Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(4):390-403. doi:10.7150/ijbs.10930 This issue Cite
Research Paper
MicroRNA-23a Participates in Estrogen Deficiency Induced Gap Junction Remodeling of Rats by Targeting GJA1
Ning Wang1,†, ![]() , Lu-Yao Sun1,†, Shou-Chen Zhang3, Ran Wei1, Fang Xie1,2, Jing Liu1, Yan Yan1, Ming-Jing Duan1, Lin-Lin Sun1, Ying-Hui Sun1, Hui-Fang Niu1, Rong Zhang1, Jing Ai1
, Lu-Yao Sun1,†, Shou-Chen Zhang3, Ran Wei1, Fang Xie1,2, Jing Liu1, Yan Yan1, Ming-Jing Duan1, Lin-Lin Sun1, Ying-Hui Sun1, Hui-Fang Niu1, Rong Zhang1, Jing Ai1 ![]()
1. Department of Pharmacology, Harbin Medical University (the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China), Harbin, People's Republic of China, 150081
2. Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education, Harbin, People's Republic of China, 150081
3. Electron Microscopy Center, Harbin Medical University, Harbin, People's Republic of China, 150081
† Authors with equal contributions to the work.
Received 2014-10-28; Accepted 2015-1-21; Published 2015-2-15
Abstract

Increased incidence of arrhythmias in women after menopause has been widely documented, which is considered to be related to estrogen (E2) deficiency induced cardiac electrophysiological abnormalities. However, its molecular mechanism remains incompletely clear. In the present study, we found cardiac conduction blockage in post-menopausal rats. Thereafter, the results showed that cardiac gap junctions were impaired and Connexin43 (Cx43) expression was reduced in the myocardium of post-menopausal rats. The phenomenon was also observed in ovariectomized (OVX) rats, which was attenuated by E2 supplement. Further study displayed that microRNA-23a (miR-23a) level was significantly increased in both post-menopausal and OVX rats, which was reversed by daily E2 treatment after OVX. Importantly, forced overexpression of miR-23a led to gap junction impairment and Cx43 downregulation in cultured cardiomyocytes, which was rescued by suppressing miR-23a by transfection of miR-23a specific inhibitory oligonucleotide (AMO-23a). GJA1 was identified as the target gene of miR-23a by luciferase assay and miRNA-masking antisense ODN (miR-Mask) assay. We also found that E2 supplement could reverse cardiac conduction blockage, Cx43 downregulation, gap junction remodeling and miR-23a upregulation in post-menopausal rats. These findings provide the evidence that miR-23a mediated repression of Cx43 participates in estrogen deficiency induced damages of cardiac gap junction, and highlights a new insight into molecular mechanism of post-menopause related arrhythmia at the microRNA level.
Keywords: Connexin43, Post-menopause, Estrogen, MicroRNA-23a, Arrhythmia
Introduction
The incidence of cardiovascular diseases (CVD) is increasing in women after menopause [1-2]. Comparing with reproductive age women, post-menopausal women show lower heart rate, longer PR intervals and QRS durations [3-4]. Prolongation of PR intervals, which is attributed to slower Atrial-His and His-ventricular conduction velocity, may indicate atrio-ventricular block [5]. Widen of QRS durations which represent intra-ventricular conduction delay is associated with increasing risk of arrhythmic death [6]. Previous studies reported that estrogen could rescue the electrophysiological blockage through up-regulating inward Ca2+ currents [7-8]. However, whether E2 could prevent electrophysiological blockage by protecting gap junctions, another important factor for cardiac electrical propagation, is still unknown.
Gap junctions in the heart are composed by three principal connexins which are connexin43 (Cx43), Cx40, and Cx45 [9]. Remodeling of gap junctions due to reductions of connexins (Cxs) are important signs of arrhythmic tendency [9]. Especially, reduction of Cx43, the predominant type of Cxs in the heart [10], has been implicated in slowing conduction velocity and raising arrhythmia susceptibility [11-12]. Heterozygous Cx43-/+ mice exhibited significantly prolonged QRS intervals [13]. Interestingly, E2 has been shown to protect against Cx43 reduction following myocardial infarction [14-15], suggesting the involvement of Cx43 in E2 deficiency induced alternation of electrophysiological property.
However, the molecular mechanisms of how E2 regulates Cx43 are far from determined. MicroRNAs are a group of essential post-transcriptional regulators [16] and their participations have been implicated in various cardiovascular diseases [17-19]. It has been demonstrated that Cx43 could be post-transcriptionally regulated by microRNAs in myocardium [17], and E2 could exert its effect via influencing microRNA expression profiles of various cell types [20-22]. For example, Zhao, et al. reported that miR-203 contributed to E2 induced inhibition of vascular smooth muscle cell proliferation [23]. These studies implied that microRNAs might participate in E2 deprivation induced abnormal expression of Cx43.
In the present study, we found gap junction disorganizations as well as Cx43 protein reduction in the myocardium of both post-menopausal and ovariectomized (OVX) rats which could be restored by E2 supplement. The further in vitro study showed that upregulation of miR-23a negatively regulated Cx43 expression via targeting GJA1, the gene encoding Cx43, which was prevented by E2.
Materials and Methods
Animals
Female young Wistar rats (6 months) and post-menopausal Wistar rats (18 months) were obtained from Animal Center of the Second Affiliated Hospital of Harbin Medical University was housed at Animal Facility of Harbin Medical University following institutional guidelines. The 17β-estradiol was dissolved in ethanol and subcutaneous injected using oil as the carrier [24]. All protocols were approved by the ethic committees of Harbin Medical University and confirmed by the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Female rats at age of 6M were randomly separated into 3 groups: 1) sham operated (Sham); 2) ovariectomized (OVX); and 3) ovariectomized plus 17β-estradiol daily delivery (2 ug/kg/d) (Sigma chemical, St. Louis, MO, USA) (OVX+E2). The dosage of 17β-estradiol was selected based on previous in vivo studies to preserve physiological circulating estrogen level (Arias-Loza, Hu et al. 2007). Nine weeks after ovariectomy, before sacrificed, the standard limb lead IIECG was recorded for 30 minutes by an ECG recorder (BL-420, ChengDu TME Technology Co, Ltd, ChengDu, China). The heart tissues were collected after the experimental rats were anesthetized with sodium pentobarbital (100 mg/kg, i.p.). All animal procedures were approved by the Institutional Animal Care and Use Committee at Harbin Medical University (No.HMUIRB-2008-06) and the Institute of Laboratory Animal Science of China (A5655-01).
Western blotting analysis
Equal amount of protein samples from each group (100 μg) was fractionated by SDS-PAGE and electro-blotted onto PVDF membranes (Millipore, Bedford, MA). Membranes were incubated at 4°C overnight in specified primary antibody: Cx43 (Santa Cruz, sc13558, lot# k2408, 1: 200) diluted with PBS. After washing three times by PBST, membranes were probed with second antibody: Alexa Fluor® 790 anti-mouse IgG (H+L) (1: 10000). The membranes were scanned by Odyssey Infrared Imaging System (LI-COR Bioscience, Lincoln, NE). Western blot bands were measured by area × OD using Odyssey v1.2 software and normalized to GAPDH (anti-GAPDH antibody from Kangcheng Inc, Shanghai, China. 1: 5000) as a loading control.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated from myocardium, whole blood or NRVCs using TRIZOL methods and reversely transcribed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystem, USA). Levels of microRNAs and mRNAs were quantified by qRT-PCR performed on 7500 Fast Real-Time PCR Systems (Applied Biosystems, USA) using SYBR Green PCR Master Mix (Applied Biosystems, USA). The relative expression of mature miRNAs and mRNA was quantified using comparative 2-ΔΔCt method. U6 and GAPDH were used as internal controls. For measuring miR-23a, the specific and sensitive reverse-transcription primer as follow, 5'-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGGAAATC-3', the PCR primers forward, 5'-GGATCACATTGCCAGGGAT-3'; and reverse, 5'-CAGTGCGTGTCGTGGAGT-3' were used. For Cx43, the primers forward, 5'-CAGCCTCCAAGGAGTTCCAC-3'; and reverse, 5'-GAGAGATGGGGAAGGACTTGT-3' were used.
