Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(5):525-535. doi:10.7150/ijbs.10927 This issue Cite
Research Paper
NOD2 is Involved in the Inflammatory Response after Cerebral Ischemia-Reperfusion Injury and Triggers NADPH Oxidase 2-Derived Reactive Oxygen Species
Huiqing Liu1, Xinbing Wei1, Lingjun Kong1, Xiaoqian Liu1,2, Li Cheng1, Shi Yan3, Xiumei Zhang1, Lin Chen1 ![]()
1. Department of Pharmacology, School of Medicine, Shandong University, Wenhua West Road 44, Jinan, Shandong 250012, P.R. China.
2. Key Laboratory of Molecular Pharmacology and Drug Evaluation (Ministry of Education of China), School of Pharmacy, Yantai University, Qingquan Road 30, Yantai, Shandong 264005, P.R. China.
3. Department of Obstetrics and Gynaecology, Qilu Hospital of Shandong University, Wenhua West Road 107, Jinan, Shandong 250012, P.R. China.
Received 2014-10-28; Accepted 2015-2-23; Published 2015-3-25
Abstract

Background: Increasing evidences suggest that innate immunity is involved in cerebral ischemia-reperfusion (I/R) injury, but the liable innate immune receptors have not been completely elucidated. Here, we explored the role of the nucleotide-binding oligomerization domain (NOD)2, a member of the cytosolic NOD-like receptor family, in acute focal cerebral I/R injury.
Methods: An in vivo middle cerebral artery occlusion (MCAO) model that in wild type (WT) and NOD2 deficient (NOD2-/-) mice and in vitro model of oxygen glucose deprivation and reoxygenation (OGD/R) in cultured primary microglia and astrocytes were used to investigate the expression of NOD2 and explore the roles of NOD2 in ischemic stroke.
Results: Our results showed that NOD2 expression was significantly increased in microglia and astrocytes in response to the I/R insult. Pretreatment with muramyl dipeptide, an extrinsic ligand of NOD2, significantly increased the infarct volume and neurological dysfunction in mice subjected to MCAO. Genetic ablation of the NOD2 gene significantly improved stroke outcomes and reduced inflammation, as evidenced by a lower expression of the pro-inflammatory cytokines IL-1β, IL-6 and TNFα in conjunction with attenuated activation of nuclear factor κB (NF-κB), p38 mitogen activated protein kinases (MAPK) and JNK. Moreover, NOD2 deficiency prevented the upregulation of the NADPH oxidase (NOX) 2 and ROS generation induced by I/R. Mechanistically, NOD2-induced production of IL-6 in primary cultured microglia was mediated through activation of NOX2.
Conclusions: This study showed the contribution of NOD2 to inflammatory response and provided direct evidence that NOX2-mediated oxidative stress as an important target molecule linked NOD2 to inflammatory damage in ischemic stroke. Pharmacological targeting of NOD2-mediated inflammatory response at multiple levels may help design a new approach to develop therapeutic strategies for prevention of deterioration of cerebral function and for the treatment of stroke.
Keywords: Nucleotide-binding oligomerization domain (NOD)2, cerebral ischemia-reperfusion, NADPH oxidase, NF-κB, MAPK, inflammation.
Introduction
Acute ischemic stroke is the second leading cause of death and the most frequent cause of permanent disability in adults worldwide [1]. Although reperfusion is critical for ischemic brain tissue to restore normal function, it can paradoxically result in secondary damage, called ischemia-reperfusion (I/R) injury. Increasing evidences indicate that the post-ischemic inflammatory response plays a detrimental role in stroke outcome [2, 3]. Thus, modulation of inflammatory signaling may be a useful therapeutic approach to treat acute stroke.
Innate immunity is not only the first line of defense against infection, but also is implicated in a number of sterile inflammatory events, including cerebral injury [4, 5]. The innate immunity depends on transmembrane toll-like receptors (TLRs) and cytoplasmic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) to induce inflammatory responses. The NLRs have recently emerged as central regulators of immunity and inflammation associated with human diseases [6, 7]. Recently, it was reported that the inflammasome complex is formed after focal brain ischemia and inhibition of NLRP1 by a neutralizing antibody reduced the detrimental consequences of post-ischemic inflammation [8]. NLRP3 deficiency also ameliorates neurovascular damage in experimental ischemic stroke [9]. Another two members of the NLR family, NOD1 and NOD2, have also been studied with regards to inflammatory diseases recently. NOD1 and NOD2, via their caspase recruitment domain (CARD), signal to the nuclear factor κB (NF-κB) and mitogen activated protein kinases (MAPKs) pathways [10, 11]. In particular, the importance of NOD2 in inflammatory homeostasis is underscored by the observation that variants in CARD15 gene are associated with susceptibility to Crohn's disease [12, 13]. Moreover, we proved that enhanced NOD2-mediated innate immunity is involved in atherosclerosis by up-regulating the prostaglandin E2 pathway [14]. NOD2 has also been demonstrated to be up-regulated in microglia [15] and astrocytes [16] following bacterial infection [17]. However, little is known about the role of NOD2 in cerebral I/R. Therefore, in the present study, we used an in vivo middle cerebral artery occlusion (MCAO) model in wild type (WT) and NOD2 deficient mice and in vitro model of oxygen-glucose deprivation and reoxygenation (OGD/R) in cultured cells to investigate the expression of NOD2 in ischemic brains and further explore the relevance of NOD2 mediated signaling in cerebral I/R injury and inflammation.
