Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(5):536-545. doi:10.7150/ijbs.10754 This issue Cite
Research Paper
Brain-Derived Neurotrophic Factor Regulates TRPC3/6 Channels and Protects Against Myocardial Infarction in Rodents
Pengzhou Hang1#, Jing Zhao2#, Benzhi Cai3, Shanshan Tian1, Wei Huang1, Jing Guo1, Chuan Sun1, Yue Li2 ![]() , Zhimin Du1
, Zhimin Du1 ![]()
1. Institute of Clinical Pharmacology of the Second Affiliated Hospital (Key Laboratory of Drug Research, Heilongjiang Higher Education Institutions), Harbin Medical University, Harbin 150086, China.
2. Department of Cardiology of the First Affiliated Hospital (Key Laboratory of Cardiac Diseases and Heart Failure), Harbin Medical University, Harbin 150001, China.
3. Department of Pharmacology (State-Province Key Laboratories of Biomedicine- Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), Harbin Medical University, Harbin 150081, China.
# The first two authors made equal contribution to this research.
Received 2014-10-8; Accepted 2015-2-23; Published 2015-3-25
Abstract

Background: Brain-derived neurotrophic factor (BDNF) is associated with coronary artery diseases. However, its role and mechanism in myocardial infarction (MI) is not fully understood.
Methods: Wistar rat and Kunming mouse model of MI were induced by the ligation of left coronary artery. Blood samples were collected from MI rats and patients. Plasma BDNF level, protein expression of BDNF, tropomyosin-related kinase B (TrkB) and its downstream transient receptor potential canonical (TRPC)3/6 channels were examined by enzyme-linked immunosorbent assay and Western blot. Infarct size, cardiac function and cardiomyocyte apoptosis were measured after intra-myocardium injection with recombinant human BDNF. Protective role of BDNF against cardiomyocyte apoptosis was confirmed by BDNF scavenger TrkB-Fc. The regulation of TRPC3/6 channels by BDNF was validated by pretreating with TRPC blocker (2-Aminoethyl diphenylborinate, 2-APB) and TRPC3/6 siRNAs.
Results: Circulating BDNF was significantly enhanced in MI rats and patients. Protein expression of BDNF, TrkB and TRPC3/6 channels were upregulated in MI. 3 days post-MI, BDNF treatment markedly reduced the infarct size and serum lactate dehydrogenase activity. Meanwhile, echocardiography indicated that BDNF significantly improved cardiac function of MI mice. Furthermore, BDNF markedly inhibited cardiomyocyte apoptosis by upregulating Bcl-2 expression and downregulating caspase-3 expression and activity in ischemic myocardium. In neonatal rat ventricular myocytes, cell viability was dramatically increased by BDNF in hypoxia, which was restored by TrkB-Fc. Furthermore, protective role of BDNF against hypoxia-induced apoptosis was reversed by 2-APB and TRPC3/6 siRNAs.
Conclusion: BDNF/TrkB alleviated cardiac ischemic injury and inhibited cardiomyocytes apoptosis by regulating TRPC3/6 channels, which provides a novel potential therapeutic candidate for MI.
Keywords: Brain derived neurotrophic factor, Myocardial infarction, Transient receptor potential canonical channel, Apoptosis, TrkB-Fc
Introduction
Myocardial infarction (MI) is one of the leading causes of sudden cardiac death all over the world [1]. Accumulating evidence indicates that neurotrophic factors such as nerve growth factor (NGF) elicit beneficial actions on the post-MI heart by promoting neovascularization and improving myocardial blood flow and cardiac function [2]. Brain-derived neurotrophic factor (BDNF), a secreted protein in neurotrophin family, has favorable neuroprotective effect against oxygen-glucose deprivation (OGD) [3]. It has been well recognized that BDNF specifically binds to the tropomyocin-related kinase receptor B (TrkB), and further modulates downstream intracellular signaling pathways and thereby affects the development and function of the nervous system [4]. Previous studies have reported that both endogenous and exogenous BDNF exhibited promising protective effect against cerebral ischemia [5, 6]. Interestingly, several clinical studies suggested that plasma BDNF was associated with coronary atherosclerosis and unstable angina [7, 8]. Recently, more attention has been paid to the effect of BDNF on ischemic heart diseases [9, 10]. However, its role in MI heart still remains elusive. For instance, Okada et al. reported that upregulation of BDNF protected against cardiac dysfunction post-MI through a central nervous system mediated signaling pathway [11]. In contrast, in other studies, it was found that reduced BDNF levels improved post-MI survival and reduced left ventricular remodeling by attenuating inflammation and angiogenesis [12]. Moreover, Cai et al. reported that BDNF significantly enhanced the extent of myocardial injury in older rat hearts without affecting younger hearts [13].
Transient receptor potential canonic (TRPC) family is a Ca2+-permeable nonselective cation channel, which is divided into four subgroups based on their sequence similarities: TRPC1, TRPC2, TRPC4/5 and TRPC3/6/7 [14]. Previous studies have demonstrated that TRPC3 and TRPC6 channels participate in BDNF-mediated neuron survival. Moreover, overexpression of TRPC3 and TRPC6 channels protects cerebellar granule neurons against serum deprivation-induced cell death [15, 16]. It has also been found that TRPC3/6 channels could regulate intracellular calcium homeostasis and thereby activate calcium/calmodulin-dependent protein kinase (CAMK) and cAMP-response element binding protein (CREB) signaling cascades to determine the cell fate in neruons [17]. However, the potential role of BDNF/TRPC cascade in cardiac ischemia remains poorly understood.
