 expression both in vivo and in vitro, and the increased BMI1 level was correlated with reduced disease-free survival. Moreover, BMI1 silencing led to a significantly increased response to chemotherapeutic drugs in GC cells, suggesting that BMI1 should be considered as a potential therapeutic target. Our findings provide insight into the design of novel combination strategies for enhancing the chemosensitivity of GC.
Abbreviations
GC: gastric cancer; CSCs: cancer stem cells; 5Fu:5-fluorouracil; DDP: cisplatin; SGC7901-FR cells: SGC7901 5Fu resistant cells; AGS-FR cells: AGS 5Fu resistant cells
Acknowledgements
This study was supported by the Zhejiang provincial natural science foundation [Grant No. LY12H16029, LY13H160027] and key project of Provincial Administration of Traditional Chinese Medicine [Grant No. 2012ZZ002]. We thank the surgeons and nurses who kindly facilitate the recruitment and collection of patient information.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Shin HR, Bray F. et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-2917
2. Yang L. Incidence and mortality of gastric cancer in China. World J Gastroenterol. 2006;12:17-20
3. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306-313
4. Vidal SJ, Rodriguez-Bravo V, Galsky M. et al. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. 2013
5. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674
6. Reya T, Morrison SJ, Clarke MF. et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-111
7. Kim MR, Choi HK, Cho KB. et al. Involvement of Pin1 induction in epithelial-mesenchymal transition of tamoxifen-resistant breast cancer cells. Cancer Sci. 2009;100:1834-1841
8. Creighton CJ, Li X, Landis M. et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820-13825
9. Wang XQ, Ongkeko WM, Chen L. et al. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. 2010;52:528-539
10. Jagani Z, Wiederschain D, Loo A. et al. The Polycomb group protein Bmi-1 is essential for the growth of multiple myeloma cells. Cancer Res. 2010;70:5528-5538
11. Chiba T, Miyagi S, Saraya A. et al. The polycomb gene product BMI1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res. 2008;68:7742-7749
12. Li X, Lewis MT, Huang J. et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672-679
13. Kang YK, Kang WK, Shin DB. et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol. 2009;20:666-673
14. Ajani JA, Rodriguez W, Bodoky G. et al. Multicenter phase III comparison of cisplatin/S-1 with cisplatin/inFusional fluorouracil in advanced gastric or gastroesophageal adenocarcinoma study: the FLAGS trial. J Clin Oncol. 2010;28:1547-1553
15. Van Cutsem E, Moiseyenko VM, Tjulandin S. et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol. 2006;24:4991-4997
16. van Rhenen A, Feller N, Kelder A. et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11:6520-6527
17. Gerber JM, Smith BD, Ngwang B. et al. A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood. 2012;119:3571-3577
18. Yu F, Yao H, Zhu P. et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109-1123
19. Dylla SJ, Beviglia L, Park IK. et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428
20. Todaro M, Alea MP, Di Stefano AB. et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389-402
21. Diehn M, Cho RW, Lobo NA. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780-783
22. Fuchs E. The tortoise and the hair: slow-cycling cells in the stem cell race. Cell. 2009;137:811-819
23. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834-846
24. Graham SM, Jorgensen HG, Allan E. et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319-325
25. Jorgensen HG, Allan EK, Jordanides NE. et al. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109:4016-4019
26. Jorgensen HG, Copland M, Allan EK. et al. Intermittent exposure of primitive quiescent chronic myeloid leukemia cells to granulocyte-colony stimulating factor in vitro promotes their elimination by imatinib mesylate. Clin Cancer Res. 2006;12:626-633
27. Wells A, Griffith L, Wells JZ. et al. The dormancy dilemma: quiescence versus balanced proliferation. Cancer Res. 2013;73:3811-3816
28. Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012;30:372-378
29. Oishi N, Wang XW. Novel therapeutic strategies for targeting liver cancer stem cells. Int J Biol Sci. 2011;7:517-535
30. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113:175-179
31. Dimri GP, Martinez JL, Jacobs JJ. et al. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002;62:4736-4745
32. Chiba T, Miyagi S, Saraya A. et al. The polycomb gene product BMI1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res. 2008;68:7742-7749
33. Park IK, Qian D, Kiel M. et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302-305
34. Zhang S, Balch C, Chan MW. et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311-4320
35. Qin L, Zhang X, Zhang L. et al. Downregulation of BMI-1 enhances 5-fluorouracil-induced apoptosis in nasopharyngeal carcinoma cells. Biochem Biophys Res Commun. 2008;371:531-535
36. Crea F, Duhagon SM, Hurt EM. et al. BMI1 silencing enhances docetaxel activity and impairs antioxidant response in prostate cancer. Int J Cancer. 2011;128:1946-1954
Author contact
![]() Corresponding author: Name: Xiang-Dong Cheng, Department of Gastrointestinal Surgery, The First Affiliated Hospital of Zhejiang Chinese Medicine University, Youdian Road 54#, Hangzhou, 310006, Zhejiang, China. Tel: +86-571-87070965; Fax: +86-571-87070965; E-mail: abcsurgcom
Corresponding author: Name: Xiang-Dong Cheng, Department of Gastrointestinal Surgery, The First Affiliated Hospital of Zhejiang Chinese Medicine University, Youdian Road 54#, Hangzhou, 310006, Zhejiang, China. Tel: +86-571-87070965; Fax: +86-571-87070965; E-mail: abcsurgcom