Primary neonatal rat ventricular cells (NRVCs) culture and treatment with miRNA-23a and E2
Cardiomyocytes were dissociated from minced hearts of 1~2 days old Wistar rats using a 0.25% solution of trypsin and cultured as monolayers in DMEM (Gibco, Grand Island, NY) at a 37°C incubator that has humidified air containing 5% CO2. After 48h, the cultured NRVCs in every well (2×105/well) were transfected with 2.5 µg NC, miR-23a, AMO-23a as well as miR-23a plus AMO-23a by X-treme GENE siRNA transfection reagent (Cat.# 04476093001, Roche). microRNAs were synthesized by RiboBio (Guangzhou Co., Ltd.). NC is a short RNA with random nucleotide sequences that are totally different from miR-23a or AMO-23a sequence. miR-23a mimics (sense: 5'-AUCACAUUGCCAGGGAUUUCC-3', antisense: 5'-GGAAAUCCCUGGCAAUGUGAU-3'). AMO-23a is miR-23a inhibitor (antisense oligonucleotides of mature miR-23a). The 5 oligonucleotides at both ends of AMO-23a were modified with 2'-O-methyl nucleotides. The miRNA-masking antisense (ODN-23a) was designed to be fully complementary to the miR-23a targeting sequence on 3'UTR of GJA1. 48h after transfection, the NRVCs were harvested for further experiments. For examining morphological changes by electron microscope, the NRVCs (after 48h transfection) were shaved from culture-plates by cell scraper and collected into eppendorf tubes. Then, they were centrifuged at 3000 rpm/min for 15 minutes and preserved in 2.5% glutaraldehyde. In E2 treatment experiment, the E2 that was dissolved in ethanol at physiological concentration (1 nM or 10 nM) was given to NRVCs alone or before miR-23a transfection [25].
Luciferase activity assay
In order to generate reporter vectors that carry mRNA binding sites, 3'-untranslated region (3'-UTR) of GJA1 was prepared by PCR-based method. The outcomes of PCR were inserted into the multiple cloning sites of downstream the luciferase gene (HindⅢ and SpeⅠ sites) in the pMIR-REPORTTM luciferase miRNA expression reporter vector (Ambion, Inc.) following standard procedures as described previously [26]. For luciferase assay: after 24h starvation in serum-free medium, HEK293 cells were transfected with 1 µg PGL3-GJA1 3'UTR pMIR-REPORTTM (firefly luciferase vector) as well as 0.1 µg PRL-TK (TK-driven Renilla luciferase expression vector) together with LipofectamineTM 2000 (Invitrogen, USA). After 48h, luciferase activities were quantified with a dual luciferase reporter assay kit (Promega, Cat.# E1960) on a luminometer (GloMaxTM 20/20, Promega, USA).
Transmission electron microscopy
Pieces of left ventricle longitudinal myofilaments of selected samples were preserved and fixed in 2.5% glutaraldehyde dissolved in PBS (0.1 mol/L, pH 7.4). Then all specimens were prepared following standard procedures by steps: first, rinsed in buffer and post-fixed in PBS-buffered 1% OsO4 for 1-2 h; second, stained embolic in uranyl acetate and dehydrated in ethanol and finally embedded in epoxy resin. After electron stained, the ultra-thin sections were observed using a transmission electron microscope (JEM-1220, JEOL Ltd., Tokyo, Japan). Gap junction (GJ) length was measured in randomly photographed ventricular tissue to determine the relative size of GJ per unit intercalated disc (ID) [27-28]. At least 6 IDs were randomly selected for morphometric analysis from each group. Initially, ID were photographed at low magnification to measure their total length, then all portions of the ID containing GJ were photographed again for further analysis at a final print magnification of 20,000. The results were expressed as percentage of GJ length per ID length.
Immunofluorescence
Sections were first fixed in 4% paraformaldehyde for 15 min. After that they were treated with 0.1% Triton X-100 for 1h and afterwards blocked by goat serum for 1h at room temperature. Specimens were incubated with primary antibodie of Cx43 (Santa Cruz, sc13558, lot# k2408, 1: 100) diluted with PBS for 3h at 37°C. After washing 3 times with PBS, specimens were incubated with secondary antibody 1:500 Alexa Fluor 594 conjugated anti-mouse (Invitrogen) for 1h at room temperature. Finally all sections were covered with cover slip and observed under confocal microscope. Cx43 immunoreactivity at the gap junctions in each image was quantified and normalized to the area of the respective cardiomyocyte using Image-Pro Plus 6.0 (Media Cybernetics, Bethesda, MA, USA). The relative Cx43 immunoreactivity levels of 18M, OVX or OVX+E2 rats that were presented in the column were normalized to 6M or Sham group.
Statistical analysis
All data were presented as mean ± SEM and statistical analyzed by GraphPad Prism 5.0. Two-tailed T test was applied for comparisons between two groups, and ANOVA was used to compare the difference of three or more groups. P < 0.05 was recognized as statistically significant difference.
Results
E2 deficiency induced the prolongation of PR intervals and QRS durations
In the present study, we chose 18 months female old rats to mimic post-menopausal situation because rats at this age were approximately equivalent to the women around 50 years old [29] which was the average onset age of women menopause [30]. To verify whether 18 months old rats successfully imitated E2 deficiency condition, we measured the E2 levels of both young (6M) and post-menopausal rats (18M). The results showed that the average serum level of E2 was 77.68 ± 7.55 pg/mL in female rats at age of 6M. However, it was lower than 25 pg/mL in post-menopausal rats, which was the minimum detectable value by Elisa method (Fig. 1A).
E2 deficiency induced prolongation of PR intervals and QRS durations. (A) Average serum estrogen level of both young (6M) and post-menopausal (18M) rats. (B-C) Representative electrocardiograms and quantitative analysis showed prolongation of PR intervals and QRS durations of post-menopausal rats. (D) Estrogen level of Sham, OVX and OVX+E2 rats. (E-F) Representative electrocardiograms and quantitative analysis of PR intervals and QRS durations of Sham, OVX and OVX+E2 rats. n=6 in each group. * P<0.05, ** P < 0.01 and *** P < 0.001 relative to 6M or Sham. # P < 0.05 and ## P < 0.01 relative to OVX.
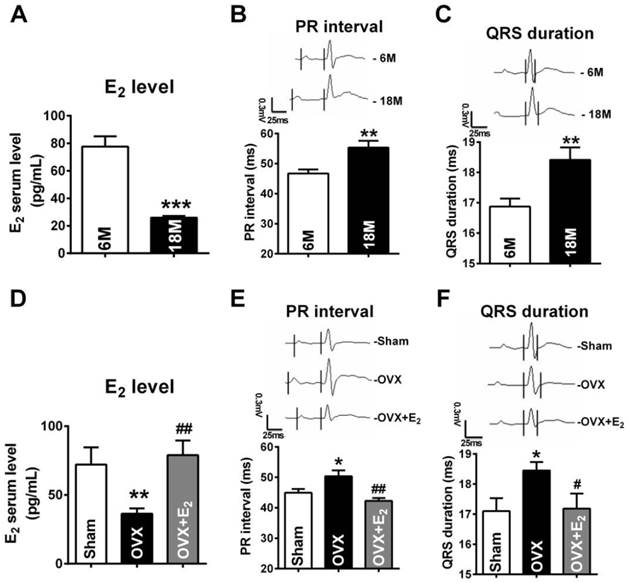
Next, by monitoring ECG, we found that both PR intervals and QRS durations in post-menopausal rats were prolonged (Fig. 1B and C). The PR intervals and QRS durations in 18 months rats were 56.34 ± 2.247 ms and 18.42 ± 0.407 ms respectively, which were significant longer than they were in rats at 6 months old rats (PR: 46.71 ± 1.341 ms, QRS: 16.87 ± 0.268 ms ). Such electrophysiological alternations are in accordance to the reports in post-menopausal women [3-4, 31].