Based on these findings, the purpose of the present study is to examine the potential cardioprotective actions of BDNF on left coronary artery occlusion model and to explore whether BDNF protects ischemic heart by regulating TRPC3/6 channels. Our hypothesis is that BDNF/TrkB protects against MI and inhibits cardiomyocyte apoptosis by regulating TRPC3/6 channels.
Materials and Methods
Human blood samples
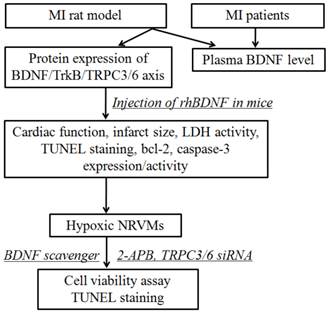
All the experimental procedures were approved by the ethics committee of Harbin Medical University, with informed consent of these patients, and performed in accordance with the principles outlined in the Declaration of Helsinki. Whole experimental design was shown in Fig. 1. The blood samples were obtained from 7 individuals with acute MI and 5 without MI from the First Affiliated Hospital of Harbin Medical University. The characteristics of MI patients and control subjects were list in Table 1. There were no statistical differences between control and MI patients in the following variables: age, sex, hypertension, diabetes mellitus, hyperlipidemia, smoking, and obesity. And the blood samples of MI patients were collected within 12 h from the onset of MI. The blood samples were immediately centrifuged at 3000 rpm for 10 min at 4°C, and the plasma was stored at -80°C until assayed.
Schematic diagram illustrating the experimental design. MI, myocardial infarction; rhBDNF, recombinant human BDNF; NRVMs, neonatal rat ventricular myocytes.
Clinical Characteristics of control and MI patients (Mean ± SD)
| Characteristics | MI group (n = 7) | Control group (n = 5) | P value |
|---|---|---|---|
| Age (years) | 59.14 ± 11.38 | 55.40 ± 8.93 | 0.555 |
| Sex (male/female) | 4/3 | 3/2 | 1.000 |
| BMI (kg/m2) | 22.29 ± 1.50 | 21.4 ± 1.95 | 0.392 |
| SBP (mmHg) | 125.29 ± 11.95 | 115.40 ± 9.04 | 0.152 |
| DBP (mmHg) | 81.71 ± 6.37 | 78.20 ± 2.59 | 0.275 |
| Glucose (mmol/L) | 5.57 ± 0.47 | 5.54 ± 0.60 | 0.919 |
| TC (mmol/L) | 4.59 ± 0.77 | 4.12 ± 0.64 | 0.292 |
| LDL (mmol/L) | 2.64 ± 0.95 | 2.79 ± 0.57 | 0.764 |
| TG (mmol/L) | 1.15 ± 0.39 | 0.85 ± 0.34 | 0.202 |
| HDL (mmol/L) | 1.06 ± 0.19 | 1.20 ± 0.20 | 0.236 |
BMI: body mass index; SBP: systolic blood pressure; DBP: diastolic blood pressure; TC: total cholesterol; LDL: low-density lipoprotein; TG: total triglyceride; HDL: high-density lipoprotein.
Animals
Male Wistar rats (200-250 g) and Kunming mice (25-30 g) were provided by Animal Center of the First and Second Affiliated Hospital of Harbin Medical University. The animals were raised under standard animal room conditions (temperature 21 ± 1 °C; humidity 55-60%) with food and water unlimited for 1 week before the experiments. All the experimental procedures were in accordance with the regulations of the ethic committees of Harbin Medical University and confirmed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (revised 2011). The surgery procedures were performed under sodium pentobarbital anesthesia.
Acute MI model
Wistar rats were randomly divided into four groups: sham, MI for 1 h, 6 h and 24 h, respectively. MI was induced by occluding the left coronary artery as described previously [18]. Remote tissues of ischemic areas in the left ventricles were quickly dissected for subsequent analysis.
Intra-myocardium BDNF microinjection
Kunming mice were randomly divided into three group: sham, MI (infarction for 3 days) and BDNF group (injected with BDNF 1 day before infarction), respectively. The infarction procedures were the same as the rats. 1 μg of recombinant human (rh)BDNF (R&D Systems, USA) in 50 μl of PBS or PBS alone was injected into the myocardium through a 26-gauge needle in two injections (25 μl/injection, 2 mm apart) in the mid-left ventricular anterior wall of the mice hearts based on previous studies [13]. On day 1 and day 3 post-MI, echocardiography was performed. The blood and ventricles were collected 3 days post-MI. After being fixed, the ventricles were embedded in paraffin and cut (cross section) about 5 μm in thickness for subsequent experiments.
Infarction area measurement
The measurement of area at risk and infarct size was according to previous study [19]. Briefly, 2% Evans Blue dye was injected into the abdominal aorta to delineate the area at risk (AAR) from not-at-risk myocardium. Then, the left ventricles were crosscutting into 2 mm thick slices and stained with 1% triphenyltetrazolium chloride (TTC) for 10 min at 37°C, the infarct area (IA) was stainless while the live area turned red. IA and AAR were assessed with Image ProPlus 5.0 software by an observer blinded to the sample identity. Myocardial infarct size is expressed as the ratio of IA to AAR.
Echocardiographic measurement
Transthoracic echocardiography with an ultrasound instrument (Vivid 7 GE Medical) equipped with a 10-MHz phased-array transducer was used to measure the left ventricular function. Left ventricular systolic volume (LVVs), left ventricular diastolic volume (LVVd) was measured, and left ventricular ejection fraction (LVEF) and fractional shortening (FS) were calculated from M-mode recording.
