Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(4):423-433. doi:10.7150/ijbs.11032 This issue Cite
Research Paper
Biased Signaling in Naturally Occurring Mutations in Human Melanocortin-3 Receptor Gene
Fan Yang1,2*, Hui Huang1*, Ya-Xiong Tao1 ![]()
1. Department of Anatomy, Physiology and Pharmacology, College of Veterinary Medicine, Auburn University, Auburn, AL, USA.
2. Current address: College of Life Sciences, Inner Mongolia Agricultural University, Hohhot, Inner Mongolia 010018, China.
*These authors contributed equally to this work.
Received 2014-11-10; Accepted 2015-1-21; Published 2015-2-23
Abstract

The melanocortin-3 receptor (MC3R) is primarily expressed in the hypothalamus and plays an important role in the regulation of energy homeostasis. Recently, some studies demonstrated that MC3R also signals through mitogen-activated protein kinases (MAPKs), especially extracellular signal-regulated kinases 1 and 2 (ERK1/2). ERK1/2 signaling is known to alter gene expression, potentially contributing to the prolonged action of melanocortins on energy homeostasis regulation. In the present study, we performed detailed functional studies on 8 novel naturally occurring MC3R mutations recently reported, and the effects of endogenous MC3R agonist, α-melanocyte stimulating hormone (MSH), on ERK1/2 signaling on all 22 naturally occurring MC3R mutations reported to date. We found that mutants D158Y and L299V were potential pathogenic causes to obesity. Four residues, F82, D158, L249 and L299, played critical roles in different aspects of MC3R function. α-MSH exhibited balanced activity in Gs-cAMP and ERK1/2 signaling pathways in 15 of the 22 mutant MC3Rs. The other 7 mutant MC3Rs were biased to either one of the signaling pathways. In summary, we provided novel data about the structure-function relationship of MC3R, identifying residues important for receptor function. We also demonstrated that some mutations exhibited biased signaling, preferentially activating one intracellular signaling pathway, adding a new layer of complexity to MC3R pharmacology.
Keywords: Melanocortin-3 receptor, naturally occurring mutations, cAMP signaling, ERK1/2 signaling, biased signaling.
Introduction
It is well known that several hypothalamic circuits are involved in the regulation of feeding behavior and energy homeostasis, especially the arcuate nucleus and the paraventricular nucleus [1]. The melanocortin system plays an important role in the regulation of energy homeostasis [1-3]. The melanocortin-3 receptor (MC3R) was the third member of melanocortin receptor (MCR) family cloned, primarily expressed in brain, especially in the arcuate nucleus [4-8]. It is a rhodopsin-like G protein-coupled receptor (GPCR), primarily coupling to Gs protein to activate adenylyl cyclase that produces cAMP. Mouse genetic studies demonstrate that the MC3R regulates feeding efficiency and nutrient partitioning [9, 10]. Mice lacking both Mc3r and melanocortin-4 receptor (Mc4r) are more obese than mice lacking only Mc4r [9]. Recent studies report that MC3R acts as a mediator of the entrainment pattern of food intake to restricted feeding [11-13]. All these findings suggest that MC3R functions differently from MC4R, playing a non-redundant role in regulating energy homeostasis.
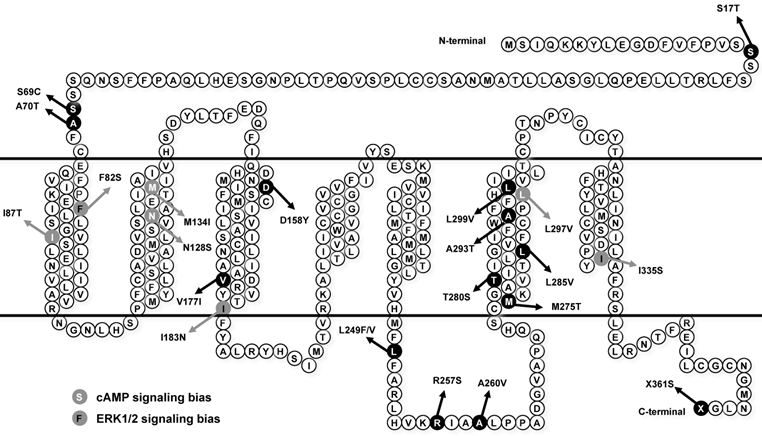
Loss-of-function mutations in MC4R are considered as pathogenic causes for human monogenic obesity, with more than 175 mutations identified [14-16] (reviewed in [17, 18]). However, few naturally occurring mutations in MC3R have been identified [19-25] (reviewed in [26]). The influence of these MC3R mutations on human obesity is controversial. The first MC3R mutation identified in two obese patients in Singapore is I183N, which results in complete loss-of-function [19, 27, 28] and co-segregates with childhood obesity [21]. Therefore I183N is considered a pathogenic mutation. Calton et al. reported that MC3R mutations are not associated with severe obesity in the two North American cohorts they studied, because the prevalence of MC3R mutations in obese subjects is not significantly different from that in control groups [23]. However, Mencarelli et al. reported that MC3R mutations with impaired function are significantly more prevalent in obese subjects of Italian and French origin [24]. Our previous studies showed that residues T280 and I335 play critical roles in different aspects of MC3R function, and T280S and I335S are potentially pathogenic for obesity [29, 30]. Collectively, the pathogenic role of MC3R in the development of obesity still needs further investigation. Therefore, in this study, detailed functional analyses were performed on 8 MC3R mutations (S17T, F82S, D158Y, V177I, L249F, R257S, L285V, and L299V) recently identified in North American cohorts [23], Italian and French subjects [24], and children and adolescents in Belgium [25] (Fig. 1).
Schematic model of the hMC3R with the mutations investigated in this study highlighted. Mutations that induce biased signaling are also indicated.
It was suggested that the complexity of feeding behavior and long-lasting effects of melanocortins on energy homeostasis regulation involves the regulation of gene expression [31], especially through mitogen-activated protein kinases. MC4R has been reported to activate extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling cascade [31-33], which directly alter gene expression [34, 35]. We have reported that in addition to agonists, antagonists including inverse agonists also activate ERK1/2 in the MC4R [36-38] (reviewed in [39]). We also suggested that defects in basal or ligand-stimulated ERK1/2 signaling might cause obesity in patients harboring MC4R mutations where no other defect was identified previously [40].