Materials and Methods
Animals
NOD2-deficient (NOD2-/-) mice on a C57BL/6 background and wild type (WT) C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed under specific pathogen free conditions with a 12 h light/dark cycle and with free access to food and water. All studies were performed following institutional approval in accordance with Shandong University Guide for the Care and Use of Laboratory Animals. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Shandong University (Permit Number: ECAESDUSM2012016). All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Model for transient focal cerebral ischemia
Transient MCAO was induced in both NOD2-/- and WT mice (25-28 g) as previously described by Jasmin [18] and Yang [9]. A successful occlusion was indicated by a severe reduction in the regional cerebral blood flow to < 20% of the baseline by a laser-Doppler flow meter (Perimed, Sweden) using a probe attached to the skull in the area of cerebral cortex supplied by the MCA. After 2 h of MCAO, the monofilament was carefully retracted to allow reperfusion confirmed by an immediate increase in regional cerebral blood flow. Rectal temperature was maintained at 36.5-37.5°C with a homeothermic blanket throughout the procedure until complete recovery of the mice from anesthesia. For muramyl dipeptide (MDP) group, an extrinsic ligand of NOD2, MDP was intraventricularly administered to mice 30 min before MCAO. MDP (200 μM in normal saline) or the vehicle (normal saline) was injected using a Hamilton syringe (3 μL, 0.4 mm posterior and 1.0 mm lateral to the bregma and 2.2 mm from the duramater). Mice were anesthetized and then decapitated after reperfusion at 2 h, 6 h, 12 h, 24 h or 48 h. The sham animals were subjected to the same procedure except for the MCAO.
Infract volume and neurological function assessment
Stroke outcomes were evaluated at 24 h after reperfusion by a blinded investigator using 4-point grade scale neurological scoring system [19]. Following neurological examination, mice were euthanized and the brains were immediately removed. 2-mm brain coronal sections were made and stained by 2,3,5-triphenyltetrazolium chloride (TTC). The staining images were recorded by a digital camera and quantified by C imaging 1280 software. The infarct volume was numerically integrated across each section and over the entire ipsilateral hemisphere[19]. Animals experienced a subarachnoid hemorrhage were excluded from the study.
Morphological examinations
The brains were fixed with 4% paraformaldehyde and subsequently embedded in paraffin and cut into 5-μm-thickness sections. The sections were stained with haematoxylin and eosin (H&E) to observe the neural morphology in the cerebral cortex and hippocampus.
Measurement of reactive oxygen species (ROS)
NADPH oxidase (NOX) activity was determined by measurement of ROS production in brain or cell homogenates. A modified dihydroethidium fluorescence spectrometric assay was used to evaluate ROS production as previously described by Yi [20].
Primary Cell Cultures and treatment
Primary microglia and astrocytes were isolated and cultured as described by del Zoppo GJ [21]. The purity of microglia and astrocytes were evaluated by immunofluorescent staining using antibodies against CD11b (Abcam, Cambridge, UK), and glial fibrillary acidic protein (GFAP, Chemicon, Temecula, CA, USA), respectively.
For OGD/R experiments, cells were incubated in OGD buffer (51 mM NaCl, 65 mM K-gluconate, 0.13 mM CaCl2, 1.5 mM MgCl2, 10 mM HEPES pH 6.8, penicillin and streptomycin). The buffer was deoxygenated using nitrogen for 30 min before the cell treatment. Cells were incubated for 1 h at 37°C in a humidified incubator set to 5% CO2 and 95% N2. Reoxygenation was induced by changing the media and returning the cells to the regular humidified incubator with 5% CO2 and ambient O2 concentration (non-hypoxic conditions). Control cells were incubated in regular media in non-hypoxic conditions. For drug treatment, MDP (10 μg/ml) was added at the beginning of reoxygenation. Apocynin (100 μM) was added 30 min before OGD.
Immunofluorescence staining
Staining was performed on brain sections and cultured cells as previously described [18]. Primary polyclonal antibody NOD2 (sc-30199, Santa Cruz Biotechnology; 1:100 dilution) was used in this study. To determine the lineage of NOD2-positive cells, an anti-neutron nuclei (NeuN) antibody (Invitrogen, Carlsbad, CA; 1:200 dilution) was used to identify neurons, An anti-GFAP antibody (Chemicon; 1:200 dilution) was used to identify astrocytes, an anti-CD11b antibody (Abcam; 1:200 ) and an anti-Iba-1 antibody (Proteintech; 1:100 dilution) were respectively used to characterize microglia/macrophages. Following immunofluorecence, an antifading solution containing 200 mg/ml of DAPI was used to mount the sections.
RNA extraction and real-time RT-PCR
RNA extraction and reverse-transcription were performed as described previously [22]. Quantitative real time-PCR analysis was performed with SYBR green (Invitrogen Applied Biosystems, Carlsbad, CA, USA) using a Bio-Rad iCycler system (Bio-Rad, Hercules, CA, USA). The mRNA levels of target gene were normalized by the housekeeping gene β-actin. Relative expression was determined by the 2-ΔΔCT method. The specific primers for target and housekeeping genes in this study are listed in Table 1.
List of primer sets for SYBR-Green based real time PCR.
| Target gene | Sequence of forward primer (5'-3') | Sequence of reverse primer (5'-3') |
|---|---|---|
| β-actin | GAATTGCTATGTGTCTGGGT | CATCTTCAAACCTCCATGATG |
| NOD2 | GCCTTCCTTCTACAGCACGT | TGGCAGGGCTCTTCTGCAAG |
| IL-1β | ACCTTCCAGGATGAGGACATGA | AACGTCACACACCAGCAGGTTA |
| IL-6 | ACAACCACGGCCTTCCCTAC | CATTTCCACGATTTCCCAGA |
| TNFα | GTGAAGGGAATGGGTGTT | GGTCACTGTCCCAGCATC |
Measurements of TNFα, IL-1β and IL-6 in the supernatants of the ipsilateral hemisphere
TNFα, IL-1β and IL-6 in brain homogenate supernatants were measured by ELISA (R&D Systems, Abingdon, UK) following the manufacturer's protocol.
