Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(5):569-586. doi:10.7150/ijbs.11039 This issue Cite
Research Paper
The effects of Insulin Pre-Administration in Mice Exposed to Ethanol: Alleviating Hepatic Oxidative Injury through Anti-Oxidative, Anti-Apoptotic Activities and Deteriorating Hepatic Steatosis through SRBEP-1c Activation
Jiangzheng Liu1, Xin Wang1, Zhengwu Peng2, Tao Zhang1, Hao Wu1, Weihua Yu1, Deqing Kong1, Ying Liu1, Hua Bai1, Rui Liu1, Xiaodi Zhang1, Chunxu Hai1 ![]()
1. Department of Toxicology, the Ministry of Education Key Lab of Hazard Assessment and Control in Special Operational Environment, Shaanxi Provincial Key Lab of Free radical biology and medicine, School of Public Health, The Fourth Military Medical University, Xi'an, 710032, P. R. China;
2. Department of Psychiatry, Xijing Hospital, The Fourth Military Medical University, Xi'an, 710032, P. R. China.
Received 2014-11-11; Accepted 2015-1-21; Published 2015-4-3
Abstract
Alcoholic liver disease (ALD) has become an important liver disease hazard to public and personal health. Oxidative stress is believed to be responsible for the pathological changes in ALD. Previous studies have showed that insulin, a classic regulator of glucose metabolism, has significant anti-oxidative function and plays an important role in maintaining the redox balance. For addressing the effects and mechanisms of insulin pre-administration on ethanol-induced liver oxidative injury, we investigated histopathology, inflammatory factors, apoptosis, mitochondrial dysfunction, oxidative stress, antioxidant defense system, ethanol metabolic enzymes and lipid disorder in liver of ethanol-exposed mice pretreatment with insulin or not. There are several novel findings in our study. First, we found insulin pre-administration alleviated acute ethanol exposure-induced liver injury and inflammation reflected by the decrease of serum AST and ALT activities, the improvement of pathological alteration and the inhibition of TNF-α and IL-6 expressions. Second, insulin pre-administration could significantly reduce apoptosis and ameliorate mitochondrial dysfunction in liver of mice exposed to ethanol, supporting by decreasing caspases-3 activities and the ratio of Bax/Bcl-2, increasing mitochondrial viability and mitochondrial oxygen consumption, inhibition of the decline of ATP levels and mitochondrial ROS accumulation. Third, insulin pre-administration prevented ethanol-mediated oxidative stress and enhance antioxidant defense system, which is evaluated by the decline of MDA levels and the rise of GSH/GSSG, the up-regulations of antioxidant enzymes CAT, SOD, GR through Nrf-2 dependent pathway. Forth, the modification of ethanol metabolism pathway such as the inhibition of CYP2E1, the activation of ALDH might be involved in the anti-oxidative and protective effects exerted by insulin pre-administration against acute ethanol exposure in mice. Finally, insulin pre-administration deteriorated hepatic steatosis in mice exposed to ethanol might be through SRBEP-1c activation. In summary, these results indicated that insulin pre-administration effectively alleviated liver oxidative injury through anti-inflammatory, anti-oxidative and anti-apoptotic activities but also deteriorated hepatic steatosis through SRBEP-1c activation in mice exposed to ethanol. Our study provided novel insight about the effects and mechanisms of insulin on ethanol-induced liver injury.
Keywords: Insulin, Alcoholic liver disease, Oxidative injury, Nrf-2, Ethanol metabolism, Inflammatory.
Introduction
The liver is the major metabolic organ for alcohol but also the main target organ for alcoholic toxicity (1). In recent years, the incidence of alcoholic liver disease (ALD) in whole world increases year by year, ALD as the most important disease caused by alcohol abuse is becoming an important public health problem in the whole world (2). Long-term and heave drinking can lead to ALD, including fatty liver, alcoholic hepatitis, liver fibrosis, liver cirrhosis and hepatocellular carcinoma. About 2/3 of alcoholics can develop into alcoholic liver disease (3). The further study of the endogenous protective mechanisms against alcoholic liver injury is worthy for our attention.