In order to explore whether the abnormal electrophysiological alternations were associated with E2 deficiency, we monitored ECG of E2 deprived 6 months old rats. Compared with age-matched sham female rats, the E2 levels were reduced to 36.35 ± 3.90 pg/mL in rats suffering from ovariectomized (OVX) surgery, which were restored to 78.94 ± 10.67 pg/mL by daily E2 delivery after OVX (Fig. 1D). It is worth noting that, in OVX rats, both PR intervals and QRS durations were significantly prolonged and E2 supplement reversed such prolongation (Fig. 1E and F).
E2 deficiency caused gap junction remodeling and Cx43 expression reduction in ventricle myocardium of rats
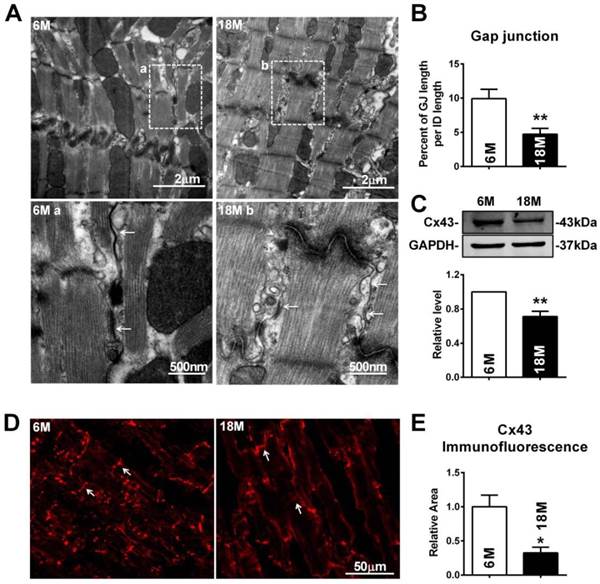
To further explore whether these electrophysiological changes were associated with cardiac gap junction remodeling, transmission electron microscope (TEM) was employed. Different from fasciae adherentes junctions and desmosome, the other two types of adhesion junction at intercalated disk, the typical characteristics of gap junction are that it is the narrowest area between cells with 2 nm gaps and displayed as dense and uniform electron-dense zones (Fig. 2A). Our results showed that the gap junctions in the myocardium of post-menopausal rats were broken with disappeared portions or enlarged cell-to-cell appositions (Fig. 2A). Quantitative analysis of image data demonstrated significantly reduced size of gap junction per intercalated disk in 18M rats hearts (Fig. 2B). Since downregulation of Cx43 level was reported to be one of the possible causes for gap junction damages, we measured Cx43 protein level in the heart. As illustrated in Figure 2C, Cx43 protein level was significantly decreased in 18M rats compared with 6M rats. The result was further verified by reduced Cx43 immunofluorescence intensity in post-menopausal rat hearts (Fig. 2D and E).
Gap junction remodeling and Cx43 reduction in post-menopausal rats. (A) The representative electron micrographs of myocardium from both 6M and 18M rats. Up: Full image of intercalated disk including fasciae adherentes junctions, desmosome and gap junctions. Down: magnified images of gap junctions from dashed boxes of above images. The gap junctions are pointed by arrows. (B) The size of gap junction per intercalated discs. (C) Cx43 protein level in myocardium of 6M and 18M rats. (n=6). (D-E) The representative confocal images and quantitative analysis of Cx43 immunofluorescence activity of 6M and 18M rats. The values in column are normalized to 6M. n=3, * P<0.05 and ** P < 0.01 relative to 6M.
Similar with the findings of gap junction remodeling in post-menopausal rats, broken and/or enlarged cell-to-cell appositions of the gap junctions were also observed in rats suffering from OVX surgery, which were repaired by daily E2 delivery after OVX (Fig. 3A). The size of gap junction per intercalated disk was also decreased in OVX rats and could be recovered by E2 delivery (Fig. 3B). Furthermore, E2 treatment reversed the decreased expression of Cx43 in OVX rats (Fig. 3C), which was further verified by immunofluorescence staining (Fig. 3D and E).
E2 deficiency caused gap junction remodeling and Cx43 reduction in OVX rats. (A) The representative electron micrographs of myocardium from Sham, OVX and OVX+E2 rats. Up: Full image of intercalated disk including fasciae adherentes junctions, desmosome and gap junctions. Down: Magnified images of gap junctions from dashed boxes of above images. The gap junctions are pointed by arrows. (B) The size of gap junction per intercalated discs. (C) Cx43 protein level in myocardium of Sham, OVX and OVX+E2 rats. (n=6). (D, E) The representative confocal images and quantitative analysis of Cx43 immunofluorescence activity of Sham, OVX and OVX+E2 rats. (F) The mRNA level of Cx43 in experimental rats' hearts. The values in column are normalized to Sham (n=3). * P<0.05, ** P < 0.01 and *** P < 0.001 relative to Sham. # P < 0.05 and ## P < 0.01 relative to OVX.
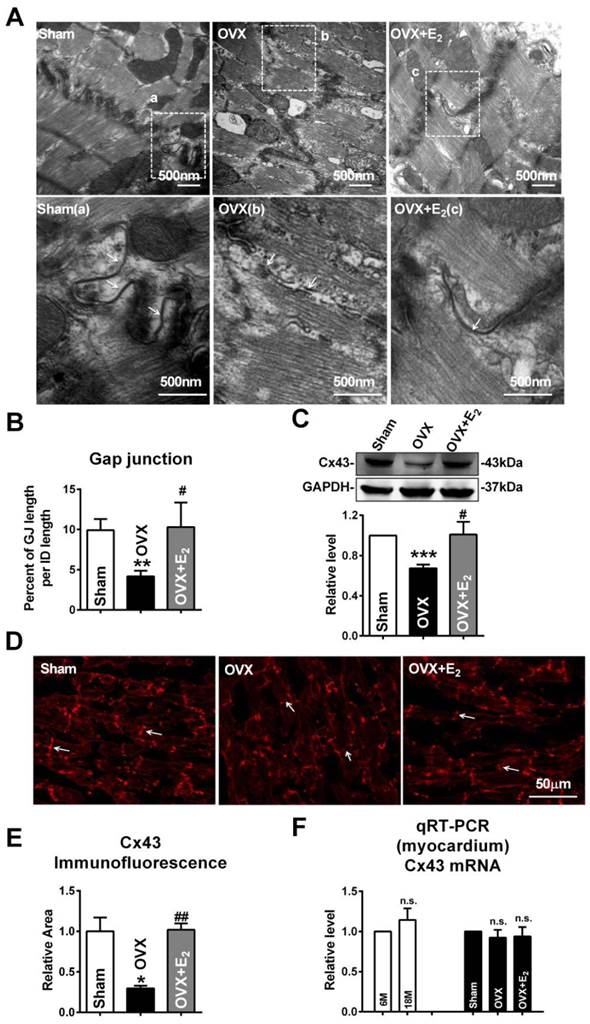
Interestingly, the mRNA level of Cx43 showed no significant differences either between 6M and 18M rats or among rats of sham, OVX and OVX+E2 (Fig. 3F). The inconsistent changes of mRNA and protein level of Cx43 implied that the reduction of Cx43 protein following E2 deprivation might happen at a post-transcriptional level.