Western blot analysis
Western blotting was performed as previously reported [23]. Primary antibodies anti-NOD2 (Santa Cruz Biotechnology, Inc. Dallas, TX, USA), anti-NADPH oxidase 2 (NOX2 , BD Transduction Laboratories, Lexington, KY, USA), anti-phospho-NF-κB p65, anti-IκBα, anti-JNK, anti-phospho-JNK, anti-p38, anti-phospho-p38 MAPK (Cell Signaling Technology, Beverly, MA, USA), anti-ERK1/2 and anti-phospho-ERK1/2 (Sigma-Aldrich, St. Louis, MO, USA) were used in this study. To document the loading controls, the membrane was reprobed with a primary antibody against the housekeeping proteins GAPDH or β-actin.
Statistics
All data are presented as mean ± S.E.M. The data of the neurological deficits and cerebral infarct volumes were analyzed by the Kruskal-Wallis test followed by Dunn's test. One-way analysis of variance (ANOVA) and Tukey's multiple comparisons test were used to evaluate the significance of differences in other data. P<0.05 was considered statistically significance.
Results
The NOD2 expression was up-regulated in the mouse brain subjected to focal cerebral ischemia reperfusion
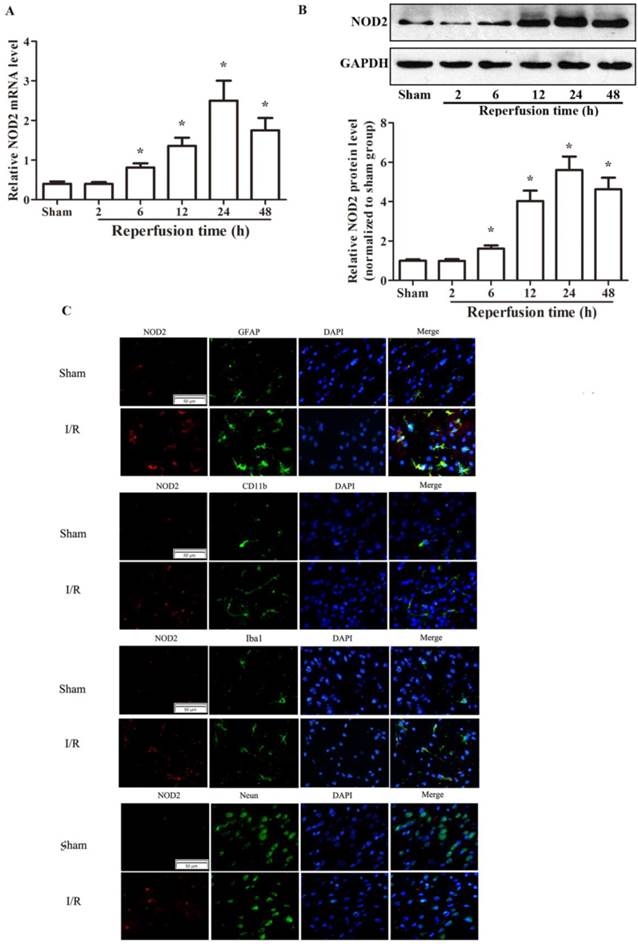
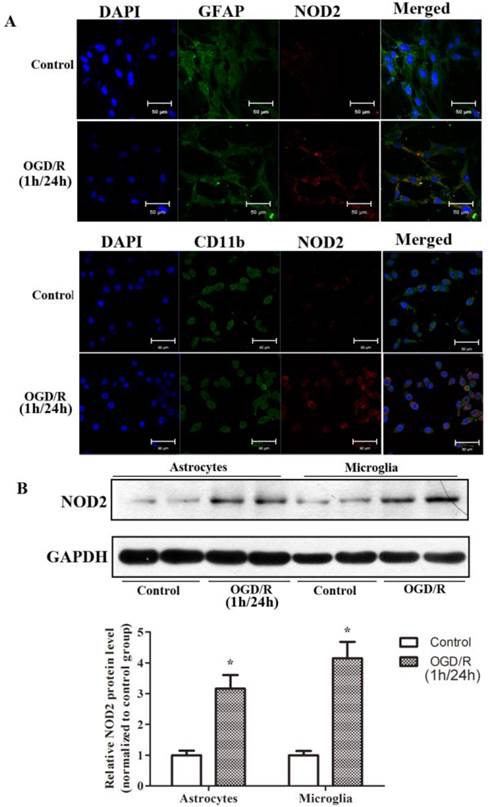
Firstly, by real-time RT-PCR (Fig. 1A) and western blotting (Fig. 1B) analyses, we found that NOD2 expression was markedly enhanced in the ischemic penumbra with a peak expression at 24 h after reperfusion. We further found that NOD2 was significant upregulated in microglia/macrophages and astrocytes by double immunofluorescent analysis (Fig. 1C), which was further confirmed by double immunofluorescence staining (Fig. 2A) and western blot analysis (Fig.2B) in primary cultured microglia and astrocytes subjected to 1 h OGD and 24 h reoxygenation in vitro.