Plasma BDNF level, serum LDH and caspase-3 activity measurement
Plasma BDNF level from rats and human were measured by sandwich ELISAs according to the manufacturer's instructions for BDNF (Promega, USA). Assays were performed on polystyrene 96-well plates. The BDNF concentration was quantified against a standard curve calibrated with known amounts of protein. The BDNF ELISA systems have low cross-reactivity (<3%) with other related neurotrophic factors. Each value is a mean of duplicated measurement. Serum LDH activity was measured using LDH detection kits (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer's instruction. Caspase-3 activity was measured using caspase-3 activity kit (Beyotime, China) according to the manufacturer's protocol.
Western blot analysis
Total protein was extracted from the left ventricles for immunoblotting analysis with the procedures described previously [20]. Briefly, proteins were separated by electrophoresis on SDS-polyacrylamide gels and transferred moist to nitrocellulose filter membranes. Membranes were incubated with primary antibodies against BDNF (1:200, Santa Cruz, USA), TrkB (1:200, Santa Cruz, USA), TRPC3 (1:500, Alomone Biolab, Israel), TRPC6 (1:200, Alomone Biolab, Israel), Bcl-2 (1:1000, Sigma, USA) overnight at 4°C. After washing, the membrane was incubated with secondary antibody for 1 h. The images were captured on the Odyssey CLx Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Western blot bands were quantified using Odyssey CLx v2.1 software and normalizing to GAPDH (anti-GAPDH antibody 1:1000, Kangcheng, China) as a loading control.
Real time RT-PCR
Total RNA were extracted from ventricles using Trizol reagent (Invitrogen, USA) according to manufacturer's protocols. Sequences of gene-specific PCR primers (Invitrogen) used were as follow: Bcl-2 F: 5- CGGTGGTGGAGGAACTCTTC -3; R: 5- TGTGCAGATGCCGGTTCA -3; Caspase-3 F: 5- CTCGCTCTGGTACGGATGTG -3; R: 5- TCCCATAAATGACCCCTTCATCA -3; TRPC3 F: 5- TTAATACCTTCACCATGCGGAG -3; R: 5- GAACTCTTGGAGGCCAACAGG- 3; TRPC6 F: 5- AGAAATTTGGAATTTTGGGAAG -3; R: 5- TCCTTATCAATCTGGGCCTGC -3; and GAPDH F: 5- AAGAAGGTGGTGAAGCAGGC -3; R: 5- TCCACCACCCAGTTGCTGTA -3. Quantitative real-time PCR was performed in 20 μl volumes with SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK) at 95°C for 10 min and 40 cycles at 95°C for 15 s, 60°C for 30 s and 72°C for 30 s, using ABI 7500 Fast Real-Time PCR system (Applied Biosystems, Warrington, UK). GAPDH mRNA was measured as an internal control. ΔΔCt was calculated for each sample, and the expression levels were indicated with 2-ΔΔCt.
Isolation and culture of cardiomyocytes
The procedures to culture neonatal rat ventricular myocytes (NRVMs) are just as described previously [21]. Briefly, the hearts of neonatal wistar rats were rapidly removed. Both ventricles were cut into l to 2 mm3 and dissociated in 0.25% trypsin at 37 °C for 1-2 min. Cardiac tissues were trypsinized until disappeared and cell suspensions were collected. Then all suspensions were pelleted by centrifugation at 2000 rpm for 3 min. The isolated cells were then resuspended in DMEM (Hyclone Laboratories) added with 10% fetal bovine serum (Gibco), transferred into culture flask and cultured at 37°C in humid air with 5% CO2. After fibroblast adherence for 90 min, neonatal cardiomyocytes were plated into 6 well plate at a density of 3×105 cells per well. After 48 h culture, NRVMs were serum starved and treated with rhBDNF (100 ng/ml, 200 ng/ml and 400 ng/ml, respectively) for 24 h according to previous study [22] in the absence or presence of 2-Aminoethyl diphenylborinate (2-APB, 30 μM, a TRPC channel blocker) [23] and TRPC3/TRPC6 siRNAs. Moreover, 4 μg/ml BDNF scavenger (TrkB-Fc chimera, R&D Systems, USA) was pretreated to block the effect of BDNF. The concentration of TrkB-Fc was calculated from its ED50 (0.1-0.4 μg/ml vs. 16 ng/ml rhBDNF) and according to previous study [24]. Cells were exposed to hypoxia (1% O2, 5% CO2 and 94% N2) for 12 h to mimic ischemic status.
Transfection of siRNA
TRPC3 siRNA (sense: 5′-CCACCCAGUUCACAUGGACAGAAAU-3′; antisense: 5′-AUUUCUGUCCAUGUGAACUGGGUGG-3′) and TRPC6 siRNA (sense: 5′-GUCCAUUCAUGAAGUUCGUTT-3′; antisense: 5′-ACGAACUUCAUGAAUGGACTT-3′) were synthesized by Invitrogen, USA. Cardiomyocytes (1 × 105 per well) were serum starved and then transfected with X-treme GENE siRNA transfection reagent (Roche, USA) according to the manufacturer's instructions. Cardiomyocytes were treated with transfection regent alone in Si-control group.
Cell viability assay
Adherent cardiomyocytes were washed and placed in fresh medium. The cells were treated as designated and were subsequently incubated with 20 µl MTT (0.5 mg/ml) for 4 h. The culture medium was carefully removed, and 200 µl DMSO was added to each well to dissolve the formazan. After the cells were rocked for 10 min, the absorbance values were detected at 490 nm using an Infinite M200 microplate spectrophotometer (Tecan, Salzburg, Austria).