E2 deficiency induced upregulation of miR-23a in rats
MicroRNA, as one of the most well established post-transcriptional regulators, can reduce target protein levels by directly targeting at the 3'UTR of mRNA [16]. Therefore, we hypothesized that microRNA might participate in the downregulation of Cx43 expression in both post-menopausal and OVX rats. Lots of studies uncovered that increased miR-23a level participated in cardiac structural remodeling in myocardial hypertrophy. We compared the differential expressed microRNAs from blood of pre- and post-menopausal women by microarray analysis. The data showed that both miR-23a and miR-584 levels were significantly increased in post-menopausal women (Fig. 4A; Pre-: S01, S02 and S03; Post-: S04, S05 and S06). By bioinformatics analysis using the TargetScan database (http://targetscan.org/), miR-23a was selected as the final target since it was miR-23a but not miR-584 that had binding sites on the GJA1 3'UTR.
Increase of miR-23a in E2 deficiency rats. (A) The microarray analysis showed differential expressed miRNAs between pre- and post-menopausal women blood. Pre-: S01, S02 and S03; Post-: S04, S05 and S06. Red: up-regulation; Green: down-regulation; Black: no change. (P < 0.01). (B-C) The miR-23a expression levels in myocardium and blood of 6M and 18M rats was determined by qRT-PCR assay. Values were presented in 2-ΔΔCT, which are normalized to 6M. (D-E) MiR-23a expression levels in the myocardium and blood of Sham, OVX and OVX+E2 rats. Values were presented in 2-ΔΔCT, and are normalized to Sham. n=6. (F) The miR-23a expression level in NRVCs treated with vehicle, 1 nM E2 or 10nM E2. n=3 * P < 0.05, ** P < 0.01 and *** P < 0.001 relative to 6M, Sham or Ctl. # P < 0.05 and ### P < 0.001 relative to OVX.
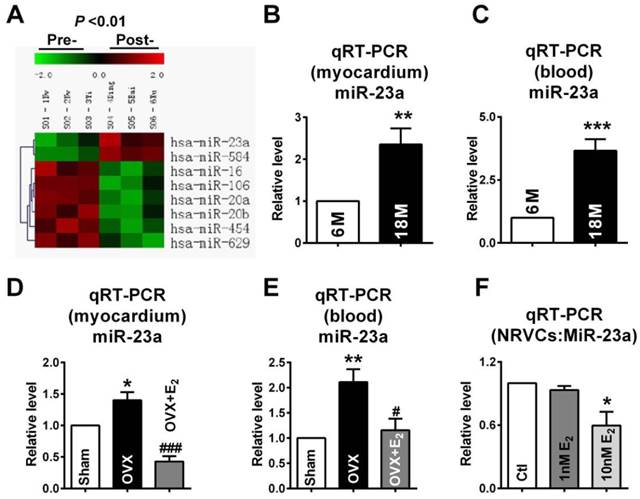
In order to confirm the involvement of miR-23a in gap junction remodeling, we observed whether miR-23a levels was increased in E2 deficiency rats. The miR-23a levels in both myocardium and blood of post-menopausal rats were measured. As predicted, compared with 6M rats, the levels of miR-23a in post-menopausal (18M) rats were significantly elevated by about 2.1 folds in the myocardium and 3.7 folds in the blood (Fig. 4B and C). Similar, miR-23a levels were also remarkably increased in both myocardium and blood of OVX rats, which were reversed by E2 supplement after OVX (Fig. 4D and E). We also found that 10 nM of E2 significantly decreased miR-23a level in NRVCs which suggested that E2 could also suppress miR-23a level in vitro (Fig. 4F).
MiR-23a directly targeted Cx43
To test whether miR-23a could directly target the 3'UTR of GJA1 (Fig. 5A), luciferase reporter assay was performed using HEK293 cells. Compared with the negative control (NC), transfection of miR-23a with the luciferase reporter gene containing the 3'UTR of GJA1 resulted in a significant decrease of luciferase activity, and joint application of miR-23a with its inhibitor (AMO-23a), attenuated the reduction of luciferase activity (Fig. 5B). MiR-23a had no effect on mutated 3'UTR of GJA1 which suggested that GJA1 was targeted by miR-23a via the predicted binding sites (Fig. 5B).
MiR-23a regulated the expression of Cx43 and induced gap junction remodeling. (A) Complementarity between miR-23a seed sequence (5'end 7 nucleotides) and the 3'UTR of rat's GJA1 mRNA predicted by a computational and bioinformatics-based approach using the TargetScan database. The miRNA-masking antisense (ODN-23a) was designed to be fully complementary to the miR-23a targeting sequence on 3'UTR of GJA1. Watson-Crick complementarity is connected by “|”. (B) Luciferase reporter assay for determining interactions between miR-23a and its binding sites at the 3'UTR of GJA1 or mutated 3'UTR of GJA1 in HEK293 cells. Cells were transfected with negative control (NC), miR-23a or miR-23a+AMO-23a. (C) The representative electron micrographs of NRVCs. Up: Full image of intercalated disk including fasciae adherentes junctions, desmosome and gap junctions. Gap junctions were pointed by arrows in dashed boxes. Down: magnified images of gap junctions from dashed boxes of above images. (D) The miR-23a expression level in cultured primary neonatal rat ventricular cells (NRVCs) after transfection. (E) The size of gap junction per intercalated discs. (F) Effects of miR-23a on endogenous Cx43 protein level in NRVCs were determined by western blot analysis. Cells were transfected with NC, miR-23a, AMO-23a or miR-23a+AMO-23a. (n=3 batches of cells for each group). * P < 0.05 and *** P < 0.001 relative to NC. # P < 0.05 and ## P < 0.01 relative to miR-23a transfected NRVCs.
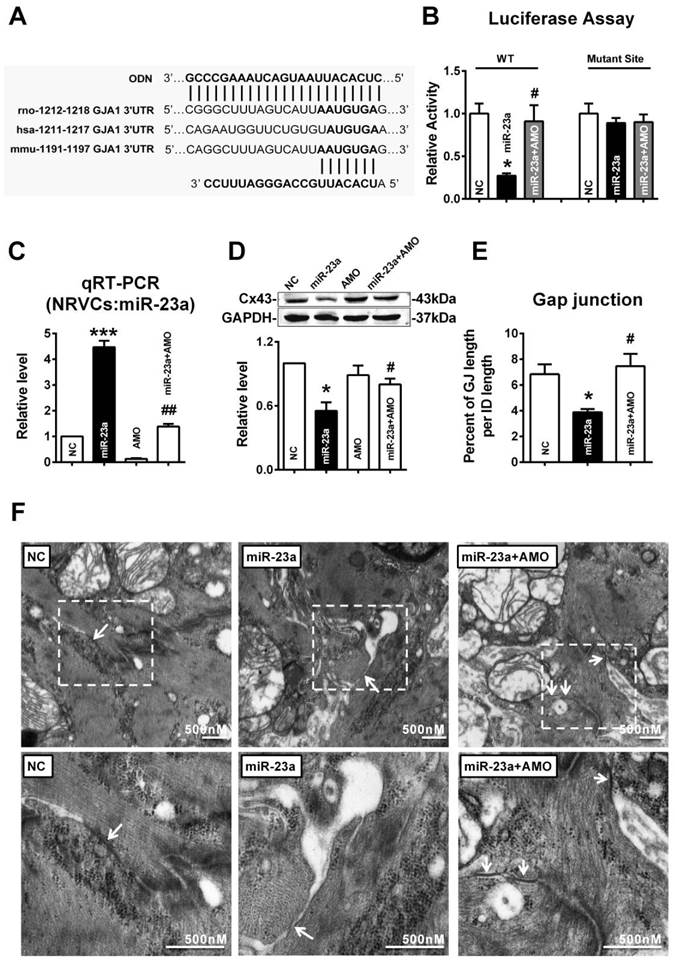
Next, we tested whether miR-23a directly reduced Cx43 protein level in cultured primary neonatal rat ventricular cells (NRVCs). The successful uptake of miR-23a into NRVCs after transfection was verified (Fig. 5C). Results showed that transfection of miR-23a induced significant downregulation of Cx43 protein level, which was rescued by co-transfection of AMO-23a, indicating that miR-23a alternations can regulate Cx43 protein level (Fig. 5D). Importantly, we observed that gap junctions in NC group displayed very close cell-to-cell appositions, and however, overexpression of miR-23a prevented gap junction development in NRVCs indicated by enlarged cell-to-cell appositions as well as blurred and decreased size of gap junctions. The phenomenon was prevented by co-transfection of AMO-23a (Fig. 5E and F).