The expression of NOD2 in wild type (WT) mice was increased after cerebral ischemia-reperfusion injury. (A) Relative quantitative mRNA levels of NOD2 were determined by real-time RT-PCR analysis in penumbral cortex from WT mice after 2 h occlusion of the middle cerebral artery (MCAO) and 2 h, 6 h, 12 h, 24 h, 48h reperfusion. (B) Western blot analysis of NOD2 protein levels in the penumbral cortex from WT mice after 2 h MCAO and 2 h, 6 h, 12 h, 24 h and 48 h reperfusion. Results are representative of six independent experiments. *P<0.05, I/R WT mice vs. sham-operated WT mice. (C) Representative images of double immunolabeling for NOD2 and glial fibrillary acidic protein antibody (GFAP, astrocyte marker), CD11b (myeloid cell marker), Iba-1 (microglial/macrophage marker) or NeuN antibody (neuron marker) in the penumbral cortex from WT mice after 2 h MCAO and 24 h reperfusion. DAPI indicates 4',6-diamidino-2-phenylindole. Scale bars: 50 μm.
The expressions of NOD2 in astrocytes and microglia cells were upregulated after 1 h oxygen-glucose deprivation (OGD) and 24 h reperfusion in vitro. (A) Immunofluorescent staining indicating that NOD2 is expressed and enhanced by OGD both in microglial cells and in astrocytes, scale bars: 50 μm. (B) Western blot analysis of NOD2 expression in astrocytes and microglial cells subjected to 1 h OGD and 24 h reperfusion in vitro. Results are representative of four independent experiments. *P<0.05, OGD/R group compare with control group.
Stimulation of NOD2 exacerbated stroke outcomes
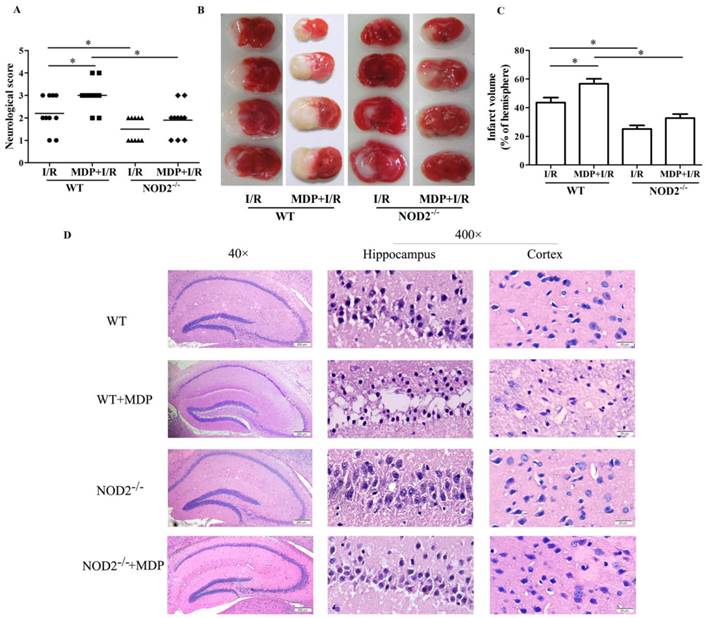
Next, we determined if the NOD2 signaling pathway was involved in I/R insult. Pretreatment with MDP showed worse neurological dysfunctions (Fig. 3A) and larger infarction volume (Fig. 3B and 3C) as compared with WT mice at 24 h after reperfusion. To further assess the role of NOD2 stimulation in ischemia-evoked neuronal injury, H&E staining was used to show morphological features of injured neurons in hippocampus and cerebral cortex. It was found that the neuronal damage was exacerbated in mice pretreated with MDP (Fig. 3D). Collectively, these results implicate that the activation of NOD2 is detrimental to the brain during ischemia.
NOD2 stimulation exacerbated stroke outcomes and NOD2 deficiency improved stroke outcomes. Wild type (WT) and NOD2 deficient (NOD2-/-) mice were suffered from 2 h occlusion of the middle cerebral artery (MCAO) and 24 h reperfusion. For muramyl dipeptide (MDP) group, MDP, an extrinsic ligand of NOD2 was intraventricularly administered to mice 30 min before MCAO. (A) Neurological deficit scores (B) Representative photographs of coronal brain sections following infarction, stained with 2, 3, 5-triphenyltetrazolium chloride. Red tissue is healthy; white tissue is infarcted. (C) Summary of cerebral infarct volume in brains. The infarct volume was expressed as the percentage of the contralateral hemispheric area. (D) Representative photomicrographs of H&E staining in the cortex and hippocampus. For magnification ×40, scale bars: 200 μm; for magnification ×400, scale bars: 20 μm. Results are representative of eight independent experiments. *P<0.05, compared with indicated groups.
NOD2 deficiency alleviated I/R injury
Genetic deletion of NOD2 resulted in a significant decrease in neurological scores in comparison with WT mice at 24 h after reperfusion (Fig. 3A). In accordance, NOD2-/- mice also had a significantly smaller infarct volume (Fig. 3B and 3C) (11.5% reductions vs the control, P<0.05); I/R-induced neuronal damage and loss was consistently attenuated in NOD2-/-mice (Fig. 3D). Moreover, there were no statistical differences in the neurological scores, infarct volume and neuronal damage between the MDP and I/R groups in NOD2-/-mice. However, compared to WT mice, NOD2-/-mice pretreated with MDP had better stroke outcomes. Collectively, these results implicate that NOD2 deficiency could protect the brain against I/R injury.