Discordant data have been reported on whether the MC3R activates ERK1/2, with both supporting [13, 41, 42] and refuting [31] data reported. Moreover, whether naturally occurring mutations in the MC3R affects the two signaling pathways differentially was unknown. In this study, we investigated the effects of endogenous MC3R agonist α-melanocyte stimulating hormone (α-MSH) on ERK1/2 signaling in all 22 naturally occurring MC3R mutations reported so far in the literature, including 8 novel mutations functionally characterized in the present study and 14 mutations (S69C, A70T, I87T, N128S, M134I, I183N, L249V, A260V, M275T, T280S, A293T, L297V, I335S, and X361S), where the binding and Gs-cAMP signaling have been previously characterized in detail [25, 27, 29, 30].
Materials and methods
Plasmid and peptides
Human MC3R (hMC3R) cDNA with 3×HA tags at the N-terminus inserted in pcDNA3.1 vector was obtained from Missouri S&T University cDNA Resource Center (http://www.cDNA.org/, Rolla, MO). [Nle4,D-Phe7]-α-melanocyte stimulating hormone (NDP-MSH) was purchased from Peptides International (Louisville, KY) and iodinated as described previously [37]. α-MSH was purchased from Phoenix Pharmaceuticals (Belmont, CA).
Site-directed mutagenesis of the hMC3R mutants
Mutations in hMC3R were generated by QuikChangeTM site-directed mutagenesis kit (Stratagene, La Jolla, CA) as described previously [43]. Plasmid DNAs were prepared by IsoPure DNA purification kits from Denville Scientific (Metuchen, NJ). Automated DNA sequencing was performed by the DNA Sequencing Facility of Cancer Research Center, University of Chicago (Chicago, IL)
Cells and transfections
Human embryonic kidney (HEK) 293T cells, purchased from American Type Culture Collection (Manassas, VA), were maintained at 37 °C in growth media (Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% newborn calf serum, 10 mM Hepes, 100 IU/ml of penicillin, 100 μg/ml of streptomycin, 50 μg/ml of gentamicin, and 0.25 μg/ml of amphotericin B), with 5% CO2. Cells were plated on gelatin-coated 35mm 6-well clusters from Corning (Corning, NY) and transient transfection was done using the calcium precipitation method [44] at 50-70% confluency. Cells were incubated for approximately 48 h after transfection before assayed.
Ligand binding assay
Ligand binding assays were performed as described previously [43]. Briefly, 48 h after transfection, cells were washed twice with warm Waymouth's MB752/1 media (Sigma-Aldrich, St. Louis, MO) containing 1 mg/ml bovine serum albumin (BSA) (referred to as Waymouth/BSA). Fresh Waymouth/BSA was added to each well, incubated with 125I-NDP-MSH (100,000 cpm) with or without different concentrations of unlabeled NDP- or α-MSH at 37 °C for 1 h. The total volume in each well was 1 ml. The final concentrations of unlabeled ligands are indicated in the figures. After incubation, cells were placed on ice and washed twice with cold Hank's balanced salt solution (Sigma-Aldrich) containing 1 mg/ml BSA to terminate the reaction. Then 100 μl of 0.5 N NaOH were added to each well to solubilize cells. Cell lysates were collected using cotton swabs and counted in a γ-counter. All determinations were performed in duplicate. The receptor occupancy (RO) and IC50 values were calculated by GraphPad Prism 4.0 (San Diego, CA) [45].
Ligand stimulated cAMP signaling
HEK293T cells transfected with WT or mutant hMC3Rs were washed twice with warm Waymouth/BSA 48 h after transfection. Then fresh Waymouth/BSA containing 0.5 mM isobutylmethylxanthine (Sigma-Aldrich) was added and cells were incubated at 37 °C for 15 min. Then, either Waymouth/BSA alone or different concentrations of ligands were added and cells were incubated for another 1 h at 37 °C. Cells were then placed on ice and intracellular cAMP was extracted with 0.5 N percholoric acid containing 180 μg/ml theophylline. The cAMP levels were measured by radioimmunoassay [46, 47]. All determinations were performed in triplicate. Maximal responses (Rmax) and EC50 values were calculated using GraphPad Prism 4.0.
Quantification of hMC3R expression by flow cytometry
Flow cytometry was performed as previously described [45, 48, 49]. Briefly, HEK293 cells were transiently transfected with 1 μg plasmid in 2ml media for each well in 6-well cluster. After 48 h incubation, cells were detached in filtered phosphate buffered saline for immunohistochemistry (PBS-IH) (137 mM NaCl, 2.7 mM KCl, 1.4 mM KH2PO4, 4.3 mM Na2HPO4, pH 7.4), and then precipitated by centrifugation at 500 × g. Cells were fixed using PBS-IH containing 4% paraformaldehyde for 30 min. 1% Triton X-100 treatment for 4 min was applied to permeabilize cells for measuring total receptor expression (this step was omitted when measuring cell surface expression). Cells were blocked with PBS-IH containing 5% BSA for 1 h and then immunostained with primary anti-HA.11 antibody (1:50, Covance, Princeton, NJ) in PBS-IH containing 0.5% BSA for 1 h. Cells were washed with PBS-IH containing 0.5% BSA and the secondary Alexa Fluor 488-conjugated goat anti-mouse IgG (1:2000, Invitrogen, Carlsbad, CA) in PBS-IH containing 0.5% BSA were added under dim light. After 1 h incubation, cells were washed and resuspended in PBS-IH with 0.5% BSA. The expression of mutant hMC3Rs in 10,000 cells from each transfection was quantified by an Accuri flow cytometer with a 488-nm wavelength laser. Cells transfected with pcDNA3.1 empty vector was used to correct for background staining. Mutant expression levels were calculated as percentage of WT hMC3R expression using the following formula: (mutant - empty vector) / (WT - empty vector) × 100% [45, 48, 49].