The pathogenesis of ALD is apparently complex. A lot of experimental and clinical evidences have revealed that oxidative stress (4) and acetaldehyde (5) play a key role in the pathogenesis of ALD, which is including hepatocytes dysfunction, dyslipidaemia, apoptosis, inflammation, fibrosis, and carcinogenesis (6). It should be noted that 80% of the consumed ethanol is metabolized in the liver, and liver is the important target organ damaged by ethanol exposure (7). In the hepatocytes, under normal condition ethanol is mainly oxidized to acetaldehyde by alcohol dehydrogenase (ADH), and then the toxic acetaldehyde oxidized to non-toxic acetic acid by acetaldehyde dehydrogenase (ALDH) (8). Ethanol intake can also induce the activation of an important alcohol metabolizing enzyme CYP2E1 with the result of a large amount of ROS generation, which in turn results in oxidative stress (9). The accumulation of high active ROS such as hydrogen peroxide (H2O2), superoxide anion (O2-·), and hydroxyl radical (·OH) in the liver during the ethanol oxidative metabolism process plays a key role in the development of alcoholic injury (10). Many studies had revealed that the ROS accumulation can overwhelm the intrinsic antioxidant defense system and then trigger oxidative stress and represent a significant cytotoxicity effects in liver and other tissues (11). During the development of ALD, the burst of ROS generation could directly cause oxidative injury to various intracellular bioactive molecules, resulting in the peroxidation modification of lipids to form lipid peroxides, the peroxidation protein to form carbonyl, and the damage of DNA, which will finally induce oxidative injury in liver (4). ROS accumulation also indirectly affect the normal cellular signal transduction processes, which facilitates the hepatotoxicity actions of ROS (12). Of the various mechanisms about the pathogenesis of alcoholic liver injury, oxidative stress is supported to be the foundational and most important mechanism (4). Acetaldehyde is the toxic by-product during the ethanol metabolism and it chemical reactivity and toxicity is more strongly than that of ethanol (5). There have been several studies showing acetaldehyde has obvious cytotoxic effect on liver cells. Acetaldehyde can combined with protein and form aldehyde-protein adduct, which will leading to protein dysfunction, and as a new antigen it is involved in inflammation action, eventually resulting in cells denaturation and death (13). The removal of acetaldehyde is considered to be the vital method in the prevention of ALD (14). In addition, liver steatosis is believed to be the typical manifestation of alcoholic liver injury especially in the early stage of ALD and SREBP-1c is the key enzyme involved in the synthesis of triglyceride in hepatocytes (15). When in the status of steatosis, the liver will becomes more susceptible to oxidative stress, which contributes the more serve liver pathogenesis (16).
Mitochondria are the main organelles for intracellular oxidation reactions and are also the main place for ROS production (17). During the ethanol metabolism process, NAD as co-enzyme is a large number of consumption, resulting in a reduction of the ratio of intracellular NAD/NADH, which can promotes mitochondrial respiratory chain circulation and generate excess ROS (12). When mitochondrial ROS generation beyond the antioxidant ability of scavenging, ROS can damage mitochondria and overflow into the cytoplasm, leading to cell damage (18). Meanwhile mitochondria are particularly susceptible to oxidative stress, excessive production of ROS could decrease the mitochondrial membrane potential (MMP) and inhibit mitochondrial function, resulting in the decrease in mitochondrial activity, mitochondrial oxygen consumption and ATP generation (19). ATP level is an important index to evaluate the function of mitochondrial energy metabolism and could determine death fate of cells (20). The presence of mitochondrial dysfunction has been shown in many studies in vivo and vitro about ALD (18). Mitochondrial damage can lead to the release of caspase-3 into cytoplasm, which could trigger the process of apoptosis. Oxidative stress and mitochondrial dysfunction can cause the occurrence of hepatocytes apoptosis (21). A growing number of studies suggest that apoptosis plays an important role in alcoholic liver disease (22). A high rate of apoptosis in the liver tissue has also been described in the ALD models in experimental studies, and it should be note that the rate of apoptosis is closely associated with liver function parameters such as ALT and AST (21). The prevention of cellular apoptosis and necrosis is an important effective strategy against alcoholic liver injury (22). Bcl-2 family proteins (including anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax) play a key regulatory role in apoptotic process. The ratio of Bax/Bcl-2 represents the degree of apoptosis in a certain extent (23).