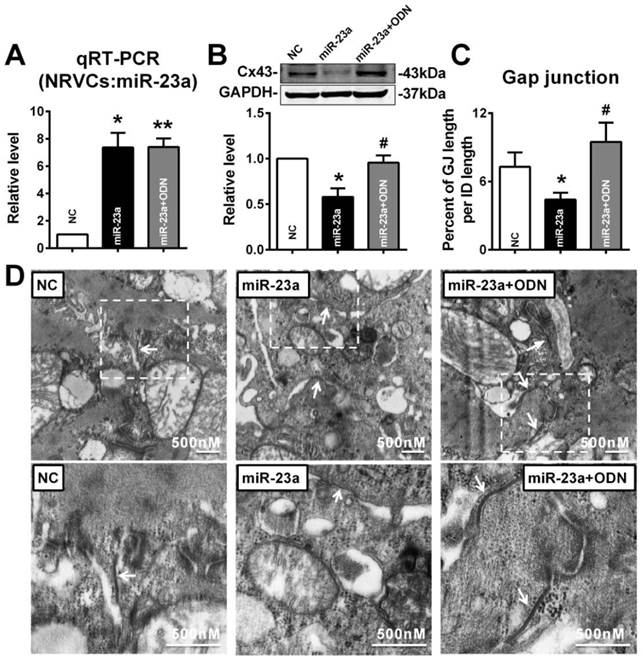
In order to confirm whether overexpression of miR-23a could reduce endogenous Cx43 expression via directly binding 3'UTR of GJA1, the miRNA-masking antisense ODN (miR-Mask) technique was employed [32]. The miR-Mask was designed to be fully complementary to the miR-23a targeting sequence on 3'UTR of GJA1 (Figure 5A). Unlike AMO-23a, co-transfected ODN with miR-23a failed to retrieve miR-23a level (Fig. 6A), but succeeded in blocking Cx43 repression (Fig. 6B). Surprisingly, co-transfection of ODN-23a with miR-23a into NRVCs prevented gap junction remodeling that was induced by transfection of miR-23a alone (Fig. 6C, and D).
MiR-23a overexpression induced Cx43 reduction and gap junction defects can be prevented by ODN. ODN was designed to be fully complementary to the miR-23a targeting sequence on 3'UTR of GJA1. (A) The level of miR-23a in NRVCs after transfection of miR-23a and ODN-23a. (B) De-repression of Cx43 protein level by ODN-23a from miR-23a in NRVCs was determined by western blot analysis. (C) The size of gap junction per intercalated discs. (D) The representative electron micrographs of NRVCs. Up: Full image of intercalated disk including fasciae adherentes junctions, desmosome and gap junctions. Gap junctions were pointed by arrows in dashed boxes. Down: magnified images of gap junctions from dashed boxes of above images. The gap junctions are pointed by arrows. n=3. * P < 0.05 and ** P < 0.01 relative to NC. # P < 0.05 relative to miR-23a transfected NRVCs.
E2 could prevent miR-23a overexpression induced Cx43 repression in vitro
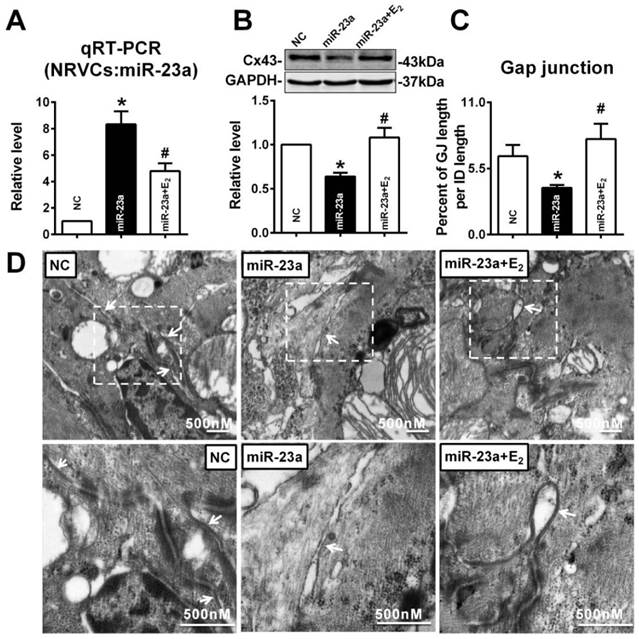
To verify whether E2 can protect against the reduction of Cx43 expression induced by miR-23a, 10 nM of E2 was given to miR-23a transfected NRVCs. As illustrated as Figure 7A, compared with miR-23a transfection group, E2 treatment significantly inhibited the increased miR-23a level in NRVCs (Fig. 7A). Along with the downregulation of miR-23a, 10 nM of E2 significantly blocked the decreased expression of Cx43 protein level induced by miR-23a transfection (Fig. 7B). Accordingly, E2 successfully reversed the enlarged cell-to-cell appositions induced by miR-23a overexpression, gap junctions in E2 pre-treated NRVCs displayed dense and uniform electron-dense zones (Fig. 7C, and D).
E2 prevented Cx43 repression and gap junction defects induced by miR-23a overexpression in NRVCs. (A) qRT-PCR assay revealed that E2 treatment inhibited the expression of miR-23a in NRVCs. (B) E2 treatment prevented the reduction of Cx43 protein level induced by miR-23a in NRVCs. (C) The size of gap junction per intercalated discs. (D) The representative electron micrographs of NRVCs. Up: Full image of intercalated disk including fasciae adherentes junctions, desmosome and gap junctions. Gap junctions were pointed by arrows in dashed boxes. The gap junctions are pointed by arrows. Down: magnified images of gap junctions from dashed boxes of above images. The gap junctions are pointed by arrows. n=3. * P < 0.05 relative to NC. # P < 0.05 relative to miR-23a transfected NRVCs.