NOD2 deficiency attenuated the I/R induced inflammation
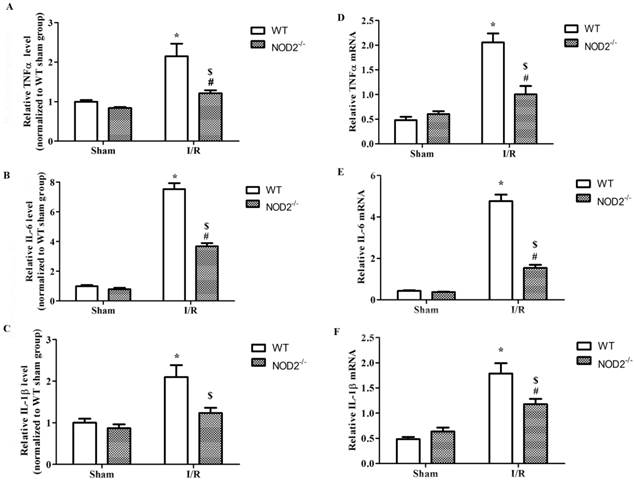
To gain the mechanistic insights into the role of NOD2 in cerebral I/R injury, the proinflammatory cytokine responses were determined. The data showed that brain TNFα, IL-1β and IL-6 concentrations (Fig. 4A-4C) and mRNA expression (Fig. 4D-4F) were induced at 24 h after the reperfusion insults in WT mice. Compared to the WT mice, the up-regulation of TNFα, IL-1β and IL-6 was significantly decreased in the NOD2-/- mice.
NOD2 deficiency reduced the production of inflammatory cytokines after cerebral ischemia-reperfusion injury. (A-C) The levels of proinflammatory cytokines, including IL-6, IL-1β and TNFα, in penumbral cortex were determined by ELISA at 24 h after reperfusion. (D-F) Relative quantitative mRNA levels of IL-6, IL-1β and TNFα in penumbral cortex were determined by real-time RT-PCR analysis at 24 h after reperfusion. Results are representative of six independent experiments. * P<0.05, vs. sham-operated WT mice; # P<0.05, vs. sham-operated NOD2-/- mice; $ P< 0.05, vs. I/R WT mice.
Activation of NF-κB and p38 MAPK, JNK mediated NOD2-induced inflammation in cerebral I/R injury
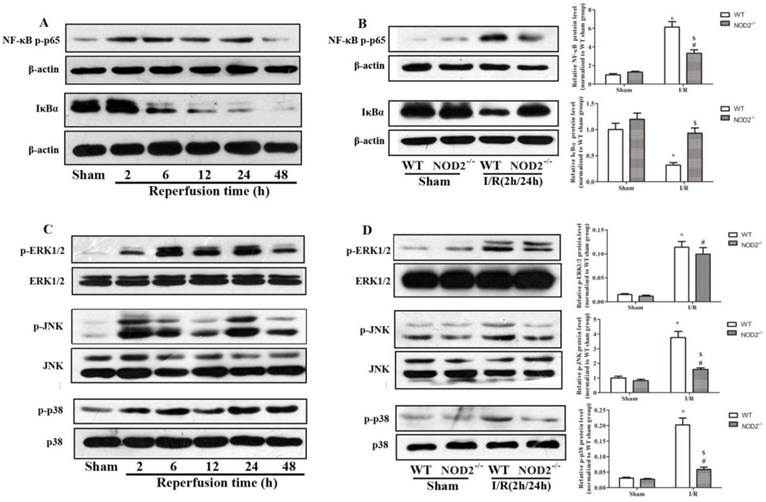
The NF-κB signaling pathway plays an important role in the inflammatory process following the I/R injury [24]. We assessed the I/R-induced activation of NF-κB in the absence of NOD2. As shown in Fig. 5A, the NF-κB pathway was markedly activated as evidenced by the phosphorylation of NF-κB subunit p65 and degradation in IκBα in the brain of WT mice in response to I/R insult. These changes in the NF-κB pathway were significantly inhibited in NOD2-/- mice subjected to 2 h of MCAO and 24 h of reperfusion (Fig. 5B). We also assessed I/R-injury induced changes in the MAPKs signaling pathway, including extracellular signal-regulated kinase (ERK) 1/2, Jun N-terminal kinase (JNK) and p38 MAPK. The results showed that cerebral I/R also resulted in strong phosphorylations of ERK1/2, JNK and p38 MAPK in the WT mice (Fig. 5C). However, I/R-induced activations of JNK and p38 MAPK were remarkably suppressed in NOD2-/- mice, and the activation of ERK was obviously unaffected (Fig. 5D).
NOD2 deficiency inhibited NF-κB and MAPK signaling activation after cerebral ischemia-reperfusion injury. (A and C) p-p65, IκBα, p-ERK , p-JNK and p-p38 MAPK protein expressions in the penumbral cortex from wild type (WT) mice at 2 h, 6 h, 12 h, 24 h and 48 h after reperfusion. (B and D) p-p65, IκBα, p-ERK , p-JNK and p-p38 MAPK protein expressions in the penumbral cortex from WT and NOD2-/- mice at 24 h after reperfusion. Statistical results from the densitometric measurements after normalization were calculated as the mean ± S.E.M. Results are representative of six independent experiments. * P<0.05, vs. sham-operated WT mice; # P<0.05, vs. sham-operated NOD2-/- mice; $ P<0.05, vs. I/R WT mice.