ERK1/2 phosphorylation assay
The phosphorylated ERK1/2 (pERK1/2) levels were measured by Western blot as described before [36-38, 40]. Briefly, HEK293T cells were plated on gelatin-coated 100 mm dishes and transfected with WT or mutant hMC3R constructs. Approximately 24 h after transfection, cells were starved overnight, and then treated with either 10-5 M α-MSH or Waymouth/BSA alone for 5 min. Proteins were extracted in lysis buffer containing phosphatase and protease inhibitors, separated by 10% SDS-PAGE, and transferred onto PVDF membrane. pERK1/2 and β-tubulin levels were detected with primary rabbit anti-pERK1/2 antibody (1:1000 ~ 1:2000, Cell Signaling Technology, Beverly, MA) and mouse anti-β-tubulin antibody (1:5000 ~ 1:10,000, Developmental Studies Hybridoma Bank at University of Iowa, Iowa City, IA) in Tris-buffered saline with Tween-20 containing 5% BSA, respectively. Then the membranes were incubated with secondary horseradish peroxidase-conjugated donkey anti-rabbit antibody (1:1000 ~ 1:2000, Jackson ImmunoResearch Laboratories, West Grove, PA) and horseradish peroxidase-conjugated donkey anti-mouse antibody (1:5000, Jackson ImmunoResearch Laboratories) in milk at room temperature for 2 h. PVDF membranes were visualized by enhanced chemiluminescence reagent (Pierce, Rockford, IL) and the signals were quantified using ImageJ 1.44 (National Institutes of Health, Bethesda, MD). ERK1/2 phosphorylation levels were expressed as a ratio of pERK1/2 over β-tubulin.
Statistical analyses
Student's t-test was used to determine the significance of differences in the expression, ligand binding, cAMP signaling and pERK1/2 levels between WT and mutant hMC3Rs. Statistical analysis was carried out using GraphPad Prism 4.0.
Results
Quantification of the expression of the novel mutant hMC3Rs using flow cytometry
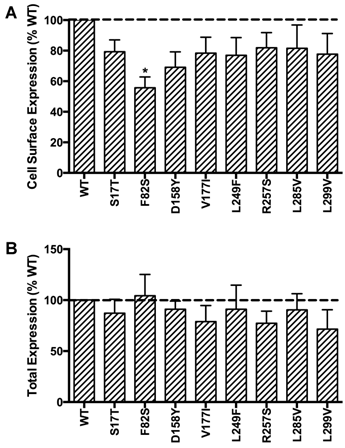
HEK293T cells, which do not express endogenous MC3R, were used as a mammalian expression system in the present study. To study the localization of the novel mutant hMC3Rs and to quantitate their expression levels, flow cytometry was performed. The data of non-permeabilized cells showed that all the mutants had similar cell surface expression as the WT hMC3R except F82S, which had significantly decreased cell surface expression to about 60% of the WT hMC3R (Fig. 2A). All mutants had similar total protein expression levels as the WT hMC3R as shown in permeabilized cells (Fig. 2B).
Quantitative measurement of cell surface (A) or total (B) expression of the novel mutant hMC3Rs by flow cytometry. In panel A, cell surface expression of mutant hMC3Rs were expressed as percentage of WT hMC3R cell surface expression after correction of the nonspecific staining in cells transiently transfected with empty vector. Data were mean ± SEM of 4-8 experiments. Star (*) indicates significantly different from WT hMC3R, P < 0.05. In panel B, total expression of mutant hMC3R were expressed as percentage of WT hMC3R total expression level after correction for the nonspecific staining. Data were mean ± SEM of at least four experiments.
Ligand binding properties of the novel mutant hMC3Rs
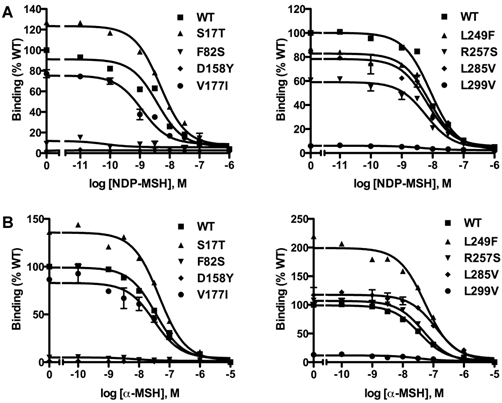
Competitive ligand binding assays were performed with the superpotent analog NDP-MSH [50] and the endogenous agonist α-MSH as competitors. When NDP-MSH was used, mutants F82S, D158Y and L299V had negligible specific binding (Fig. 3A), and their IC50 values could not be determined. All the other novel mutants had similar IC50s as the WT hMC3R (Table 1). When the endogenous agonist α-MSH was used as the competitor (Fig. 3B), the IC50 of F82S, D158Y and L299V could not be determined either. Mutants S17T, L249F and L285V had significantly increased IC50s (decreased binding affinity). The other two mutants had similar IC50s as the WT hMC3R (Table 2).
Ligand binding properties of the new mutant hMC3Rs with NDP-MSH (A) and α-MSH (B) as the ligand. Different concentrations of unlabeled NDP-MSH or α-MSH competitively displaced the binding of 125I-NDP-MSH to hMC3Rs on intact cells. Results are expressed as percentage of WT binding ± range from duplicate determinations within one experiment. All experiments were performed at least three times.
Binding and signaling properties of the 8 new mutant hMC3Rs with NDP-MSH as the ligand.
| hMC3R | NDP-MSH binding | Basal signaling (% WT) | NDP-MSH-stimulated cAMP | ||
|---|---|---|---|---|---|
| IC50 (nM) | RO (% WT) | EC50 (nM) | Rmax (% WT) | ||
| WT | 6.45 ± 0.87 | 100 ± 0 | 100 ± 0 | 1.28 ± 0.49 | 100 ± 0 |
| S17T | 5.73 ± 0.48 | 136.00 ± 15.43 | 107.50 ± 24.68 | 1.24 ± 0.68 | 81.87 ± 17.35 |
| F82S | ND | 8.07 ± 1.45c | 84.05 ± 19.86 | ND | 22.99 ± 2.26c |
| D158Y | ND | ND | 54.05 ± 8.81b | ND | ND |
| V177I | 4.33 ± 1.61 | 82.96 ± 11.96 | 88.01 ± 21.52 | 0.62 ± 0.14 | 61.81 ± 10.02 |
| L249F | 7.74 ± 1.99 | 128.39 ± 23.49 | 69.48 ± 20.70 | 4.98 ± 2.54 | 50.02 ± 2.74b |
| R257S | 5.88 ± 0.12 | 73.38 ± 8.36a | 78.80 ± 13.97 | 3.26 ± 1.62 | 44.05 ± 12.03a |
| L285V | 6.07 ± 1.02 | 93.68 ± 13.78 | 52.40 ± 11.86b | 5.76 ± 2.64 | 29.71 ± 3.22b |
| L299V | ND | 8.65 ± 1.51c | 78.55 ± 15.31 | 14.84 ± 4.29a | 38.53 ± 5.31b |
a Significantly different from the WT hMC3R, P < 0.05.
b Significantly different from the WT hMC3R, P < 0.01.
c Significantly different from the WT hMC3R, P < 0.001.