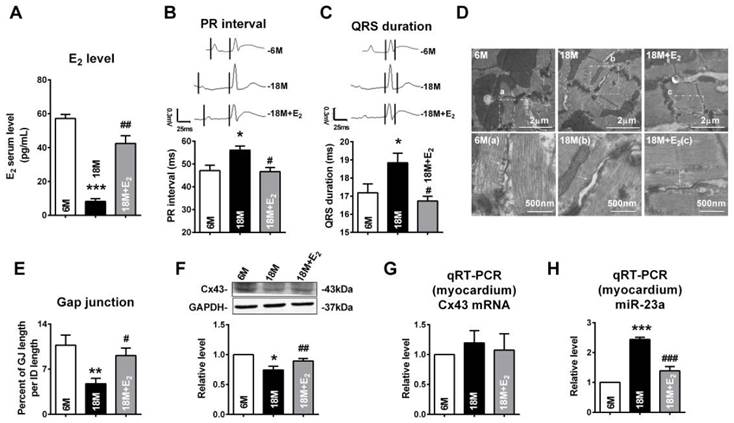
We also determined whether E2 delivery could reverse gap junction remodeling and Cx43 reduction in left ventricle of post-menopausal rats via downregulation of miR-23a. Along with the reviving of estrogen (Fig 8A), we found that QRS durations and PR intervals of postmenopausal rats were shorten (Fig 8B and C). We also found that E2 supplement could protect the structure and the size of gap junctions in postmenopausal rats' hearts (Fig 8D and E). The protein level of Cx43 in postmenopausal rats was also restored to normal level after E2 supplement (Fig 8F). Consistent with the finding in young OVX and OVX+E2 rats, the mRNA levels of Cx43 in 6M, 18M and 18M+E2 are comparative which suggested a post-transcriptional regulation of Cx43 protein level (Fig 8G). The miR-23a level in postmenopausal rats hearts was also suppressed to normal level by E2 supplement (Fig 8H) suggested that estrogen deficiency may regulate gap junction remodeling as well as Cx43 downregulation via miR-23a in the hearts.
E2 delivery improved gap junction remodeling as well as Cx43 downregulation via regulating miR-23a. (A) Average serum estrogen level of experimental animals. (B-C) Representative electrocardiograms and quantitative analysis of PR intervals and QRS durations. (D) The representative electron micrographs of myocardium from 6M, 18M and 18M+E2 rats. Up: Full image of intercalated disk including fasciae adherentes junctions, desmosome and gap junctions. Gap junctions were pointed by arrows in dashed boxes. Down: magnified images of gap junctions from dashed boxes of above images. The gap junctions are pointed by arrows. (E) The size of gap junction per intercalated discs. (F) The Cx43 protein level of experimental rat hearts. (G) The mRNA level of Cx43 in experimental rats. (H) The miR-23a level in left ventricular tissue of experimental rats. n=6 * P<0.05, ** P < 0.01 and *** P < 0.001 relative to 6M. # P < 0.05, ## P < 0.01 and ### P < 0.001 relative to 18M.
Discussion
In the present study, we reported the prolongation of both PR intervals and QRS durations, damages of gap junction accompanied by decreased Cx43 expression in hearts of 18M old female rats and OVX rats. Moreover, our study identified that miR-23a was increased in both the hearts and blood of post-menopausal and OVX rats. Further study demonstrated that miR-23a post-transcriptionally regulated the expression of Cx43 via targeting 3'UTR of GJA1, and E2 treatment reversed gap junction remodeling and Cx43 reduction by inhibiting miR-23a upregulation.
In contrast to age-matched men, women before 40s have a lower prevalence of heart diseases including coronary heart disease, heart failure, stroke as well as hypertension [33]. However, the advantage of women vanished after menopause [34]. E2 is believed to play a critical role in these clinical changes. Deprivation of E2 can induce considerable changes to cardiovascular system, such as stiffness increasing, extracellular matrix deposition, eNOs activity declining, inflammation, apoptosis as well as hypertrophy [1]. Moreover, E2 level is also considered to have great influence on cardiac conduction system, because gender differences of electrocardiogram parameters diminished by E2 deprivation after menopause and studies in animal models suggested that estrogen could protect against ventricular arrhythmia [4]. In the present study, we chose 18 months old rats as the animal model to imitate post-menopausal condition because female rats at age of 18M old have ceased estrus cycles and exhibit extremely low serum E2 level [35-36]. These changes are consistent with E2 secretion alternations in the post-menopausal women. On the other hand, in terms of lifespan, 18 months old rats are equivalent to human around 50 years old and that is the average menopausal onset age of women [37-38]. In the present study, we confirmed that rats at age of 18M had PR and QRS prolongations, which is consistent with electrophysiological property alternations in post-menopausal women [31, 39]. We also measured the cardiac function of experimental rats by a pressure-volume (PV) loops (Scisense, Ontario, Canada) and did not find significant changes in cardiac contractile function, revealed by comparative ejection fraction between different groups (Supplementary Material: Fig. S1). Studies have found that ovariectomy augmented pressure-overload induced hypertrophy [40] which suggested that although estrogen deficiency did not alter cardiac function parameters at rest, but make the cardiovascular system vulnerable to extrinsic stimulations.
Because Cx43 reduction could slow conduction velocity and induce structural remodeling of gap junction [41-42], we assumed that the reduction of Cx43 may be the underlying reason for PR and QRS prolongation in E2 deficiency rats. We observed enlarged cell-to-cell appositions as well as blurred and decreased size of gap junctions in the heart of 18M rats. By examining the electron micrographs, we did not find evident alternations in myofilament structure in estrogen deficiency rats. Although we did find that estrogen deprivation induced several evident changes in cardiomyocytes ultrastructure, including endoplasmic reticulum swelling and mitochondrial damages, and may worth further investigating in the future study.
In order to identify whether these changes were due to E2 deficiency, we established animal models of rats that suffered from ovariectomy (OVX) and rats receiving E2 supplement after OVX. We found that Cx43 protein level declined in OVX rats and was restored by E2 supplement. Previous studies reported that E2 could reduce infarct size [14] and lessen arrhythmias vulnerability [15] via preserving Cx43 expression, and the existence of putative estrogen response element sites at GJA1 promoter region indicated that E2 might directly upregulate Cx43 expression [43]. However, it could not explain how E2-deificiency decreases Cx43 expression completely.
MicroRNAs are a group of small non-coding RNAs that can reduce protein expression via repressing target mRNAs translation or degradation of target mRNAs directly [44]. In recent years, a large body of researches have implicated that microRNAs might play important roles in the pathological processes of various heart diseases [45]. For example, overexpression of miR-1 could induce arrhythmogenesis via repressing kir2.1 and Cx43 [17, 46]. Most recently, overexpression of miR-1 has been found to influence the expression of CaMKII which is a key regulator of arrhythmias [18, 47]. miR-26 could protect against atrial fibrillation in mice through targeting kir2.1 [48]. In our study, E2 was reported to change microRNA expression profiles of many animals and tissues, including zebrafish [49], mouse uterus [50], chicken gonads [51], and vascular smooth muscle cells [23]. Cell lines, such as MCF-7 also exhibit E2 associated microRNAs differential expressions [52]. These studies provided a possibility that E2 might exert its cardiac protective effects through microRNAs.
In the present study, we chose miR-23a as the potential Cx43 targeted microRNA for two reasons: the first reason was that the miR-23a level was significantly increased in post-menopausal women blood; the second reason was that miR-23a had potential binding sites on 3'UTR of GJA1. By Real-time PCR, we found that miR-23a was significantly increased in both post-menopausal and OVX rats, which can be prevented by E2 supplement. By luciferase assay and the miRNA-masking antisense ODN (miR-Mask) technique, we demonstrated that miR-23a could directly reduce endogenous Cx43 expression via binding the 3'UTR of GJA1. And E2 treatment could prevent miR-23a overexpression induced Cx43 repression.
The mechanism of estrogen regulating miR-23a might be complicated. The regulatory regions of miR-23a contained the estrogen receptor α (ERα) binding sites [53]. Interestingly, researches suggested that most of estrogen suppressed microRNAs had ERα binding sties at their promoters [54] and estrogen may repress microRNA via ERα [55]. However, the detailed mechanism is far from clear. A possible explanation is that the E2 -ERα may regulate microRNA expression via upregulating c-myc [56]. Previous study reported that E2 administration could reverse the OVX induced c-myc decreasing in rats uterus [57] and c-myc could suppress miR-23a expression in human cancer cell lines [58]. The research found that c-myc directly bound to the transcriptional unit, C9orf3 which encompasses miR-23a [58-59]. However, whether the increased miR-23a expression induced by E2 deficiency is mediated by changes of c-myc needs to be studied further. We also measured the level of other miR-23a cluster members, including miR-27a and miR-24-2. We found that both miR-27a and miR-24-2 showed upregulation tendency in estrogen deficiency rats and these changes were reversed by estrogen complementary (Supplementary Material: Fig. S2). The results suggested that estrogen might regulate the whole miR-23a~27a~24-2 cluster.