Under normal physiological conditions, oxidative stress damage can be prevented by antioxidant defense system, which is consists of non-enzymatic chain breaking antioxidants (GSH, Vitamin C etc.) to scavenge of ROS and antioxidase (SOD, CAT, GR ,HO-1 etc.) to catalyze the elimination of ROS accumulation (24). It has been documented that serves ethanol exposure could impair the non-enzymatic and enzymatic antioxidative system in crowd with drinking and experimental animals. Recent evidences showed that enhancing antioxidative activities by supplement with antioxidant could effectively relieve alcoholic liver injury (12). GSH as the important endogenous antioxidant plays a key role in the antioxidative response by directly scavenging of ROS and recycling other antioxidants (25). The transcription factor Nrf-2 has been proved to play a key regularly role in antioxidative response. Under the stimulation of oxidative stress, Nrf-2 can dissociate with keap-1 and translocate to the nuclear. In the nuclear, Nrf-2 binds on the antioxidant-response elements (ARE) of gene promoters and then up-regulates the expressions of antioxidative defense genes, including SOD-2 (located in mitochondrial ), heme oxygenase-1 (HO-1) and glutathione reductase (GR) (26) and etc. The activation of Nrf-2 is important for maintaining intracellular and mitochondrial GSH balance and increasing the activities of antioxidative enzymes to protect cells from oxidative insult mediated by ethanol (27).
Inflammation is an important feature of ALD (28). Alcohol exposure can cause the activation of inflammatory cells such as Kupffer cell and macrophages in liver, resulting in the increase of the generation of inflammatory cytokines such as TNF-α and IL-6 (29). TNF-α can bind to the corresponding membrane receptor and then further increase the generation of ROS and deteriorate oxidative stress. Excessive generation of inflammatory cytokines could also induce cell apoptosis, which can be inhibited by the supplement with antioxidants (28). The anti-inflammatory therapy has a certain therapeutic effect on ALD (30). Adiponectin as an endogenous bioactive peptides secreted by adipose cells is proved to have the functions of enhancing insulin sensitivity, anti-inflammatory and antioxidative abilities (31). The secretion disorder of Adiponectin also participates in the occurrence and development process of ALD (32).
In recent years, insulin resistance is found to be closely related to ALD. High dose of ethanol exposure could suppress insulin signaling pathway in the liver (33). And it should be noted that there have been many researches revealing that insulin possesses various biological effects including anti-oxidative, anti-inflammatory, anti-apoptosis and etc (34). Recently research in our laboratory also confirmed that insulin plays an important role in the regulation of the cellular redox balance via Nrf-2 dependent pathway (34, 35). However, what are the effects of insulin on alcoholic liver injury and the mechanisms under these responses remains largely unclear. In the present study, mice exposed to ethanol were pre-administration with insulin or not to evaluate the actions and mechanisms of insulin against ethanol toxicity. Our study might provide significant and novel information about the role and underlying mechanism of insulin pre-administration on the pathogenesis of ALD and deepen our understanding of the development of ALD.