TUNEL staining
Apoptosis of cardiomyocytes was detected by staining with the In situ Cell Death Detection Kit (TUNEL fluorescence FITC kit, Roche, USA) according to the manufacturer's instruction. After TUNEL staining, the ventricular specimens (3 days post-MI) or cardiomyocytes (12 h after hypoxia) were immerged into Hoechst (Sigma-Aldrich) solution to stain nuclei. Fluorescence staining was viewed by a laser scanning confocal microscope (FV1000, Olympus, Japan). The apoptosis content was calculated as TUNEL-positive cells per field.
Statistical analysis
Quantitative data were expressed as mean ± SEM (Clinical characteristics were expressed as mean ± SD) and analyzed by GraphPad Prism 5.0 software. Two-group comparisons were performed by Student's t test. Multiple-group comparisons were carried out using one-way ANOVA followed by Dunnet's t-test. Enumeration data were analyzed with the chi-squared test. A two-tailed P < 0.05 was considered to be statistically significant.
Results
BDNF level was elevated in MI
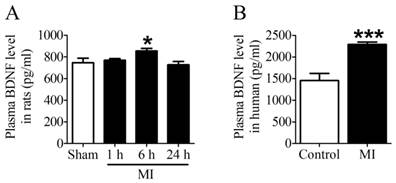
First of all, we examined the plasma BDNF level in MI rats and human. ELISA assay showed that plasma BDNF level did not change at 1 h post-MI but significantly increased after MI for 6 h, and returned toward 1 h values by 24 h (Fig. 2A). Meanwhile, we observed that plasma BDNF level in MI patients was markedly enhanced compared with non-MI patients (Fig. 2B). Thus, these results suggested that circulating BDNF level was elevated in acute MI.
Plasma BDNF concentration in myocardial infarction (MI) rats and human. (A) Plasma BDNF levels were measured in sham and ischemia for 1 h, 6 h and 24 h by ELISA. (B) Plasma BDNF concentration was measured in control and MI patients. *p <0.05 vs. sham, n = 4 each group, ***p<0.001 vs. control, n = 5 in control group while n = 7 in MI group.
BDNF/TrkB/TRPC3/6 axis was upregulated in MI
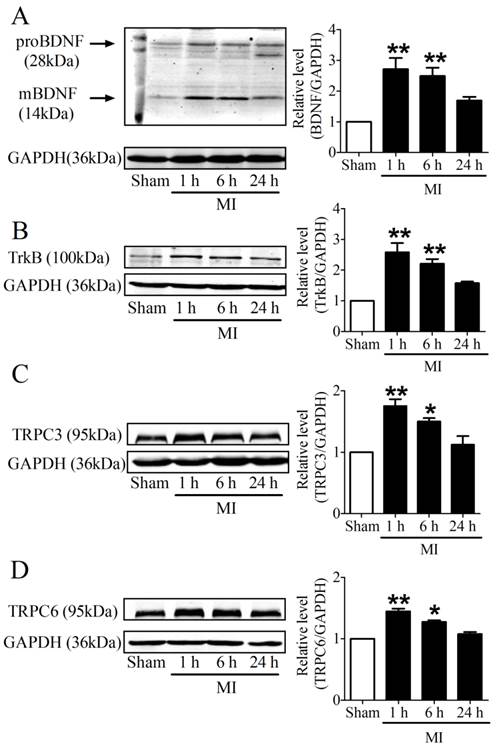
It is recognized that 28 kDa Pro-BDNF form is cleaved extracellularly by proteases to generate an active mature 14 kDa BDNF form [25]. To evaluate the time course of BDNF expression in the infarcted ventricles at 1, 6 and 24 h post-MI, we used an anti-BDNF antibody that recognizes both the 28 kDa proform and 14 kDa active form of BDNF. We found that both pro-BDNF and mature BDNF expression was upregulated in MI for 1 h and 6 h, and recovered after MI for 24 h (Fig. 3A). In parallel, protein expression of TrkB was also increased in MI for 1 h and 6 h, while reduced in MI for 24 h (Fig. 3B). Consistently, protein levels of TRPC3/6 channels were significantly increased after MI for 1 h and 6 h, then recovered after 24 h (Fig. 3C, D). Thus, these data together supported that BDNF/TrkB/TRPC3/6 axis was upregulated in MI.
Expression of BDNF/TrkB and downstream TRPC3/6 channels. (A) Representative bands of protein expression of proBDNF and mature BDNF by western blot analysis. (B) Representative bands of protein expression of TrkB. (C, D) Representative bands of protein expression of TRPC3 and 6 channels. Values given are normalized to band intensity of GADPH (anti-GAPDH antibody) used as internal control. *p<0.05, **p<0.01 vs. sham, n = 4 rats each group.
BDNF ameliorated ischemic heart function
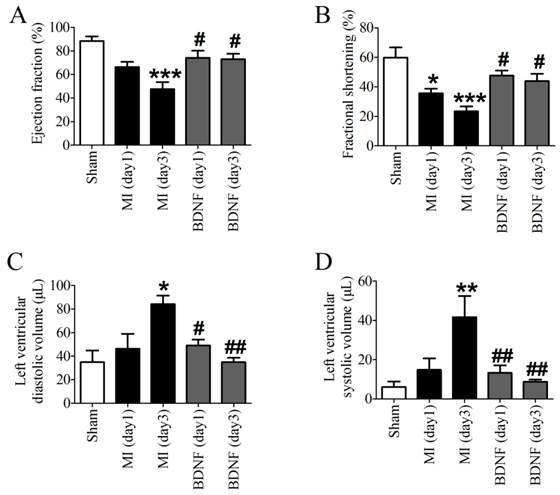
Echocardiographic data indicated that both EF and FS were decreased in MI, which was dramatically augmented by BDNF (Fig. 4A, B). Moreover, LVVd and LVVs were restored by BDNF in MI (Fig. 4C, D). These data revealed that BDNF pretreatment could significantly alleviate cardiac dysfunction during MI.