ND, not detected.
The data are expressed as the mean ± SEM of at least three independent experiments for IC50, EC50, and Rmax, and at least six independent experiments for RO and basal signaling. The basal cAMP was 36.88 ± 5.89 pmol/106 cells and the Rmax was 2526.00 ± 502.11 pmol cAMP/106 cells for the WT hMC3R.
Binding and signaling properties of the 8 new mutant hMC3Rs with α-MSH as the ligand.
| hMC3R | α-MSH binding | α-MSH-stimulated cAMP | ||
|---|---|---|---|---|
| IC50 (nM) | EC50 (nM) | Rmax (% WT) | ||
| WT | 28.49 ± 2.86 | 2.91 ± 1.42 | 100.00 | |
| S17T | 50.89 ± 4.58a | 13.92 ± 11.42 | 102.74 ± 10.19 | |
| F82S | ND | ND | 21.18 ± 6.31b | |
| D158Y | ND | ND | ND | |
| V177I | 51.28 ± 10.25 | 3.10 ± 1.96 | 80.18 ± 11.26 | |
| L249F | 58.66 ± 2.57b | 1.31 ± 0.55 | 55.99 ± 13.71 | |
| R257S | 40.81 ± 3.79 | 3.62 ± 0.35 | 61.78 ± 16.14 | |
| L285V | 124.86 ± 20.05b | 18.90 ± 10.64 | 40.95 ± 11.19a | |
| L299V | ND | 115.71 ± 50.16 | 29.03 ± 8.73a | |
a Significantly different from the WT hMC3R, P < 0.05.
b Significantly different from the WT hMC3R, P < 0.01.
ND, not detected.
The data are expressed as the mean ± SEM of at least three independent experiments. The Rmax was 2188.03 ± 638.76 pmol cAMP/106 cells for the WT hMC3R.
As shown in Table 1, F82S, D158Y and L299V were almost completely devoid of specific binding. R257S exhibited significantly decreased RO to about 70% of WT hMC3R. All the other new mutants had similar RO as the WT hMC3R (Table 1).
cAMP signaling activity of the novel mutant hMC3Rs in response to NDP-MSH or α-MSH stimulation
To investigate whether these eight new missense mutations would affect MC3R signaling, the intracellular cAMP levels stimulated by different ligands were measured. First, cAMP signaling property in response to NDP-MSH was tested. Fig. 4A showed the result of one representative experiment. For mutants F82S and D158Y, their EC50s could not be determined. L299V had significantly increased EC50. S17T and V177I had similar maximal signaling as the WT hMC3R, while the other mutants had significantly reduced maximal signaling, especially D158Y, which was completely inactive (Table 1).
Cyclic AMP signaling properties of the new mutant hMC3Rs with NDP-MSH (A) and α-MSH (B) as the ligand. Intracellular cAMP concentrations were measured as described in Materials and Methods. Results are expressed as the mean ± SEM of triplicate determinations from one experiment. All experiments were performed at least three times.
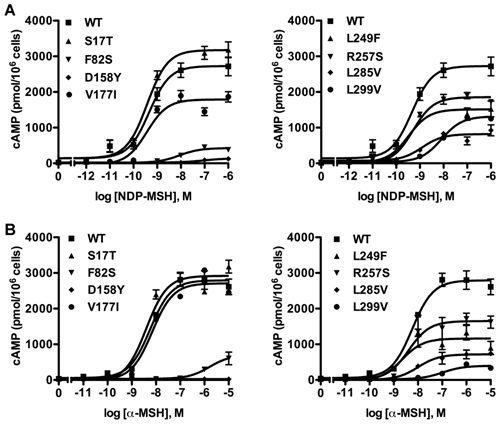
Then the cAMP signaling property was investigated with α-MSH as the ligand (Fig. 4B). Similarly, EC50 of mutants F82S and D158Y could not be determined. D158Y was totally devoid of cAMP signaling. Three mutants (F82S, L285V and L299V) displayed significantly decreased maximal signaling compared to the WT hMC3R. The signaling properties of the remaining mutants were similar to that of the WT hMC3R (Table 2).
Different from the MC4R, the MC3R has little constitutive activity [29]. In the current study, none of the 8 novel mutants had increased basal cAMP levels. Indeed, two mutants (D158Y and L285V) had significantly decreased basal activity compared to the WT hMC3R (Table 1).
ERK1/2 signaling efficacies to endogenous agonist α-MSH
HEK293T cells transiently transfected with all the 22 naturally occurring missense mutations in MC3R identified to date, excluding the two single nucleotide polymorphisms (K6T and I81V), were used to explore the effect of endogenous MC3R agonist α-MSH on ERK1/2 signaling. We found that α-MSH acted as an agonist for MAPK pathway, resulting in a significantly increased pERK1/2 level with a 5.6-fold maximal response for the WT hMC3R (Fig. 5). Fourteen mutants (S69C, A70T, F82S, V177I, I183N, L249F, L249V, R257S, A260V, M275T, L285V, A293T, L299V and I335S) had significant activation of ERK1/2 upon α-MSH stimulation for 5 min (Fig. 5 and Table 3). Although not statistically significant, two mutants (S17T and X361S) had more than 5-fold increase in pERK1/2 levels. The other six mutants (I87T, N128S, M134I, D158Y, T280S, and A297V) had no significant change in pERK1/2 levels. These data demonstrated that α-MSH, known as an agonist for MC3R in the conventional Gs-cAMP pathway, exhibited agonist activity causing significantly elevated pERK1/2 levels for WT and some mutant hMC3Rs. α-MSH caused balanced agonism in Gs-cAMP and ERK1/2 pathways for WT and 15 mutant hMC3Rs, while for the other 7 mutants, α-MSH induced divergent activities (Table 3).