Methods and Materials
Materials and kits
Ethanol, Trizol reagent, MTT, MitoSOX, rhodamine 123, DHE, DMSO, Hochest-33342 were purchased from Sigma Chemical Corp (St. Louis, USA). Insulin was purchased from Novo Nordisk Corp (Copenhagen, Denmark). The ATP content and Caspases-3, CYP2E1, ALDH activity assay kits were purchased from Beyotime Institute of Biotechnology (Nanjing, China). Detection kits for ALT, AST, CAT, SOD, GR, ADH activities, and MDA, GSH, GSSG levels were provided from Jianchen Corp (Nanjing, China). ELISA kit for Adiponectin level was purchased from HUAMEI Biotechnology Corp (Wuhan, China). Cytoplasmic and nuclear fractionation isolation kit was purchased from Beyotime Institute of Biotechnology (Nanjing, China). RT-PCR primers for β-actin, IL-6, TNF-α, CYP2E1 and SREBP-1 were synthesized by AuGCT Biotechnology (Beijing, China). RT-PCR one step cDNA synthesis kit and RT-PCR amplification kit was purchased from TaKaRa Biotechnology Corp (Tokyo, Japan). Nrf-2, GR, CAT, SOD-2, and β-action antibodies were purchased from Santa Cruz Biotechnology Corp (CA, USA). Bcl-2, Bax, ADH and Lamin B antibodies were purchased from Cell Signaling Biotechnology Corp (MA, USA). SREBP-1c antibody was purchased from Boster Biotechnology Corp (Wuhan, China). Tubulin, CYP2E1 and ALDH2 antibodies were purchased from Abcam Biotechnology Corp (Cambridge, USA). Anti-rabbit IgG and anti-mouse IgG were purchased from Boster Biotechnology Corp (Wuhan, China). All other chemicals and materials in our study were obtained from Tianjin Chemical Company (Tianjin, China) with highest purity commercially available.
Experimental protocols and preparation of samples
Adult male C57BL/6 mice at 12-14 weeks of age, weighing 33-34 g, were purchased from the Laboratory Animal Center of Fourth Military Medical University. They were housed in plastic cages with controlled temperature (24 ± 2°C) and humidity (60%, range 55-65%) under a 12h/12h light/dark cycle and were free access to tap commercial rodent chow and water. Experimental mice were randomly divided into four groups as follows: (A) Control group, negative control mice (n=8), were given saline (5 mg/kg, intraperitoneal injection) 4 h-prior to saline (10 mg/kg, intragastrical) twice a day for 7 days; (B) Insulin group, insulin control mice (n=8), were given insulin (5IU/kg, intraperitoneal injection) 4h-prior to saline (10mg/kg, intragastrical) twice a day for 7 days; (C) EtOH group, positive control mice (n=8), were given saline (5mg/kg, intraperitoneal injection) 4 h-prior to ethanol (5 g/kg, intragastrical) twice a day for 7 days; (D) Ins+EtOH group, insulin pre-administration mice (n=8), were given insulin (5 IU/kg, intraperitoneal injection) 4 h-prior to ethanol (5 g/kg, intragastrical) twice a day for 7 days. The dosage of insulin was based on our preliminary studies and previous work published by us and others. Ethanol was diluted to 51.3% to prevent acute gastric irritation. Insulin was fresh prepared each day. After ethanol exposure for 7 days, the final body weights of the mice were examined. All animal experiments were performed with the approval of the Animal Care and Use Committee of Fourth Military Medical University.
The mice were killed about 8h after the last ethanol challenge under anesthetized with sodium pentobarbital (50 mg/kg, intragastrical). Fresh blood was immediately collected from the canthus vein clump just prior to sacrifice. The blood was then centrifugated at 2000 g for 20 min to obtain the plasma, which was then stored at -20°C for further analysis such as AST, ALT activities and Adiponectin level. The livers were weighed and the values of liver weight/body weight were calculated. First, portions of livers were used in the preparation of fresh mitochondria for determination mitochondrial viability, MMP, mitochondrial oxygen consumption and mitochondrial ROS accumulation. Second, portions of livers were made into hepatic cryosections for detection of hepatic ROS accumulation. Third, portions of liver tissues were homogenized in ice cold PBS using a potter type homogenizer (Biospec, USA). The homogenate was then centrifuged at 3000 g for 15 min and then the supernatant was stored at -20°C for further biochemical analysis such as MDA level, SOD, CAT, GR, CYP2E1, ADH and ALDH activity. Above assays is manipulated according to the protocols described as below. Fourth, portions of liver tissues were stored at -20°C for further analysis such as TG, MPO, ATP, GSH, GSSG level using corresponding kits. Fifth, portions of live tissues were preserved in paraformaldehyde for the further pathological histology like H-E stain examination. Finally, portions of livers were immediately frozen in liquid nitrogen for western blot and RT-PCR analysis. The quantization of protein was analysis by a commercial protein quantification assay kit from Thermo Biotechnology Corp (MA, USA) using the bovine serum albumin (BSA) as a standard.