Effects of BDNF on cardiac function of MI mice. (A) Ejection Fractions, EF; (B) Fractional shortening, FS; (C) Left ventricle diastolic volume, LVVd; (D) LVVs; Left ventricle systolic volume. *p<0.05, **p<0.01, ***p<0.001 vs. sham, #p<0.05 vs. MI (day 3), n = 6 mice each group.
BDNF decreased serum LDH activity and reduced infarct size in MI
We next detected the functional role of BDNF in infarct heart, and found that BDNF could significantly reduce the infarct size in MI (Fig. 5A, B). LDH is a important marker for cardiac injury, therefore, serum LDH activity was further detected. We found that serum LDH activity was increased at 3 days post-MI, and was significantly decreased by BDNF (Fig. 5C). Thus, BDNF displayed protective effect against ischemic-injury.
Effect of BDNF on cardiac infarct area, lactate dehydrogenase (LDH) activity and cardiomyocyte apoptosis in mice 3 days post-MI. (A) Representative images showing infarct areas in cross section slices, scale bar: 5 mm; (B) Statistical analysis of IA/AAR ratio. IA, infarct area; AAR, area at risk. Data are expressed as mean ± SEM; *p<0.05 vs. MI, n = 4 mice each group. (C) Serum LDH activity in MI and BDNF pretreated mice. ***p<0.001 vs. sham, ###p<0.001 vs. MI, n = 4 mice each group. (D) Effect of BDNF on cardiomyocytes apoptosis 3 days post-MI by TUNEL staining, scale bar: 200 μm. (E) TUNEL positive cell (%). **p<0.01 vs. sham, ##p<0.01 vs. MI, n = 5 mice each group.
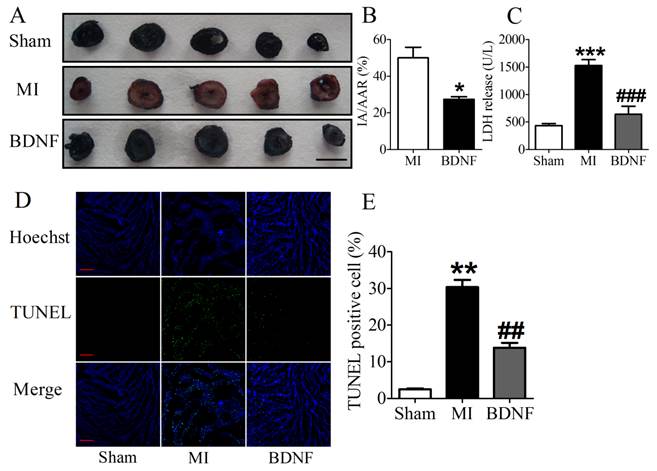
BDNF inhibited apoptosis in ischemic myocardium
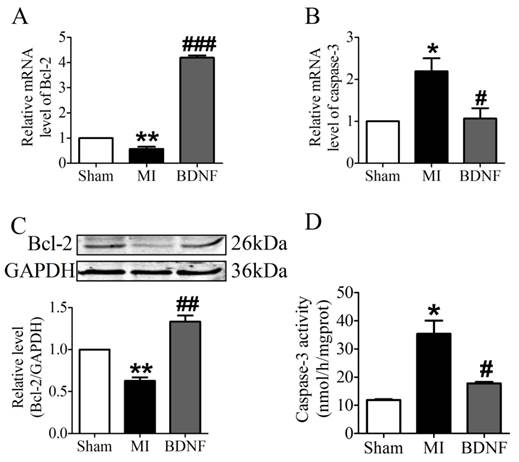
We further examined the apoptosis of cardiomyocyte by TUNEL staining. Compared with sham mice, TUNEL positive cells were increased in myocardium of MI mice. BDNF markedly inhibited the apoptosis of cardiomyocytes (Fig. 5D, E). Moreover, we detected mRNA level of caspase-3 and Bcl-2 and found that caspase-3 was downregulated while Bcl-2 was upregulated by BDNF (Fig. 6A, B). Meanwhile, protein expression of Bcl-2 was increased and caspase-3 activity was decreased in BDNF treated mice (Fig. 6C, D). These data suggested that BDNF inhibited ischemic-induced apoptosis in cardiomyocytes. Therefore, our results indicated that BDNF inhibited apoptosis in cardiac ischemic injury.
Effect of BDNF on cardiomyocytes apoptosis in mice 3 days post-MI. (A, B) The mRNA level of Bcl-2 and caspase-3. (C) Representative bands of protein expression of Bcl-2. (D) Caspase-3 activity. *p<0.05 vs. sham, #p<0.05 vs. MI, n = 4 mice each group.