Moreover, since miR-23a is predicted to have hundreds of targets (http://targetscan.org/), whether miR-23a participates in E2 deficiency related other cardiac disorders through different targets is worthy studying in the future.
As is known, the hormone replacement therapy (HRT) was controversial in clinic. In late 90s, several clinical trials indicated that HRT increased the cardiovascular risk [60-61]. However, in recent years, scientists found that many factors including the timing of HRT initiation, the age of patients and preexisting cardiovascular disease might contribute to the unfavorable effect of HRT [62-65]. The administration method and the estrogen mimics used in HRT are also important risk factors. In the present study, we used the major and the most potent natural form of estrogen [66], 17β-estradiol, to investigate the physiological effect of estrogen and we found that estrogen could protect cardiac conduction system. We believed the gap between lab researches on estrogen and clinical use of HRT might narrow down in the future.
Conclusion
These findings provide the evidence that miR-23a mediated repression of Cx43 may participate in estrogen deficiency induced cardiac gap junction damages, and highlight a new insight into molecular mechanism of post-menopause related arrhythmia at the microRNA level.
Supplementary Material
Figure S1 and S2.
Abbreviations
E2, Estrogen; Cx43, Connexin43; OVX , Ovariectomized; 3'UTR, 3' Untranslated Region; NC, Negative control; AMO-23a, miR-23a-specific antisense inhibitory oligoribonucleotide; qRT-PCR, Quantitative reverse transcription PCR.
Acknowledgements
This work was supported by Creative Research Groups of The National Natural Science Foundation of China (81121003), Natural Science Foundation of China (81271207, 31100825 and 81470523), and the funding from Key Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education (to Fang Xie)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stice JP, Lee JS, Pechenino AS, Knowlton AA. Estrogen, aging and the cardiovascular system. Future cardiology. 2009;5:93-103 doi:10.2217/14796678.5.1.93
2. Barrett-Connor E. Menopause, atherosclerosis, and coronary artery disease. Curr Opin Pharmacol. 2013 doi:10.1016/j.coph.2013.01.005
3. Suenari K, Hu YF, Tsao HM, Tai CT, Chiang CE, Lin YJ. et al. Gender differences in the clinical characteristics and atrioventricular nodal conduction properties in patients with atrioventricular nodal reentrant tachycardia. J Cardiovasc Electrophysiol. 2010;21:1114-9 doi:10.1111/j.1540-8167.2010.01779.x
4. Gowd BM, Thompson PD. Effect of female sex on cardiac arrhythmias. Cardiol Rev. 2012;20:297-303 doi:10.1097/CRD.0b013e318259294b
5. John AD, Fleisher LA. Electrocardiography: the ECG. Anesthesiol Clin. 2006;24:697-715 v-vi
6. Aro AL, Anttonen O, Tikkanen JT, Junttila MJ, Kerola T, Rissanen HA. et al. Intraventricular conduction delay in a standard 12-lead electrocardiogram as a predictor of mortality in the general population. Circ Arrhythm Electrophysiol. 2011;4:704-10 doi:10.1161/CIRCEP.111.963561
7. Chen G, Yang X, Alber S, Shusterman V, Salama G. Regional genomic regulation of cardiac sodium-calcium exchanger by oestrogen. J Physiol. 2011;589:1061-80 doi:10.1113/jphysiol.2010.203398
8. Yang X, Chen G, Papp R, Defranco DB, Zeng F, Salama G. Oestrogen upregulates L-type Ca(2)(+) channels via oestrogen-receptor- by a regional genomic mechanism in female rabbit hearts. J Physiol. 2012;590:493-508 doi:10.1113/jphysiol.2011.219501
9. Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9-19 doi:10.1093/cvr/cvn133
10. van Kempen MJ, ten Velde I, Wessels A, Oosthoek PW, Gros D, Jongsma HJ. et al. Differential connexin distribution accommodates cardiac function in different species. Microsc Res Tech. 1995;31:420-36 doi:10.1002/jemt.1070310511
11. O'Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res. 2011;108:704-15 doi:10.1161/CIRCRESAHA.110.235747
12. Chen SC, Kennedy BK, Lampe PD. Phosphorylation of connexin43 on S279/282 may contribute to laminopathy-associated conduction defects. Exp Cell Res. 2013;319:888-96 doi:10.1016/j.yexcr.2012.12.014
13. Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA. et al. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest. 1997;99:1991-8 doi:10.1172/JCI119367
14. Lee TM, Lin MS, Chou TF, Tsai CH, Chang NC. Adjunctive 17beta-estradiol administration reduces infarct size by altered expression of canine myocardial connexin43 protein. Cardiovasc Res. 2004;63:109-17 doi:10.1016/j.cardiores.2004.03.009
15. Chen CC, Lin CC, Lee TM. 17beta-Estradiol decreases vulnerability to ventricular arrhythmias by preserving connexin43 protein in infarcted rats. Eur J Pharmacol. 2010;629:73-81 doi:10.1016/j.ejphar.2009.11.050
16. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102-14 doi:10.1038/nrg2290
17. Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B. et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486-91 doi:10.1038/nm1569
18. Ai J, Zhang R, Gao X, Niu HF, Wang N, Xu Y. et al. Overexpression of microRNA-1 impairs cardiac contractile function by damaging sarcomere assembly. Cardiovasc Res. 2012;95:385-93 doi:10.1093/cvr/cvs196
19. Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013;123:11-8 doi:10.1172/JCI62876
20. Pan Q, Luo X, Chegini N. Differential expression of microRNAs in myometrium and leiomyomas and regulation by ovarian steroids. J Cell Mol Med. 2008;12:227-40 doi:10.1111/j.1582-4934.2007.00207.x
21. Pan Q, Luo X, Toloubeydokhti T, Chegini N. The expression profile of micro-RNA in endometrium and endometriosis and the influence of ovarian steroids on their expression. Mol Hum Reprod. 2007;13:797-806 doi:10.1093/molehr/gam063
22. Paris O, Ferraro L, Grober OM, Ravo M, De Filippo MR, Giurato G. et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor beta in hormone-responsive breast cancer. Oncogene. 2012 doi:10.1038/onc.2011.583
23. Zhao J, Imbrie GA, Baur WE, Iyer LK, Aronovitz MJ, Kershaw TB. et al. Estrogen receptor-mediated regulation of microRNA inhibits proliferation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2013;33:257-65 doi:10.1161/ATVBAHA.112.300200
24. Jazbutyte V, Arias-Loza PA, Hu K, Widder J, Govindaraj V, von Poser-Klein C. et al. Ligand-dependent activation of ER{beta} lowers blood pressure and attenuates cardiac hypertrophy in ovariectomized spontaneously hypertensive rats. Cardiovasc Res. 2008;77:774-81 doi:10.1093/cvr/cvm081
25. Babiker FA, De Windt LJ, van Eickels M, Thijssen V, Bronsaer RJ, Grohe C. et al. 17beta-estradiol antagonizes cardiomyocyte hypertrophy by autocrine/paracrine stimulation of a guanylyl cyclase A receptor-cyclic guanosine monophosphate-dependent protein kinase pathway. Circulation. 2004;109:269-76 doi:10.1161/01.CIR.0000105682.85732.BD
26. Sun LY, Wang N, Ban T, Sun YH, Han Y, Sun LL. et al. MicroRNA-23a mediates mitochondrial compromise in estrogen deficiency-induced concentric remodeling via targeting PGC-1alpha. Journal of molecular and cellular cardiology. 2014;75:1-11 doi:10.1016/j.yjmcc.2014.06.012