Liver histological examination by Haematoxylin and Eosin Stain
Parts of liver tissues were fixed in paraformaldehyde (4% in PBS) for 24h, embedded in paraffin, and then serially sectioned at 5 μm. Standard haematoxylin and eosin staining according to the conventional method was performed on these sections. A Nikon microscope (Melville, USA) was used to capture images and analyze the H&E stained tissue sections at 40× and 80×magnification.
Determination of AST and ALT activity in serum
AST and ALT activity in serum was measured by corresponding commercial activity assay kits (Jianchen, China) using DNPH as coloring reagent according to the manufacturer's instruction. Briefly, enzymes activities of ALT and AST were determined spectrophotometrically by monitoring the changes of absorbance (340 nm) in 3 min after the addition of reaction mix to each sample at 37 °C, and one unit of enzymes activity is defined as the amount of enzyme that catalyses the formation of 1 µmol of NAD+ in one minute. Result of AST and ALT activity in serum was expressed as U/L.
Determination of triglycerides level in liver
Triglycerides (TG) level in liver was measured by a commercial TG concentration assay kit for tissue (Jianchen, China) according to the manufacturer's instruction. Briefly, portions of liver tissues were homogenized in Tris-acetate buffer supplied by the kit and detected by triglyceride reagents. Result of TG level in liver was expressed as mg/g protein.
Determination of MPO activity in liver homogenate
MPO activity in liver was measured by a commercial MPO activity assay kit (Jianchen, China) according to the manufacturer's instruction. Briefly, portions of liver tissues were homogenized in the buffer supplied by the kit using a potter type homogenizer (Biospec, USA). Result of MPO activity in liver was expressed as U/mg protein. Protein quantification was measured by the Bradford reagent (Thermo, USA).
Determination of Adiponectin level in serum
Adiponectin level in serum was measured by a commercial Adiponectin ELISA assay kit (HUAMEI, China) according to the manufacturer's instruction. Result of Adiponectin level in serum was expressed as μg/ml.
Determination of mRNA expression by RT-PCR
Real-time-PCR (RT-PCR) analysis for mRNA determination was performed as following. Briefly, portions of liver tissues frozen in liquid nitrogen were pounded to pieces and homogenized in Trizol reagent. Total RNA from liver tissue was isolated according to the standard procedures. Then the quantization and purity of nucleic acids were determinate by the ratio of A260/A280 using an ultraviolet spectrophotometer (Bio-Rad, USA). A 2 μg of total RNA was used for cDNA synthesis using a commercial RT-PCR one step cDNA synthesis kit (TaKaRa, Japan) following to the manufacturer's instruction. Then 2 μl of reverse transcription product cDNA was added in SYBR Green Supermix to amplification using a commercial RT-PCR amplification kit (TaKaRa, Japan) according to the manufacturer's instruction in a RT-PCR Cycler (Bio-Rad, USA). The primers for target genes were synthesized by AuGCT Biotechnology Company (AuGCT, China) as shown in Table 1. The program used in the thermal cycling was 10 min at 95 °C followed by a standard procedure as: 45 cycles of a 2-step PCR process (92°C for 15 sec, 60°C for 1 min) in a final volume of 20 μl. The melting points of PCR products were assessed by incubating at 65 °C for 1 min followed by a 1 °C per minute rise over 30 min. The quantization of mRNA was calculated according to the attached software (Bio-Rad, USA) and normalized to the quality of β-actin.
The Sequences of sense and anti-sense primers for target genes.
| Gene | Sense primer(5' to 3') | Anti-sense primer(5' to 3') |
|---|---|---|
| β-actin | CTGTCCCTGTATGCCTCT | ATGTCACGCACGATTTCC |
| IL-6 | GAAATCGTGGAAATGAG | TAGGTTTGCCGAGTAGA |
| TNF-α | GTGCTCCTCACCCACACC | GGAAGACTCCTCCCAGGT |
| CYP2E1 | CGAGGGGACATTCCTGTG | CCAGGAAGTGTGCCTCTC |
| SREBP-1 | CGACATCAACATGCTTCAG | GGAAGCTTCAAGAGGAGC |
Determination of caspase-3 activity in liver homogenate.