NOX2-mediated oxidative stress contributed to NOD2-induced inflammatory responses in cerebral I/R injury
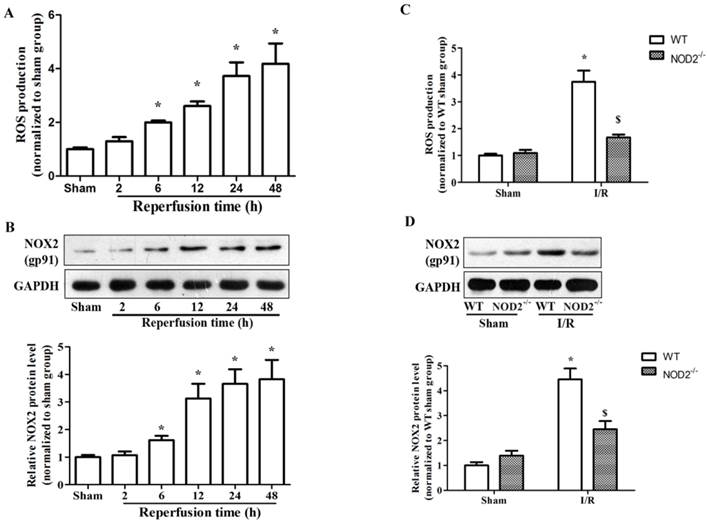
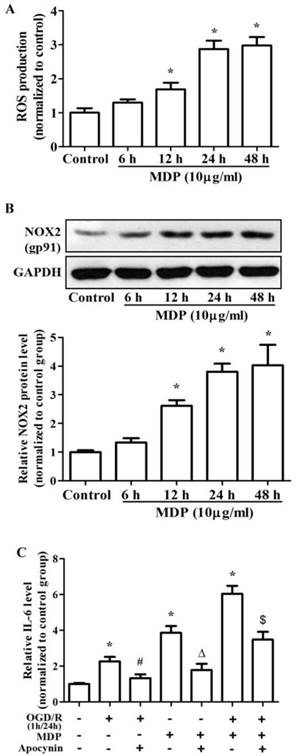
It has been demonstrated that intracellular ROS is required in TLRs - dependent inflammatory responses [25-27]. However, it is unclear whether NOD2 signaling mediates ROS production and whether this mechanism contributes to cerebral I/R injury. In an in vivo study, consistent with the up-regulation of NOD2, the ROS production and expression of NADPH oxidase subunit NOX2 were dramatically increased after reperfusion (Fig. 6A and 6B). Interestingly, we found that the expression and activation of NOX2 were significantly inhibited in the NOD2-/- mice compared to WT mice (Fig. 6C and 6D). In an in vitro study, WT microglia was treated with MDP, a selective agonistic ligand of NOD2, and we found that MDP significantly induced NOX2 expression and ROS production in a time-dependent manner (Fig. 7A and 7B). To elucidate the role of NADPH oxidase on the NOD2 induced IL-6 production further, a well-known NADPH oxidase activity inhibitor, apocynin was used in this study. As shown in Fig. 7C, IL-6 production was markedly enhanced by OGD and MDP, and apocynin significantly inhibited the increase of IL-6 induced by OGD and MDP. The results suggest that NADPH oxidase activation is involved in the NOD2-mediated inflammatory response after cerebral I/R injury.
NOD2 deficiency inhibited NADPH oxidase activity and NOX2 expression after cerebral ischemia-reperfusion injury. (A) Summarized data showing NADPH oxidase activity (reactive oxygen species production) in the penumbral cortex from WT mice after 2 h MCAO and 2 h, 6 h, 12 h, 24 h and 48 h reperfusion. (B) Western blot analysis of NOX2 protein levels in the penumbral cortex from WT mice after 2 h MCAO and 2 h, 6 h, 12 h, 24 h, 48 h reperfusion. (C) Summarized data showing NADPH oxidase activity in the penumbral cortex from NOD2 deficiency mice at 24 h after reperfusion. (D) Western blot analysis of NOX2 protein levels in the penumbral cortex from NOD2 deficiency mice at 24 h after reperfusion. Results are representative of six independent experiments. * P<0.05, vs. sham-operated WT mice; # P<0.05, vs. sham-operated NOD2-/- mice; $ P< 0.05, vs. I/R WT mice.
The NADPH oxidase inhibitor apocynin attenuates IL-6 generation in response to MDP/OGD in microglia. (A) Summarized data showing that MDP (10 μg/ml) significantly activated NADPH oxidase in a time-dependent manner. (B) Representative Western blot and quantified data showing that MDP (10 μg/ml) significantly induced NOX2 expression in a time-dependent manner. (C) Summarized data showing that apocynin attenuated MDP/OGD stimulated IL-6 generation in microglia. Results are representative of four independent experiments. *P<0.05, vs. control group; # P<0.05, vs. OGD/R group; ΔP<0.05, vs. MDP group, $ P<0.05, vs. OGD+MDP group.
Discussion
Here, we reported that NOD2 contributed to cerebral injury by NOD2-mediated inflammatory signaling in mice after ischemic stroke. We further demonstrated that NOX2-dependent oxidative stress was an important target molecule links NOD2 to inflammatory damage in ischemic stroke.
There is an acute inflammation phase and a subsequent persistent phase of inflammation following cerebral I/R injury. The acute inflammation is characterized by rapid activation of resident cells (mainly microglia), production of pro-inflammatory mediators, and infiltration of various types of hematopoietic inflammatory cells into the ischemic brain tissue [28]. Although infiltrated immune cells contribute to inflammatory responses in the brain, growing evidences suggest that resident glial cells play important roles in the initiation and progression of inflammation following I/R insult [29, 30]. NOD2 has been demonstrated to be up-regulated in microglia and astrocytes and play an important role in the inflammatory responses following central nervous system bacterial infection [17]. We wondered whether NOD2 is also induced in cerebral resident cells after I/R. We then detected the expression of NOD2 in three major types of cerebral cells including microglia, astrocytes and neurons. In this study, our data clearly showed that the expression of NOD2 was rapidly and markedly up-regulated in response to the I/R insult in microglia and astrocytes in ischemic brain rather than in neurons, indicating that NOD2 from resident glial cells, microglia and astrocytes may play an important role in the regulation of immune responses in cerebral ischemia injury.