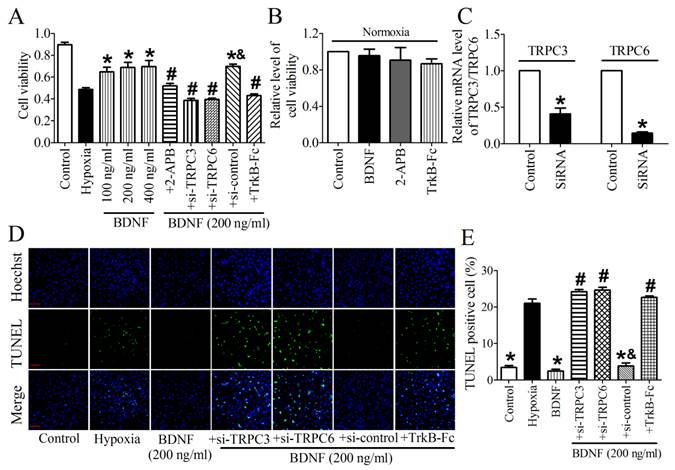
TPRC3/6 channels were required for cardioprotective effect of BDNF
We then validated the protective effect of BDNF on NRVMs and found that the cell viability was dose-dependently increased by BDNF from 100 ng/ml to 400 ng/ml (Fig. 7A). Thus, 200 ng/ml was chosen for the following study. In addition, we found that TrkB-Fc successfully decreased cell viability and increased TUNEL-positive cells (Fig. 7A, D). Thus, these evidence confirmed the cytoprotection effect of BDNF against cardiomyocytes apoptosis. To examine the role of TRPC3/6 channels in this process, we tested cell viability after treating with TRPC channel block, 2-APB. We found that cell viability was significantly recovered by 2-APB (Fig. 7A). And we also examined the effect BDNF, 2-APB and TrkB-Fc on cell viability under normoxia and found that all of them did not alter the viability of cardiomyocytes (Fig. 7B). Furthermore, because 2-APB is not a specific inhibitor for TRPC3/6, TRPC3/6 siRNAs were used to confirm the role of TRPC3/6 channels in cardioprotective role of BDNF. The efficiency of TRPC3/TRPC6 siRNAs was validated by Real-time RT-PCR analysis (Fig. 7C). We found that cell viability was reduced while apoptotic cardiomyocytes increased by TRPC3/TRPC6 siRNAs (Fig. 7A, D). Taken together, these data supported that TRPC3/6 channels were required in the protective role of BDNF against cardiomyocyte apoptosis.
Effect of BDNF and 2-APB on cell vialibity and apoptosis in NRVMs. (A) Effect of BDNF on cell viability in hypoxia. *p<0.05 vs. hypoxia, #p<0.05 vs. 200 ng/ml BDNF, &p<0.05 vs. 200 ng/ml BDNF+Si-TRPC3 or 200 ng/ml BDNF +Si-TRPC6, n = 6 each group. (B) Effect of BDNF (200 ng/ml), 2-APB and TrkB-Fc on cell vialibity in normoxia. (C) mRNA level of TRPC3/TRPC6 in control and TRPC3/TRPC6 siRNA treated cardiomyocytes. (D) Effect of BDNF on cardiomyocytes apoptosis subject to hypoxia by TUNEL staining. Nuclei are stained in blue while apoptotic cells are stained in green, scale bar: 100 μm. (E) TUNEL positive cell (%), *p<0.05 vs. hypoxia, #p<0.05 vs. 200 ng/ml BDNF, &p<0.05 vs. 200 ng/ml BDNF+Si-TRPC3 or 200 ng/ml BDNF +Si-TRPC6, n = 5 each group.
Discussion
The main finding of the present study was that: (1) BDNF/TrkB/TRPC axis was activated in acute MI; (2) upregulation of BDNF reduced infarct size and improved cardiac function in infarcted adult murine hearts; (3) BDNF inhibited apoptosis by increasing Bcl-2 and decreasing caspase-3 expression in ischemic cardiomyocytes, which could be antagnized by TrkB-Fc and TRPC3/6 siRNAs. Therefore, the present study is the first report examining the effect of BDNF/TRPC axis on cardiac ischemic apoptosis.
Previous studies have uncovered many neurotrophic factors with protective effect against ischemic damage, such as NGF [2], glial cell line-derived neurotrophic factor (GDNF) [26], neurotrophins [27]. These factors affect angiogenesis and sympathetic/vagal innervation of ischemic tissues. Similarly, previous studies documented that BDNF could stimulate the formation of new vessels by increasing vascular endothelial growth factor (VEGF) [28]. BDNF knockout mice are postnatal lethal due to an impaired endothelial cell adhesion that leads to cardiac hemorrhage, which indicates a role of BDNF in angiogenic processes [29]. However, the potential role of BDNF in apoptosis of cardiomyocytes remain unclear. Therefore, the present study mainly focused on the effect of BDNF on apoptosis of cardiomyocytes. And it was found that BDNF could significantly inhibit cardiomyocytes apoptosis based on our results. Accordingly, BDNF not only promotes angiogenesis but also represses cell death during ischemic injury.
Several previous studies suggested that plasma BDNF was increased in coronary artery diseases [7] and myocardial ischemia [11], although few studies observed reduced plasma level of BDNF in patients with acute coronary syndrome (ACS) [30]. In our study, we found both ventricular and plasma BDNF increased in response to MI. However, the effects of BDNF on infarcted heart remain vague. Therefore, both BDNF and its scavenger were employed to examine its effect on ischemic cardiomyocytes. Interestingly, Okada et al. proposed that elevated plasma levels of BDNF was associated with the upregulation of BDNF expression in the brain, but not in the heart [11]. Nevertheless, we found that the change of BDNF level in ventricles is more sensitive than the plasma in MI rats. And it is worth noting that the beneficial effect in vivo in this study was induced by intra-myocardium injection with exogenous BDNF. Thus, overexpression of BDNF in the myocardium has positive effect against cardiac ischemia.