Caspase-3 activity in liver was measured by a commercial Caspase-3 activity detection kit (Beyotime, China) according to the manufacturer's instruction. Portions of liver tissues were homogenized in the lysis buffer supplied by the kit using a potter type homogenizer (Biospec, USA) for 30 s, and the caspase-3 activity in the supernatant was detected following the manufacturer's instructions. The optical density was measured at 405 nm using a microplate spectrophotometer (Bio-Rad, USA). Result of caspase-3 activity in liver was expressed as U/mg protein. Protein quantification was measured by the Bradford reagent (Thermo, USA).
Preparation of total, nuclear and cytoplasm protein extract and determination of protein expression by Western blotting
For preparation of total protein extract, portions of liver tissues frozen in liquid nitrogen were pounded to pieces and homogenized in ice-cold RIPA protein lysis buffer (Beyotime, China) containing protease inhibitor PMSF following lysis for 30 min to obtain total proteins. Then the lysis was centrifuged at 20,000×g for 20 min, and the supernatants were added with same volume of 2×SDS buffer and 100mM DTT and then boiled for 5 min. Then the mixture was centrifuged at 10,000×g for 10 min. The supernatants were stored at - 80°C and used for detection the total protein expressions by western blotting. For preparation of nuclear and cytoplasm extracts, a commercially nuclear fractionation isolation kit (Beyotime, China) was conducted by a differential centrifugation method following to the manufacturer's instructions. Briefly, portions of liver tissues frozen in liquid nitrogen were pounded to pieces and homogenized with the lysis supplied by the kit for 15 min at 4°C and then the homogenate centrifuged at 13,500g for 5 min at 4°C. The supernatant part was geared to the cytoplasm extract and precipitation part was geared to the nuclear extracts. Finally, the samples of corresponding extracts were stored at - 80°C and used for detection the nuclear and cytoplasm protein expressions by western blotting.
For determination of protein expression, Western blotting analysis was conducted following the standard method. Protein quantification was measured by the Bradford reagent (Thermo, USA). 40ng protein extraction was separated using 10% or 15% polyacrylamide gels and transferred to nitrocellulose membrane (Millipore, USA). For immune-detection, the membrane was blocked with 5% skimmed milk in TBST buffer (TBS with 0.5% Tween-20) for 1 h and then incubated with the following primary antibodies diluted in 4% BSA as indicated: Bcl-2 (Rat anti-human IgG, 1:1000), Bax (Goat anti-human IgG, 1:2000), HO-1 (Rat anti-human IgG, 1:1000), SOD-1 (Rat anti-human IgG, 1:500), GR (Rat anti-human IgG, 1:500), Nrf-2 (Rat anti-human IgG, 1:2000), CYP2E1 (Goat anti-human IgG, 1:2000), ADH (Rat anti-human IgG, 1:2000), ALDH (Rat anti-human IgG, 1:2000), SREBP-1c (Rat anti-human IgG, 1:2000), Lamin B (Rat anti-human IgG, 1:2000) , β-actin (Rat anti-human IgG, 1:2000) and Tubulin (Rat anti-human IgG, 1:2000) respectively at 4°C for about 24h. After washed with TBS-T triple for 15min, membrane was incubated with the following horseradish peroxidase-conjugated secondary antibodies diluted in TBST as indicated: anti-rabbit IgG (1:5000) and anti-mouse IgG (1:5000). The measurement of specific antibody binding was conducted using an image analyzer Quantity One System and analyzed by the attached software (Bio-Rad, USA) from three independent observations. The quantifications of target protein levels in samples were adjusted for Lamin B, β-actin and Tubulin, which were used as internal reference for nuclear, cytoplasm and total protein extracts respectively.
Preparation of fresh liver mitochondria
For preparation of fresh liver mitochondria, a commercial mitochondrial fractionation isolation kit (Beyotime, China) was conducted following the manufacturer's instruction. After the animal was sacrificed, portion of liver was immediately removed and used in the preparation of fresh mitochondria. Briefly, liver was homogenized with the lysis buffer provide by the kit for 30 min at 4°C using a homogenizer (Biospec, USA). The nuclear fraction was removed by centrifugation at 1000g for 20 min at 4°C. Then the supernatant was centrifuged at 10,000 g for 20 min at 4°C to obtain the pellet part belonged to fresh mitochondrial fraction. The determination of mitochondrial viability, MMP, mitochondrial oxygen consumption and mitochondrial ROS accumulation was done on the same day by the following methods. The quantification of mitochondria was measured by the protein quantification using the Bradford reagent (Thermo, USA).