To define the role of NOD2 in the brain ischemia, we further compared cerebral I/R injury induced in WT mice and NOD2-/- mice with or without MDP, an extrinsic ligand of NOD2, stimulation. Pretreatment with MDP to stimulate NOD2 significantly increased the infarct volume and neurological dysfunction in WT mice subjected to I/R injury. However, there were no statistical differences in stroke outcome between the MDP and I/R groups in NOD2-/- mice. Convincingly, mice lacking NOD2 had a significantly better stroke outcome than mice that expressed normal levels of NOD2 after cerebral I/R injury.
Inflammation contributes to secondary cerebral ischemic injury [21], and NOD2 play a critical role in the innate immunity and inflammation [7], therefore, NOD2 may also be involved in the inflammatory responses to I/R insult and the associated brain damage. Our results showed that fewer pro-inflammatory mediators including IL-1β, IL-6 and TNFα, were produced in NOD2 deficient mice compared with WT substrains subjected to cerebral I/R. It has been shown that NOD2 activates NF-κB and MAPKs signaling pathways, which induce the transcription of many pro-inflammatory genes [10]. Moreover, cerebral I/R injury triggers the activation of several transcription factors involved in the inflammatory response. Among them, the activation of NF-κB and subsequent degradation of IκBα are the key events in cerebral I/R[24]. The activated NF-κB further induces the expression of genes encoding many pro-inflammatory mediators, thereby triggering and exacerbating inflammatory injury in ischemic stroke [31]. Our results also showed that I/R injury increased the phosphorylation of p65 and promoted the degradation of its endogenous inhibitor IκBα. Consistent with the down-regulations of IL-1β, IL-6 and TNFα levels in the brain, the activation of NF-κB was significantly inhibited in NOD2 deficient mice. MAPKs signaling pathways also regulate transcription of inflammatory genes in the subsequent ischemic brain damage [32]. The MAPK family includes the following major members: ERK, p38, and JNK, representing three different signaling cascades. The role of ERKs in cell growth and proliferation is well established. In contrast, JNKs and p38 MAPKs are regarded as stress-induced kinases and these kinases are implicated in regulation of cytokine release and apoptosis. Increasing evidences have demonstrated that inhibition of MAPKs could be of benefit for the inhibition of neuroinflammation [33]. JNK and p38 MAPK have been identified as specific markers in neuroinflammatory signaling pathways. Inhibition of p38 and JNK, could lead to a reduction in pro-inflammatory molecule production by inflammatory cells after an ischemic injury [34, 35]. Consistently, we found that MAPKs were activated after cerebral I/R injury in WT mice, while NOD2 deficiency significantly restrained the activation of p38 MAPK and JNK. These data suggested that activation of NF-κB, p38 MAPK and JNK mediated NOD2-dependent immune activation in brain damage after ischemia.
NOX-derived ROS is central to the oxidative stress induced by cerebral ischemia and NOX2 is the most important subtype for mediating cerebral injury [36, 37]. Emerging evidence suggests that ROS contribute to innate immunity mediated signaling processes [38]. It has been demonstrated that NOX2 plays an essential role in TLR2/3-dependent inflammatory responses [25, 27]. Additionally, TLR4-dependent ROS generation is essential for innate immunity via the TRAF6/ASK1/p38 axis [26].The NOX family member DUOX2 was recently demonstrated to involve in NOD2-dependent ROS production in intestinal epithelial cells [39]. Therefore, it is important to determine whether NOX2-derived ROS signaling is involved in NOD2-induced inflammatory responses in cerebral I/R. Indeed, our results showed that cerebral I/R-induced NOX2 expression and ROS generation were significantly diminished in NOD2 deficient mice. MDP induced a robust burst of ROS production and increased the expression of NOX2 in WT microglia. However, inhibition of NOX activity significantly reduced the production of the inflammatory mediator IL-6 in microglia via OGD or MDP stimulation. Our results indicated that NOX2 was an important source of NOD2-induced ROS and NOX2-derived ROS was essential for the generation of inflammatory mediators such as IL-6. It should be noted that the present study did not further explore the regulatory mechanisms of NOD2 activating NOX2, and further studies are needed to address this issue.
In summary, this study for the first time showed NOD2 was a novel innate immune signaling participant in brain inflammatory response and provided direct evidence that NOX2-mediated oxidative stress as an important target molecule linked NOD2 to inflammatory damage in ischemic stroke. Pharmacological targeting of NOD2-mediated inflammatory response at multiple levels may help design a new approach to develop therapeutic strategies for prevention of deterioration of cerebral function and for the treatment of stroke.
Abbreviations
I/R: ischemia-reperfusion; NOD2: nucleotide-binding oligomerization domain 2; MDP: muramyl dipeptide; NLR: nucleotide-binding oligomerization domain-like receptors; TLR: Toll-like receptor; MCAO: middle cerebral artery occlusion; WT: wild type; NOX2: NADPH oxidase 2; rCBF: regional cerebral blood flow; TTC: triphenyltetrazolium chloride; ROS: reactive oxygen species; OGD: oxygen-glucose deprivation; NF-κB: nuclear factor κB; MAPK: mitogen activated protein kinases.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No.81200904 to H. Liu, No.81100874 to L. Chen) and Shandong Province Science and Technology Plan (No.2014GSF118003).
Competing interests
The authors have declared that no competing interest exists.