27. Thomas SP, Bircher-Lehmann L, Thomas SA, Zhuang J, Saffitz JE, Kleber AG. Synthetic strands of neonatal mouse cardiac myocytes: structural and electrophysiological properties. Circ Res. 2000;87:467-73
28. Kostin S, Hein S, Bauer EP, Schaper J. Spatiotemporal development and distribution of intercellular junctions in adult rat cardiomyocytes in culture. Circ Res. 1999;85:154-67
29. Andreollo NA, Santos EF, Araujo MR, Lopes LR. Rat's age versus human's age: what is the relationship? Arq Bras Cir Dig. 2012;25:49-51
30. Grodstein F, Stampfer MJ, Manson JE, Colditz GA, Willett WC, Rosner B. et al. Postmenopausal estrogen and progestin use and the risk of cardiovascular disease. N Engl J Med. 1996;335:453-61 doi:10.1056/NEJM199608153350701
31. Taneja T, Mahnert BW, Passman R, Goldberger J, Kadish A. Effects of sex and age on electrocardiographic and cardiac electrophysiological properties in adults. Pacing Clin Electrophysiol. 2001;24:16-21
32. Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271-4 doi:10.1126/science.1147535
33. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB. et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2-e220 doi:10.1161/CIR.0b013e31823ac046
34. Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89:157-61
35. Lu KH, Hopper BR, Vargo TM, Yen SS. Chronological changes in sex steroid, gonadotropin and prolactin secretions in aging female rats displaying different reproductive states. Biol Reprod. 1979;21:193-203
36. Fortepiani LA, Zhang H, Racusen L, Roberts LJ 2nd, Reckelhoff JF. Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension. 2003;41:640-5 doi:10.1161/01.HYP.0000046924.94886.EF
37. Treloar AE. Menstrual cyclicity and the pre-menopause. Maturitas. 1981;3:249-64
38. Walker ML, Herndon JG. Menopause in nonhuman primates? Biol Reprod. 2008;79:398-406 doi:10.1095/biolreprod.108.068536
39. Rautaharju PM, Zhou SH, Wong S, Calhoun HP, Berenson GS, Prineas R. et al. Sex differences in the evolution of the electrocardiographic QT interval with age. Can J Cardiol. 1992;8:690-5
40. Bhuiyan MS, Shioda N, Fukunaga K. Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in female rats. American journal of physiology Endocrinology and metabolism. 2007;293:E1606-14 doi:10.1152/ajpendo.00246.2007
41. Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J. et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333-9
42. de Jong S, van Veen TA, van Rijen HV, de Bakker JM. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011;57:630-8 doi:10.1097/FJC.0b013e318207a35f
43. Lefebvre DL, Piersanti M, Bai XH, Chen ZQ, Lye SJ. Myometrial transcriptional regulation of the gap junction gene, connexin-43. Reprod Fertil Dev. 1995;7:603-11
44. Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653-62 doi:10.1242/dev.02073
45. Mishra PK, Tyagi N, Kumar M, Tyagi SC. MicroRNAs as a therapeutic target for cardiovascular diseases. J Cell Mol Med. 2009;13:778-89 doi:10.1111/j.1582-4934.2009.00744.x
46. Diaz RJ, Zobel C, Cho HC, Batthish M, Hinek A, Backx PH. et al. Selective inhibition of inward rectifier K+ channels (Kir2.1 or Kir2.2) abolishes protection by ischemic preconditioning in rabbit ventricular cardiomyocytes. Circ Res. 2004;95:325-32 doi:10.1161/01.RES.0000137727.34938.35
47. Rokita AG, Anderson ME. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation. 2012;126:2125-39 doi:10.1161/CIRCULATIONAHA.112.124990
48. Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N. et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest. 2013;123:1939-51 doi:10.1172/JCI62185
49. Cohen A, Shmoish M, Levi L, Cheruti U, Levavi-Sivan B, Lubzens E. Alterations in micro-ribonucleic acid expression profiles reveal a novel pathway for estrogen regulation. Endocrinology. 2008;149:1687-96 doi:10.1210/en.2007-0969
50. Nothnick WB, Healy C. Estrogen induces distinct patterns of microRNA expression within the mouse uterus. Reprod Sci. 2010;17:987-94 doi:10.1177/1933719110377472
51. Bannister SC, Smith CA, Roeszler KN, Doran TJ, Sinclair AH, Tizard ML. Manipulation of estrogen synthesis alters MIR202* expression in embryonic chicken gonads. Biol Reprod. 2011;85:22-30 doi:10.1095/biolreprod.110.088476
52. Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C. et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2011;30:1082-97 doi:10.1038/onc.2010.487
53. Bhat-Nakshatri P, Wang G, Collins NR, Thomson MJ, Geistlinger TR, Carroll JS. et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic acids research. 2009;37:4850-61 doi:10.1093/nar/gkp500
54. Maillot G, Lacroix-Triki M, Pierredon S, Gratadou L, Schmidt S, Benes V. et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009;69:8332-40 doi:10.1158/0008-5472.CAN-09-2206
55. Wickramasinghe NS, Manavalan TT, Dougherty SM, Riggs KA, Li Y, Klinge CM. Estradiol downregulates miR-21 expression and increases miR-21 target gene expression in MCF-7 breast cancer cells. Nucleic acids research. 2009;37:2584-95 doi:10.1093/nar/gkp117
56. Katchy A, Edvardsson K, Aydogdu E, Williams C. Estradiol-activated estrogen receptor alpha does not regulate mature microRNAs in T47D breast cancer cells. The Journal of steroid biochemistry and molecular biology. 2012;128:145-53 doi:10.1016/j.jsbmb.2011.10.008
57. Weisz A, Bresciani F. Estrogen induces expression of c-fos and c-myc protooncogenes in rat uterus. Mol Endocrinol. 1988;2:816-24
58. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762-5 doi:10.1038/nature07823
59. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM. et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43-50 doi:10.1038/ng.2007.30
60. Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B. et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-13
61. Hulley S, Furberg C, Barrett-Connor E, Cauley J, Grady D, Haskell W. et al. Noncardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA. 2002;288:58-66
62. Rosano GM, Vitale C, Fini M. Cardiovascular aspects of menopausal hormone replacement therapy. Climacteric. 2009;12(Suppl 1):41-6 doi:10.1080/13697130903012306
63. Hodis HN. Assessing benefits and risks of hormone therapy in 2008: new evidence, especially with regard to the heart. Cleve Clin J Med. 2008;75(Suppl 4):S3-12
64. Haines CJ, Farrell E. Menopause management: a cardiovascular risk-based approach. Climacteric. 2010;13:328-39 doi:10.3109/13697130903450154
65. Harman SM. Estrogen replacement in menopausal women: recent and current prospective studies, the WHI and the KEEPS. Gend Med. 2006;3:254-69 doi:S1550-8579(06)80214-7
66. Booth EA, Lucchesi BR. Estrogen-mediated protection in myocardial ischemia-reperfusion injury. Cardiovascular toxicology. 2008;8:101-13 doi:10.1007/s12012-008-9022-2
Author contact
![]() Corresponding authors: Jing Ai: aijinghrbmu.edu.cn or Ning Wang: wangninghrbmu.edu.cn. Department of Pharmacology (the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China), Harbin Medical University, Harbin, P.R. China 150081; Tel.: +86 451 8667-1354; Fax: +86 451 8667-1354
Corresponding authors: Jing Ai: aijinghrbmu.edu.cn or Ning Wang: wangninghrbmu.edu.cn. Department of Pharmacology (the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China), Harbin Medical University, Harbin, P.R. China 150081; Tel.: +86 451 8667-1354; Fax: +86 451 8667-1354