The effect of α-MSH on ERK1/2 signaling in mutant hMC3Rs. HEK293T cells were stimulated with or without 10-5 M α-MSH for 5 min. ERK1/2 phosphorylation was measured as described in Materials and Methods. Panel A showed the representative image of one experiment. Panel B showed the densitometry results of ERK1/2 phosphorylation of the WT and mutant hMC3Rs. Results are expressed as the percentage of the value obtained in non-stimulated cells transfected with the WT hMC3R. Shown are mean ± SEM of at least four experiments. Star (*) indicates significantly different from WT basal level, P < 0.05.
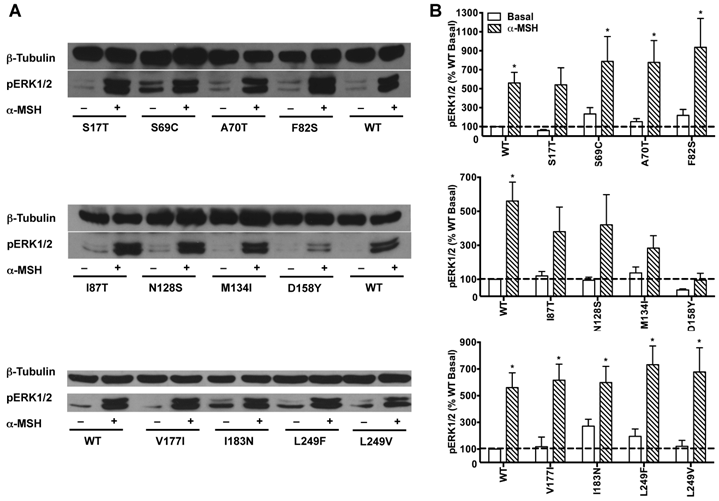
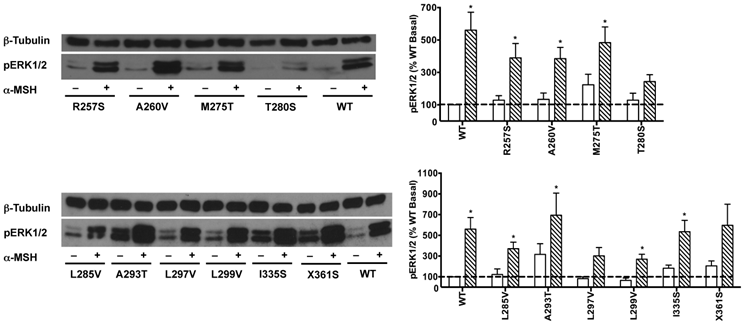
The effect of α-MSH on cAMP and pERK1/2 signaling of WT and mutant hMC3Rs.
| hMC3R | α-MSH | ||
|---|---|---|---|
| cAMP | pERK1/2 | Bias | |
| WT | ↑ | ↑ | No |
| S17T | ↑ | ↑ | No |
| S69C | ↑ | ↑ | No |
| A70T | ↑ | ↑ | No |
| F82S | — | ↑ | Yes |
| I87T | ↑ | — | Yes |
| N128S | ↑ | — | Yes |
| M134I | ↑ | — | Yes |
| D158Y | — | — | No |
| V177I | ↑ | ↑ | No |
| I183N | — | ↑ | Yes |
| L249F | ↑ | ↑ | No |
| L249V | ↑ | ↑ | No |
| R257S | ↑ | ↑ | No |
| A260V | ↑ | ↑ | No |
| M275T | ↑ | ↑ | No |
| T280S | — | — | No |
| L285V | ↑ | ↑ | No |
| A293T | ↑ | ↑ | No |
| L297V | ↑ | — | Yes |
| L299V | ↑ | ↑ | No |
| I335S | — | ↑ | Yes |
| X361S | ↑ | ↑ | No |
“↑”: Denotes increased activity compared to the basal level. “−”: denotes no significant changes between basal and maximal signaling levels. “Bias” column is designed to demonstrate whether α-MSH-stimulated cAMP and pERK1/2 signaling pathways were divergent. “Yes” denotes biased activation of either signaling pathway; “No” denotes balanced cAMP and pERK1/2 signaling.
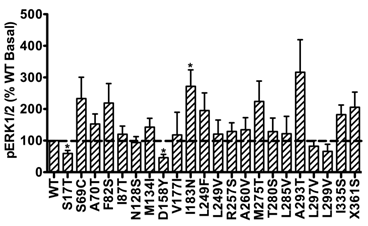
In addition, the basal pERK1/2 levels of the majority of mutant hMC3Rs were similar as that of WT hMC3R. However, S17T and D158Y had significantly decreased basal pERK1/2 levels, while mutant I183N exhibited significantly increased basal pERK1/2 (Fig. 6), indicating constitutive activity in the ERK1/2 signaling cascade.
Basal pERK1/2 signaling of mutant hMC3Rs. Results are expressed as percentage of WT basal pERK1/2 level. Shown are mean ± SEM of at least four experiments. Star (*) indicates significantly different from WT hMC3R, P < 0.05.
Discussion
In this study, we first performed detailed functional characterization of 8 new naturally occurring MC3R mutations (S17T, F82S, D158Y, V177I, L249F, R257S, L285V, and L299V). Among these mutations, S17T, D158Y, V177I, and L299V were identified in unrelated obese carriers; F82S, R257S, and L285V were identified in lean subjects; L249F was identified in both obese and lean subjects [23-25]. All 8 mutants were expressed on the cell surface, but F82S had significantly decreased cell surface expression compared to the WT hMC3R (Fig. 2A). All mutants had similar total expression levels as the WT hMC3R (Fig. 2B). These results showed that F82S was partially retained intracellularly, which is the common cause for the majority of GPCR mutations leading to human diseases [51, 52], and most of the MC4R mutations belong to this class [53].
In addition to partial intracellular retention, F82S was expressed on the cell surface. Ligand binding and cAMP signaling data showed that it had significant defects in both ligand binding and agonist-stimulated cAMP generation (Figs. 3-4 and Tables 1-2). A naturally occurring mutation in MC4R identified from obese patients with binge eating disorder substitutes the cognate Phe with Leu at codon 51, leading to unchanged ligand binding and cAMP signaling [54]. Therefore changing Phe to Leu, which has small change in size, does not affect the MC4R function [54], whereas changing hydrophobic Phe to Ser with polar side chain significantly impaired MC3R trafficking and function, suggesting that the hydrophobic feature of codon 82 might be of importance for MC3R trafficking, ligand binding and cAMP signaling.