Many previous studies have documented the mechanistic link between BDNF and TRPC3/6 channels in distinct cells [31, 32]. In addition, Wang et al. documented that BDNF protected retinal ganglion cells (RGCs) in retinal ischemia/reperfusion-induced cell death by regulating TRPC6 channel. They found that activation TRPC channels before ischemia has early neuroprotective effects on RGCs [33]. Similarly, previous study has found that TRPC6 channel was required for BDNF-induced chemo-attraction of axonal growth cone attraction [16]. Consistently, our study confirmed the cardioprotective role of BDNF in myocardial ischemia, and also indicated TRPC channels participation in this process. Therefore, these findings demonstrated the beneficial role of BDNF/TRPC pathway in ischemic injured heart. Additionally, we demonstrated that BDNF reduced the infarct area of MI in adult murine heart. Conversely, Cai and colleagues reported that BDNF pretreatment increased the infarct area and enhanced the inflammation and injury in aged rats [13]. Just like Halade et al. [12] mentioned, the main difference between them is the age. They used 2-year aging rats whereas we used adult rats and mice.
Previous studies have reported that TRPC3/6 channel-induced neuroprotection was executed by p-CREB. Overexpression of TRPC6 channel markedly increased CREB phosphorylation and enhanced CREB-dependent transcription. A modest level of Ca2+ influx through TRPC6 channels lead to activation of both ERK and CaMK, which converge on CREB and promoted neuronal survival [15]. Furthermore, TRPC6-MEK-CREB and TRPC6-CaMKIV-CREB pathways have been uncovered in neuroprotective role of Resveratrol against ischemia/reperfusion injury [34]. Likewise, in our study, we may assume that BDNF regulates TRPC3/6 channels and further activates CREB signaling pathway, and eventually inhibits cardiomyocytes apoptosis. Of course, it should be noted that excessive activation of TRPC channel may induce calcium overload and cause cell death, so future experiments are needed to validate it. In addition, it should be mentioned that both rats and mice were used in our study. By checking the amino acid sequences, we found that BDNF is high conserved between rat and mouse. Even though, it should be concerned and taken into consideration when interpreting our results.
In summary, the present work demonstrated that BDNF/TrkB protected against cardiac dysfunction and attenuated ischemic-induced apoptosis by regulating TRPC3/6 channels in infarct adult murine hearts. Our study implies that BDNF may be a new therapeutic candidate in the treatment of ischemic heart diseases.
Acknowledgements
This work was supported by Major Program of National Natural Science Foundation of China (81230081), National Natural Science Foundation of China (81200077, 81300080 and 81470462), Specialized Research Fund for the Doctoral Program of Higher Education of China (20112307130004), Natural Science Foundation of Heilongjiang Province (QC2013C087) and Scientific Research Fund of Heilongjiang Provincial Education Department (12531253).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ. et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28-e292
2. Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, Spillmann F, Campesi I, Madeddu P, Quaini F, Emanueli C. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275-1284
3. Van Kanegan MJ, He DN, Dunn DE, Yang P, Newman RA, West AE, Lo DC. BDNF mediates neuroprotection against oxygen-glucose deprivation by the cardiac glycoside oleandrin. J Neurosci. 2014;34:963-968
4. Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF function and intracellular signaling in neurons. Histol Histopathol. 2010;25:237-258
5. Larsson E, Nanobashvili A, Kokaia Z, Lindvall O. Evidence for neuroprotective effects of endogenous brain-derived neurotrophic factor after global forebrain ischemia in rats. J Cereb Blood Flow Metab. 1999;19:1220-1228
6. Ploughman M, Windle V, MacLellan CL, White N, Doré JJ, Corbett D. Brain-derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke. 2009;40:1490-1495
7. Ejiri J, Inoue N, Kobayashi S, Shiraki R, Otsui K, Honjo T, Takahashi M, Ohashi Y, Ichikawa S, Terashima M, Mori T, Awano K, Shinke T, Shite J, Hirata K, Yokozaki H, Kawashima S, Yokoyama M. Possible role of brain-derived neurotrophic factor in the pathogenesis of coronary artery disease. Circulation. 2005;112:2114-2120
8. Jiang H, Liu Y, Zhang Y, Chen ZY. Association of plasma brain-derived neurotrophic factor and cardiovascular risk factors and prognosis in angina pectoris. Biochem Biophys Res Commun. 2011;415:99-103
9. Liu Y, Sun L, Huan Y, Zhao H, Deng J. Application of bFGF and BDNF to improve angiogenesis and cardiac function. J Surg Res. 2006;136:85-91
10. Cao L, Zhang L, Chen S, Yuan Z, Liu S, Shen X, Zheng X, Qi X, Lee KK, Chan JY, Cai D. BDNF-mediated migration of cardiac microvascular endothelial cells is impaired during ageing. J Cell Mol Med. 2012;16:3105-3115
11. Okada S, Yokoyama M, Toko H, Tateno K, Moriya J, Shimizu I, Nojima A, Ito T, Yoshida Y, Kobayashi Y, Katagiri H, Minamino T, Komuro I. Brain-derived neurotrophic factor protects against cardiac dysfunction after myocardial infarction via a central nervous system-mediated pathway. Arterioscler Thromb Vasc Biol. 2012;32:1902-1909