Determination of mitochondrial viability and oxygen consumption
Mitochondrial viability was determinate using a microculture tetrazolium (MTT) assay according to the literature's methods. Briefly, 100μl fresh liver mitochondria (2mg protein/ml) were rinsed with 100 μg/ml of MTT. After 15 min incubation at 37°C, the mix was centrifuged at 1,000 g for 20 min to obtain the MTT formazan crystals, which were then dissolved in 150 μl DMSO. Optical density of the dissolving liquid was detected by a microtiter plate reader (Bio-Rad, USA) at 492 nm. Result of mitochondrial viability in liver was expressed as folds of control. The oxygen consumption of fresh mitochondria was detected by Clark electrode method. Briefly, 100μl fresh prepared liver mitochondria (20mg protein/ml) were mixed with oxygen saturated saline and assayed for oxygen consumption at 37 °C in a thermostatically controlled oxygen apparatus equipped with a Clark electrode instrument (Hansatech, UK). The result of mitochondrial oxygen consumption was analysis by attached hansatech oxygen consumption detection software and expressed as folds of control. Protein quantification was measured by the Bradford reagent (Thermo, USA).
Determination of mitochondrial ROS accumulation and mitochondrial membrane potential
Mitochondrial ROS accumulation and mitochondrial membrane potential (MMP) was determined using specific fluorescence probe MitoSOX and rhodamine 123 respectively. Briefly, 100μl fresh liver mitochondria (2mg protein/ml) were incubated with 10 μM MitoSOX or 10 μM rhodamine 123 in the dark for 20 min at 37°C. Then mitochondria were washed with ice-cold PBS and mitochondrial ROS accumulation was measured using a flow cytometer (Accuri, USA) immediately. Result of mitochondrial ROS accumulation and MMP in liver was expressed as folds of control. Protein quantification was measured by the Bradford reagent (Thermo, USA).
Determination of hepatic ATP level
For determination of ATP level in liver, a commercial ATP determination kit (Beyotime, China) was conducted. Briefly, portions of liver tissues were homogenized with the lysis buffer supplied by the kit using a potter type homogenizer (Biospec, USA), and the ATP level in the supernatant was detected following the manufacturer's instructions. Result of hepatic ATP level was expressed as μmol/g protein. Protein quantification was measured by the Bradford reagent (Thermo, USA).
Determination of hepatic ROS accumulation in cryosections
Hepatic ROS accumulation in cryosections was determined using a specific ROS fluorescence probe DHE. After the animal was sacrificed, portion of liver was immediately removed and used in the preparation of cryosections of liver (10μm), which were then stained with 10 μM DHE and Hoechst 33342 in the dark for 20 min at 37°C, washed twice, and examined with a laser scanning confocal microscopy (Olympus, Japan) at 200× magnification immediately.
Determination of MDA, GSH and GSSG level and SOD, CAT, GR, CYP2E1, ADH and ALDH activity
For determination of MDA, GSH and GSSG level and SOD, CAT, GR, CYP2E1, ADH, ALDH activity in liver homogenate or determination of MDA in mitochondria homogenate, corresponding commercial determination kits were conducted according to the standard procedures in the manufacturer's instructions. Results of MDA, GSH and GSSG level were expressed as µmol/mg-protein (in the homogenates), results of SOD, CAT, GR, CYP2E1, ADH and ALDH activity were expressed as U/mg-protein (in the homogenates). Protein quantification was measured by the Bradford reagent (Thermo, USA).
Statistical analysis
The results about the figures and tables in our study were expressed as mean ± SD, and all statistical comparisons were made by means of one-way analysis of variance (ANOVA) followed by Student's t-test using a commercially available statistic software package (SPSS, USA). Statistical significance was established at a p value less than 0.05.