D158Y was almost totally devoid of ligand binding and cAMP signaling (Figs. 3-4 and Tables 1-2). The cognate D126 in the MC4R is important for ligand-receptor interaction [55] and our previous site-directed mutagenesis study demonstrated that D158 in the MC3R plays an important role in different aspects of receptor function [45]. In that study, mutating Asp to Glu or Gln at codon 158 resulted in undetectable ligand binding and cAMP signaling in response to NDP-MSH stimulation, and mutating Asp to Glu led to significantly decreased cell surface expression [45]. Combined with the results in the present study, we conclude that D158 was important for receptor expression, ligand binding and cAMP signaling. D158Y might be the pathogenic cause to obesity for the patient harboring the mutation.
L299V had similar expression (Fig. 2) but significantly reduced ligand binding and cAMP signaling compared to the WT hMC3R (Figs. 3-4 and Tables 1-2). Previously, L299V was reported to result in decreased cell surface expression and impaired function of this mutant, suggested to be due to intracellular retention [25]. Zegers et al. used enhanced green fluorescent protein (EGFP)-MC3R fusion proteins to investigate the localization [25]. The addition of GFP can interrupt the intracellular trafficking of receptors [21]. This might be the possible explanation for the abnormal cell surface expression of L299V in the original publication.
According to the alignment of the amino acid sequences of MCRs, the Leu at codon 299 in MC3R is a conserved residue among MCRs (except melanocortin-2 receptor). This residue corresponds to L265 in MC4R [3] and is considered to probably contribute to a hydrophobic binding pocket for agonist to dock [55]. The substitution of L265 in MC4R dramatically alters the affinity and potencies of NDP-MSH and two other agonists [56]. L299 might also be important for ligand binding and subsequent cAMP signaling in the MC3R.
Although the expression (Fig. 2) and binding (Table 1) of L285V were similar to the WT hMC3R, it had significantly decreased maximal cAMP signaling (Tables 1-2), suggesting that this was a cAMP signaling-defective mutant. Mutants L249F and R257S had significantly decreased cAMP generation in response to NDP-MSH but not α-MSH (Tables 1-2). Hence the defect in cAMP signaling pathway for these two mutants was ligand-dependent. In our previous study, we showed another naturally occurring mutation, L249V, has decreased cell surface expression but ligand binding and cAMP signaling is not affected [30]. Altogether, the combined results indicated that L249 might be important for MC3R expression and cAMP signaling.
We also studied ERK1/2 signaling of all 22 naturally occurring MC3R mutations upon α-MSH stimulation. We found that α-MSH induced significant activation of ERK1/2 in the WT hMC3R (Fig. 5), consistent with previous studies when the WT MC3R was stimulated with either NDP-MSH or D-Trp8-γ-MSH as well as in mouse dorsal medial nucleus and ventromedial nucleus [13, 41, 42], although not consistent with another report [31]. The reason for this discrepancy is not clear.
Among the 22 mutations we studied, α-MSH exhibited balanced activity in Gs-cAMP and ERK1/2 cascades in 15 mutants. D158Y and T280S could not activate any of the two signaling pathways (Table 3 and Fig. 5). The other 7 mutants either selectively increased cAMP generation, or selectively activated ERK1/2 phosphorylation upon α-MSH stimulation (Table 3). α-MSH selectively elevated cAMP production for I87T, N128S, M134I, and L297V without significant activation of ERK1/2 cascade, whereas α-MSH selectively induced ERK1/2 phosphorylation for F82S, I183N, and I335S, without increasing cAMP production. Therefore these mutant receptors were biased receptors, in which agonist only stabilizes a restricted subset of conformations activating one signaling pathway [57-59]. In the literature, there are only a few examples of biased receptors from naturally occurring mutations in GPCRs, including the melanocortin-1 receptor and MC4R [40, 60-64]. Our data add to this expanding field.
We also found that I183N displayed significantly increased basal pERK1/2 level (Fig. 6), suggesting constitutive activity in basal pERK1/2 signaling. Traditionally, the constitutive activity of GPCRs is mainly associated with G protein-mediated signaling [32, 65]. Unlike the MC4R, the WT hMC3R has low constitutive activity [29]. No constitutively active naturally occurring mutations in MC3R have been reported. Only one laboratory-generated mutation of MC3R (F347A) was observed to have increased basal cAMP level [47]. However, in the current study, we found I183N, the first MC3R mutation considered to have causal relationship to human obesity, had constitutive ERK1/2 activity. Very few reports exist in the literature on constitutive activation of ERK1/2 by GPCRs. A naturally occurring α1a-adrenergic receptor variant constitutively activates ERK pathway with physiological consequences [66, 67]. Similarly, we observed constitutive ERK1/2 signaling in naturally occurring and lab-generated MC4R mutants [37, 38, 40] (reviewed in [39]). Our current findings added a new layer of the understanding of structure-function relationship of mutant hMC3Rs. Constitutive ERK activity could regulate multiple functions including proliferation and differentiation [68, 69]. The in vivo relevance of the constitutive ERK activity in I183N deserves further investigation.
In summary, detailed functional studies of 8 new naturally occurring mutations in MC3R were performed. D158Y and L299V might be pathogenic for human obesity. F82, D158, L249 and L299 played important roles in different aspects of MC3R functions. The ERK1/2 signaling pathway of WT and mutant hMC3Rs was explored. The results showed that WT hMC3R activated ERK1/2 signaling upon stimulation by α-MSH, and that some mutant hMC3Rs were biased to different intracellular signaling cascades upon α-MSH stimulation, contributing to a better understanding of MC3R pharmacology.
Abbreviations
BSA, bovine serum albumin; ERK1/2, extracellular signal-regulated kinases 1 and 2; GPCR, G protein-coupled receptor; HEK, human embryonic kidney; hMC3R, human MC3R; MAPK, mitogen-activated protein kinase; MC3R, melanocortin-3 receptor; MC4R, melanocortin-4 receptor; MCR, melanocortin receptor; MSH, melanocyte stimulating hormone; NDP-MSH, [Nle4,D-Phe7]-α-melanocyte stimulating hormone; PBS-IH, phosphate buffered saline for immunohistochemistry; RO, receptor occupancy; WT, wild type.