12. Halade GV, Ma Y, Ramirez TA, Zhang J, Dai Q, Hensler JG, Lopez EF, Ghasemi O, Jin YF, Lindsey ML. Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2013;305:H1830-H1842
13. Cai D, Holm JM, Duignan IJ, Zheng J, Xaymardan M, Chin A, Ballard VL, Bella JN, Edelberg JM. BDNF-mediated enhancement of inflammation and injury in the aging heart. Physiol Genomics. 2006;24:191-197
14. Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H. TRP channel and cardiovascular disease. Pharmacol Ther. 2008;118:337-351
15. Jia Y, Zhou J, Tai Y, Wang Y. TRPC channels promote cerebellar granule neuron survival. Nat Neurosci. 2007;10:559-567
16. Li Y, Jia YC, Cui K, Li N, Zheng ZY, Wang YZ, Yuan XB. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894-898
17. Sossin WS, Barker PA. Something old, something new: BDNF-induced neuron survival requires TRPC channel function. Nat Neurosci. 2007;10:537-538
18. Yang B, Lin H, Xu C, Liu Y, Wang H, Han H, Wang Z. Choline produces cytoprotective effects against ischemic myocardial injuries: evidence for the role of cardiac m3 subtype muscarinic acetylcholine receptors. Cell Physiol Biochem. 2005;16:163-174
19. Pan Z, Sun X, Ren J, Li X, Gao X, Lu C, Zhang Y, Sun H, Wang Y, Wang H, Wang J, Xie L, Lu Y, Yang B. miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLoS One. 2012;7:e50515
20. Zhang Y, Sun L, Zhang Y, Liang H, Li X, Cai R, Wang L, Du W, Zhang R, Li J, Wang Z, Ma N, Wang X, Du Z, Yang B, Gao X, Shan H. Overexpression of microRNA-1 causes atrioventricular block in rodents. Int J Biol Sci. 2013;9:455-462
21. Tan X, Li J, Wang X, Chen N, Cai B, Wang G, Shan H, Dong D, Liu Y, Li X, Yang F, Li X, Zhang P, Li X, Yang B, Lu Y. Tanshinone IIA protects against cardiac hypertrophy via inhibiting calcineurin/NFATc3 pathway. Int J Biol Sci. 2011;7:383-389
22. Clark CG, Hasser EM, Kunze DL, Katz DM, Kline DD. Endogenous brain-derived neurotrophic factor in the nucleus tractus solitarius tonically regulates synaptic and autonomic function. J Neurosci. 2011;31:12318-12329
23. Chu W, Wan L, Zhao D, Qu X, Cai F, Huo R, Wang N, Zhu J, Zhang C, Zheng F, Cai R, Dong D, Lu Y, Yang B. Mild hypoxia-induced cardiomyocyte hypertrophy via up-regulation of HIF-1α-mediated TRPC signalling. J Cell Mol Med. 2012;16:2022-2034
24. Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582-7587
25. Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487-491
26. Kaengkan P, Baek SE, Kim JY, Kam KY, Do BR, Lee ES, Kang SG. Administration of mesenchymal stem cells and ziprasidone enhanced amelioration of ischemic brain damage in rats. Mol Cells. 2013;36:534-541
27. Zhang J, Shi Q, Yang P, Xu X, Chen X, Qi C, Zhang J, Lu H, Zhao B, Zheng P, Zhang P, Liu Y. Neuroprotection of neurotrophin-3 against focal cerebral ischemia/reperfusion injury is regulated by hypoxia-responsive element in rats. Neuroscience. 2012;222:1-9
28. Nakamura K, Martin KC, Jackson JK, Beppu K, Woo CW, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia-inducible factor-1alpha in neuroblastoma cells. Cancer Res. 2006;66:4249-4255
29. Donovan MJ, Lin MI, Wiegn P, Ringstedt T, Kraemer R, Hahn R, Wang S, Ibanez CF, Rafii S, Hempstead BL. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development. 2000;127:4531-4540
30. Manni L, Nikolova V, Vyagova D, Chaldakov GN, Aloe L. Reduced plasma levels of NGF and BDNF in patients with acute coronary syndromes. Int J Cardiol. 2005;102:169-171
31. Su B, Ji YS, Sun XL, Liu XH, Chen ZY. Brain-derived neurotrophic factor (BDNF)-induced mitochondrial motility arrest and presynaptic docking contribute to BDNF-enhanced synaptic transmission. J Biol Chem. 2014;289:1213-1226
32. Vohra PK, Thompson MA, Sathish V, Kiel A, Jerde C, Pabelick CM, Singh BB, Prakash YS. TRPC3 regulates release of brain-derived neurotrophic factor from human airway smooth muscle. Biochim Biophys Acta. 2013;1833:2953-2960
33. Wang X, Teng L, Li A, Ge J, Laties AM, Zhang X. TRPC6 channel protects retinal ganglion cells in a rat model of retinal ischemia/reperfusion-induced cell death. Invest Ophthalmol Vis Sci. 2010;51:5751-5758
34. Lin Y, Chen F, Zhang J, Wang T, Wei X, Wu J, Feng Y, Dai Z, Wu Q. Neuroprotective effect of resveratrol on ischemia/reperfusion injury in rats through TRPC6/CREB pathways. J Mol Neurosci. 2013;50:504-513
Author contact
![]() Corresponding authors: Prof. Zhimin Du, PhD; Institute of Clinical Pharmacology of the Second Affiliated Hospital, Harbin Medical University, Xuefu Road 246#, Nangang District, Harbin 150086, China. Fax: +86 451 86665559; Tel: +86 451 86605353; Email: dzm1956com or Prof. Yue Li, MD; Department of Cardiology, the First Affiliated Hospital, Harbin Medical University, Youzheng Street 23#, Nangang District, Harbin 150001, China. Fax: +86 451 53675733; Tel: +86 451 85555673; E-mail address: ly99ly163.com
Corresponding authors: Prof. Zhimin Du, PhD; Institute of Clinical Pharmacology of the Second Affiliated Hospital, Harbin Medical University, Xuefu Road 246#, Nangang District, Harbin 150086, China. Fax: +86 451 86665559; Tel: +86 451 86605353; Email: dzm1956com or Prof. Yue Li, MD; Department of Cardiology, the First Affiliated Hospital, Harbin Medical University, Youzheng Street 23#, Nangang District, Harbin 150001, China. Fax: +86 451 53675733; Tel: +86 451 85555673; E-mail address: ly99ly163.com