References
1. Poisson SN, Schardt TQ, Dingman A, Bernard TJ. Etiology and treatment of arterial ischemic stroke in children and young adults. Current treatment options in neurology. 2014;16:315. doi:10.1007/s11940-014-0315-4
2. Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Current opinion in neurology. 2007;20:334-42 doi:10.1097/WCO.0b013e32813ba151
3. Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. The FEBS journal. 2009;276:13-26 doi:10.1111/j.1742-4658.2008.06766.x
4. Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nature reviews Neuroscience. 2002;3:216-27 doi:10.1038/nrn752
5. Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Current medicinal chemistry. 2014;21:2076-97
6. Inohara Chamaillard, McDonald C Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annual review of biochemistry. 2005;74:355-83 doi:10.1146/annurev.biochem.74.082803.133347
7. Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. Journal of leukocyte biology. 2008;83:13-30 doi:10.1189/jlb.0607402
8. Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:534-44 doi:10.1038/jcbfm.2008.143
9. Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y. et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014 doi:10.1038/jcbfm.2013.242
10. Kufer TA. Signal transduction pathways used by NLR-type innate immune receptors. Molecular bioSystems. 2008;4:380-6 doi:10.1039/b718948f
11. Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286-90 doi:10.1126/science.1184004
12. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603-6 doi:10.1038/35079114
13. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599-603 doi:10.1038/35079107
14. Liu HQ, Zhang XY, Edfeldt K, Nijhuis MO, Idborg H, Back M. et al. NOD2-mediated innate immune signaling regulates the eicosanoids in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:2193-201 doi:10.1161/ATVBAHA.113.301715
15. Sterka D Jr, Marriott I. Characterization of nucleotide-binding oligomerization domain (NOD) protein expression in primary murine microglia. Journal of neuroimmunology. 2006;179:65-75 doi:10.1016/j.jneuroim.2006.06.009
16. Sterka D Jr, Rati DM, Marriott I. Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia. 2006;53:322-30 doi:10.1002/glia.20286
17. Chauhan VS, Sterka DG Jr, Furr SR, Young AB, Marriott I. NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia. 2009;57:414-23 doi:10.1002/glia.20770
18. Jasmin JF, Malhotra S, Singh Dhallu M, Mercier I, Rosenbaum DM, Lisanti MP. Caveolin-1 deficiency increases cerebral ischemic injury. Circulation research. 2007;100:721-9 doi:10.1161/01.RES.0000260180.42709.29
19. Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG. et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circulation research. 2006;99:209-17 doi:10.1161/01.RES.0000232544.90675.42
20. Yi F, Zhang AY, Janscha JL, Li PL, Zou AP. Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney international. 2004;66:1977-87 doi:10.1111/j.1523-1755.2004.00968.x
21. del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain pathology. 2000;10:95-112
22. Ketelhuth DF, Rios FJ, Wang Y, Liu H, Johansson ME, Fredrikson GN. et al. Identification of a danger-associated peptide from apolipoprotein B100 (ApoBDS-1) that triggers innate proatherogenic responses. Circulation. 2011;124:2433-43 1-7. doi:10.1161/CIRCULATIONAHA.111.051599
23. Liu HQ, Wei XB, Sun R, Cai YW, Lou HY, Wang JW. et al. Angiotensin II stimulates intercellular adhesion molecule-1 via an AT1 receptor/nuclear factor-kappaB pathway in brain microvascular endothelial cells. Life sciences. 2006;78:1293-8 doi:10.1016/j.lfs.2005.06.049
24. Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158:995-1006 doi:10.1016/j.neuroscience.2008.07.007
25. Yang CS, Shin DM, Kim KH, Lee ZW, Lee CH, Park SG. et al. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. Journal of immunology. 2009;182:3696-705 doi:10.4049/jimmunol.0802217
26. Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S. et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nature immunology. 2005;6:587-92 doi:10.1038/ni1200
27. Yang CS, Kim JJ, Lee SJ, Hwang JH, Lee CH, Lee MS. et al. TLR3-triggered reactive oxygen species contribute to inflammatory responses by activating signal transducer and activator of transcription-1. Journal of immunology. 2013;190:6368-77 doi:10.4049/jimmunol.1202574
28. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. Journal of leukocyte biology. 2010;87:779-89 doi:10.1189/jlb.1109766
29. Aloisi F. Immune function of microglia. Glia. 2001;36:165-79
30. Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends in immunology. 2007;28:138-45 doi:10.1016/j.it.2007.01.005
31. Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, Little S. et al. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2000;20:592-603 doi:10.1097/00004647-200003000-00017
32. Nozaki K, Nishimura M, Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Molecular neurobiology. 2001;23:1-19 doi:10.1385/MN:23:1:01
33. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy--from molecular mechanisms to therapeutic benefits. Biochimica et biophysica acta. 2005;1754:253-62 doi:10.1016/j.bbapap.2005.08.017
34. Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S. et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Medicinal research reviews. 2001;21:129-45
35. Murata Y, Fujiwara N, Seo JH, Yan F, Liu X, Terasaki Y. et al. Delayed inhibition of c-Jun N-terminal kinase worsens outcomes after focal cerebral ischemia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:8112-5 doi:10.1523/JNEUROSCI.0219-12.2012
36. Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:1262-72 doi:10.1038/jcbfm.2009.47
37. Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R. et al. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke; a journal of cerebral circulation. 2007;38:3000-6 doi:10.1161/STROKEAHA.107.489765
38. Lam GY, Huang J, Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Seminars in immunopathology. 2010;32:415-30 doi:10.1007/s00281-010-0221-0
39. Lipinski S, Till A, Sina C, Arlt A, Grasberger H, Schreiber S. et al. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. Journal of cell science. 2009;122:3522-30 doi:10.1242/jcs.050690
Author contact
![]() Corresponding author: Lin Chen, Phone: 86-0531-88382036 Fax: 86-0531-88383146 E-mail: linchenedu.cn.
Corresponding author: Lin Chen, Phone: 86-0531-88382036 Fax: 86-0531-88383146 E-mail: linchenedu.cn.