Acknowledgements
The β-tubulin antibody was obtained from Developmental Studies Hybridoma Bank, developed under the auspices of NICHD and maintained by University of Iowa Department of Biology. We thank Allison Church Bird at the Auburn University College of Veterinary Medicine Flow Cytometry Core Facility for her assistance with flow cytometry experiments. This research was funded by the National Institutes of Health grant R15DK077213, American Diabetes Association Grant 1-12-BS212, Auburn University Intramural Grant Program, and Animal Health and Diseases Research Program and Interdisciplinary Grant from College of Veterinary Medicine, Auburn University. Fan Yang was supported by the fellowship from China Scholarship Council of the People's Republic of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gantz I, Fong TM. The melanocortin system. Am J Physiol. 2003;284:E468-E74
2. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571-8
3. Tao YX. The melanocortin-4 receptor: Physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31:506-43
4. Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G. et al. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246-50
5. Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB. et al. Identification of a receptor for γ melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci U S A. 1993;90:8856-60
6. Bagnol D, Lu XY, Kaelin CB, Day HE, Ollmann M, Gantz I. et al. Anatomy of an endogenous antagonist: relationship between Agouti-related protein and proopiomelanocortin in brain. J Neurosci. 1999;19:RC26
7. Jegou S, Boutelet I, Vaudry H. Melanocortin-3 receptor mRNA expression in pro-opiomelanocortin neurones of the rat arcuate nucleus. J Neuroendocrinol. 2000;12:501-5
8. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL. et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480-4
9. Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H. et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97-102
10. Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J. et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518-21
11. Sutton GM, Begriche K, Kumar KG, Gimble JM, Perez-Tilve D, Nogueiras R. et al. Central nervous system melanocortin-3 receptors are required for synchronizing metabolism during entrainment to restricted feeding during the light cycle. FASEB J. 2010;24:862-72
12. Sutton GM, Perez-Tilve D, Nogueiras R, Fang J, Kim JK, Cone RD. et al. The melanocortin-3 receptor is required for entrainment to meal intake. J Neurosci. 2008;28:12946-55
13. Begriche K, Marston OJ, Rossi J, Burke LK, McDonald P, Heisler LK. et al. Melanocortin-3 receptors are involved in adaptation to restricted feeding. Genes Brain Behav. 2012;11:291-302
14. Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113-4
15. Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111-2
16. Hinney A, Schmidt A, Nottebom K, Heibult O, Becker I, Ziegler A. et al. Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J Clin Endocrinol Metab. 1999;84:1483-6
17. Tao YX. Mutations in melanocortin-4 receptor and human obesity. Prog Mol Biol Transl Sci. 2009;88:173-204
18. Hinney A, Volckmar AL, Knoll N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci. 2013;114:147-91
19. Lee YS, Poh LK, Loke KY. A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. J Clin Endocrinol Metab. 2002;87:1423-6
20. Feng N, Young SF, Aguilera G, Puricelli E, Adler-Wailes DC, Sebring NG. et al. Co-occurrence of two partially inactivating polymorphisms of MC3R is associated with pediatric-onset obesity. Diabetes. 2005;54:2663-7
21. Lee YS, Poh LK, Kek BL, Loke KY. The role of melanocortin 3 receptor gene in childhood obesity. Diabetes. 2007;56:2622-30
22. Mencarelli M, Walker GE, Maestrini S, Alberti L, Verti B, Brunani A. et al. Sporadic mutations in melanocortin receptor 3 in morbid obese individuals. Eur J Hum Genet. 2008;16:581-6
23. Calton MA, Ersoy BA, Zhang S, Kane JP, Malloy MJ, Pullinger CR. et al. Association of functionally significant Melanocortin-4 but not Melanocortin-3 receptor mutations with severe adult obesity in a large North American case-control study. Hum Mol Genet. 2009;18:1140-7
24. Mencarelli M, Dubern B, Alili R, Maestrini S, Benajiba L, Tagliaferri M. et al. Rare melanocortin-3 receptor mutations with in vitro functional consequences are associated with human obesity. Hum Mol Genet. 2011;20:392-9
25. Zegers D, Beckers S, de Freitas F, Peeters AV, Mertens IL, Verhulst SL. et al. Identification of three novel genetic variants in the melanocortin-3 receptor of obese children. Obesity (Silver Spring). 2011;19:152-9
26. Tao YX. Mutations in the melanocortin-3 receptor (MC3R) gene: Impact on human obesity or adiposity. Curr Opin Investig Drugs. 2010;11:1092-6
27. Tao YX, Segaloff DL. Functional characterization of melanocortin-3 receptor variants identify a loss-of-function mutation involving an amino acid critical for G protein-coupled receptor activation. J Clin Endocrinol Metab. 2004;89:3936-42
28. Rached M, Buronfosse A, Begeot M, Penhoat A. Inactivation and intracellular retention of the human I183N mutated melanocortin 3 receptor associated with obesity. Biochim Biophys Acta. 2004;1689:229-34
29. Tao YX. Functional characterization of novel melanocortin-3 receptor mutations identified from obese subjects. Biochim Biophys Acta. 2007;1772:1167-74
30. Yang F, Tao YX. Functional characterization of nine novel naturally occurring human melanocortin-3 receptor mutations. Biochim Biophys Acta. 2012;1822:1752-61
31. Daniels D, Patten CS, Roth JD, Yee DK, Fluharty SJ. Melanocortin receptor signaling through mitogen-activated protein kinase in vitro and in rat hypothalamus. Brain Res. 2003;986:1-11
32. Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381-416
33. Patten CS, Daniels D, Suzuki A, Fluharty SJ, Yee DK. Structural and signaling requirements of the human melanocortin 4 receptor for MAP kinase activation. Regul Pept. 2007;142:111-22
34. Pearson G, Robinson F, Beers Gibson T, Xu B, Karandikar M, Berman K. et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153-83
35. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911-2
36. Huang H, Tao YX. Pleiotropic functions of the transmembrane domain 6 of human melanocortin-4 receptor. J Mol Endocrinol. 2012;49:237-48
37. Mo XL, Yang R, Tao YX. Functions of transmembrane domain 3 of human melanocortin-4 receptor. J Mol Endocrinol. 2012;49:221-35
38. Mo XL, Tao YX. Activation of MAPK by inverse agonists in six naturally occurring constitutively active mutant human melanocortin-4 receptors. Biochim Biophys Acta. 2013;1832:1939-48
39. Tao YX. Constitutive activity in melanocortin-4 receptor: biased signaling of inverse agonists. Adv Pharmacol. 2014;70:135-54
40. He S, Tao YX. Defect in MAPK signaling as a cause for monogenic obesity caused by inactivating mutations in the melanocortin-4 receptor gene. Int J Biol Sci. 2014;10:1128-37
41. Chai B, Li JY, Zhang W, Ammori JB, Mulholland MW. Melanocortin-3 receptor activates MAP kinase via PI3 kinase. Regul Pept. 2007;139:115-21
42. Huang H, Tao YX. Functions of the DRY motif and intracellular loop 2 of human melanocortin 3 receptor. J Mol Endocrinol. 2014;53:319-30
43. Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544-51
44. Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745-52
45. Wang SX, Fan ZC, Tao YX. Functions of acidic transmembrane residues in human melanocortin-3 receptor binding and activation. Biochem Pharmacol. 2008;76:520-30
46. Fan ZC, Sartin JL, Tao YX. Molecular cloning and pharmacological characterization of porcine melanocortin-3 receptor. J Endocrinol. 2008;196:139-48
47. Tao YX, Huang H, Wang ZQ, Yang F, Williams JN, Nikiforovich GV. Constitutive activity of neural melanocortin receptors. Methods Enzymol. 2010;484:267-79
48. Fan ZC, Tao YX. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J Cell Mol Med. 2009;13:3268-82
49. Wang ZQ, Tao YX. Functional studies on twenty novel naturally occurring melanocortin-4 receptor mutations. Biochim Biophys Acta. 2011;1812:1190-9
50. Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB. et al. 4-Norleucine, 7-D-phenylalanine-α-melanocyte-stimulating hormone: a highly potent α-melanotropin with ultralong biological activity. Proc Natl Acad Sci U S A. 1980;77:5754-8
51. Tao YX. Inactivating mutations of G protein-coupled receptors and diseases: Structure-function insights and therapeutic implications. Pharmacol Ther. 2006;111:949-73
52. Tao YX, Conn PM. Chaperoning G protein-coupled receptors: From cell biology to therapeutics. Endocr Rev. 2014;35:602-47
53. Tao YX. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol Cell Endocrinol. 2005;239:1-14
54. Tao YX, Segaloff DL. Functional analyses of melanocortin-4 receptor mutations identified from patients with binge eating disorder and nonobese or obese subjects. J Clin Endocrinol Metab. 2005;90:5632-8
55. Yang YK, Fong TM, Dickinson CJ, Mao C, Li JY, Tota MR. et al. Molecular determinants of ligand binding to the human melanocortin-4 receptor. Biochemistry. 2000;39:14900-11
56. Pogozheva ID, Chai BX, Lomize AL, Fong TM, Weinberg DH, Nargund RP. et al. Interactions of human melanocortin 4 receptor with nonpeptide and peptide agonists. Biochemistry. 2005;44:11329-41
57. Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP. et al. β-arrestin- but not G protein-mediated signaling by the "decoy" receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628-32
58. Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17:126-39
59. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179-97
60. Grasberger H, Van Sande J, Hag-Dahood Mahameed A, Tenenbaum-Rakover Y, Refetoff S. A familial thyrotropin (TSH) receptor mutation provides in vivo evidence that the inositol phosphates/Ca2+ cascade mediates TSH action on thyroid hormone synthesis. J Clin Endocrinol Metab. 2007;92:2816-20
61. Herraiz C, Jimenez-Cervantes C, Zanna P, Garcia-Borron JC. Melanocortin 1 receptor mutations impact differentially on signalling to the cAMP and the ERK mitogen-activated protein kinase pathways. FEBS Lett. 2009;583:3269-74
62. Büch TR, Heling D, Damm E, Gudermann T, Breit A. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines agouti-related protein as a biased agonist. J Biol Chem. 2009;284:26411-20
63. Leach K, Wen A, Davey AE, Sexton PM, Conigrave AD, Christopoulos A. Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology. 2012;153:4304-16
64. Sbai O, Monnier C, Dode C, Pin JP, Hardelin JP, Rondard P. Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations. FASEB J. 2014;28:3734-44
65. Tao YX. Constitutive activation of G protein-coupled receptors and diseases: Insights into mechanism of activation and therapeutics. Pharmacol Ther. 2008;120:129-48
66. Oganesian A, Yarov-Yarovoy V, Parks WC, Schwinn DA. Constitutive coupling of a naturally occurring human alpha1a-adrenergic receptor genetic variant to EGFR transactivation pathway. Proc Natl Acad Sci U S A. 2011;108:19796-197801
67. Kleine-Brueggeney M, Gradinaru I, Babaeva E, Schwinn DA, Oganesian A. Alpha1a-adrenoceptor genetic variant induces cardiomyoblast-to-fibroblast-like cell transition via distinct signaling pathways. Cell Signal. 2014;26:1985-97
68. Bourcier C, Griseri P, Grepin R, Bertolotto C, Mazure N, Pages G. Constitutive ERK activity induces downregulation of tristetraprolin, a major protein controlling interleukin8/CXCL8 mRNA stability in melanoma cells. Am J Physiol Cell Physiol. 2011;301:C609-C18
69. Monick MM, Powers LS, Barrett CW, Hinde S, Ashare A, Groskreutz DJ. et al. Constitutive ERK MAPK activity regulates macrophage ATP production and mitochondrial integrity. J Immunol. 2008;180:7485-96
Author contact
![]() Corresponding author: Ya-Xiong Tao, PhD. Department of Anatomy, Physiology and Pharmacology, 212 Greene Hall, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, United States. Tel: 01-334-844-5396 FAX: 01-334-844-5388 Email: taoyaxiedu.
Corresponding author: Ya-Xiong Tao, PhD. Department of Anatomy, Physiology and Pharmacology, 212 Greene Hall, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, United States. Tel: 01-334-844-5396 FAX: 01-334-844-5388 Email: taoyaxiedu.