Results
Insulin pre-administration alleviated acute ethanol exposure-induced liver injury and inflammation in mice
We first examined the alterations of body weight gains and the values of liver weight/body weight about the experimental mice exposed to ethanol with insulin pre-administration or not during the experimental period. As shown in Table 2, ethanol exposure significantly reduced the body weight gains and showed a high value of liver weight/body weight compared to control. Insulin administration alone had no effects on body weight gains and the values of liver weight/body weight, insulin pre-administration effectively decreased the values of liver weight/body weight but had no effects on body weight gains compared to ethanol group. These results indicated that insulin pre-treatment might attenuate ethanol-induced liver swelling in mice.
Effects of insulin pre-administration on body weight and liver weight in mice exposed to ethanol
| Group | Body weight (g) | Body weight gain (g) | Liver weight (g) | Liver weight/Body weight | |
|---|---|---|---|---|---|
| Initial | Final | Final | Final | ||
| Control | 33.4±0.8 | 35.8±1.3 | 2.1±0.7 | 1.6±0.07 | 0.047±0.002 |
| Insulin | 33.5±0.7 | 34.9±1.1 | 1.5±1.2 | 1.6±0.03 | 0.048±0.001 |
| EtOH Ins+EtOH | 33.7±0.6 33.1±0.9 | 30.1±1.6* 31.1±0.7* | -3.5±1.5* -3.0±1.1* | 1.5±0.07 * 1.4±0.05*# | 0.052±0.003 * 0.049±0.003*# |
Results are expressed as mean values of eight mice per group (± SD; n = 8). *P< 0.05, compared to control group; #P < 0.05, compared to EtOH group.
To examine whether insulin pre-administration might alleviate liver injury induced by ethanol in mice, the examination of liver histological alterations by Haematoxylin and Eosin Stain, the measurement of ALT and AST activities in serum were conducted. As shown in Fig.1A, ethanol exposure could induce the disordered arrangement of hepatocyte and the swelling of hepatocytes, indicating the typical pathological alterations of ALD. Ethanol induced histopathological disorder was effectively improved by insulin pre-administration. The protective effect of insulin was also confirmed by ALT and AST assay. As illustrated in Fig. 1B, insulin pre-administration significantly alleviated the increase of ALT and AST activities in serum mediated by ethanol exposure. Above results suggested that insulin pre-administration effectively protected the liver against the alcoholic toxicity. To investigate whether insulin pre-administration could alleviate inflammation in liver of mice exposed to ethanol, we next examined the hepatic MPO activity and hepatic mRNA expressions of TNF-α and IL-6. As noted in Fig.1C-E, insulin pre-administration significantly inhibited the increase of MPO activity, gene expressions of TNF-α and IL-6 in liver of mice exposed to ethanol. We next also detected the serum TNF-α level in these mice, the results revealed in Fig.1C showed that insulin pre-administration also alleviated the ethanol-induced high level of serum TNF-α level. As demonstrated in Fig. 1G, insulin administration alone could significantly promote the serum Adiponectin level but ethanol exposure could decline the level of serum Adiponectin, and we found that insulin pre-administration effectively inhibited the decrease of serum Adiponectin level in mice exposed to ethanol. These data indicated that insulin pre-administration had significantly anti-inflammatory effect in ALD.
Insulin pre-administration alleviated acute ethanol exposure-induced liver injury and inflammation in mice. (A) H&E staining of liver; (B) ALT and AST level in serum; (C) Hepatic MPO activity; (D) Hepatic TNF-α mRNA level determinate by RT-PCR; (E) Hepatic IL-6 mRNA level determinate by RT-PCR; (F) Serum TNF-α level determinate by ELISA assay; (G) Serum Adiponectin level determinate by ELISA assay. Data of B-C and F-G were expressed as mean ± SD of eight mice per group (n = 8). Data of D and E were expressed as the mean ± SD of three independent experiments (n = 3). *P< 0.05, compared to control group; #P < 0.05, compared to EtOH group.

Insulin pre-administration ameliorated acute ethanol exposure-induced apoptosis and mitochondrial dysfunction in liver of mice
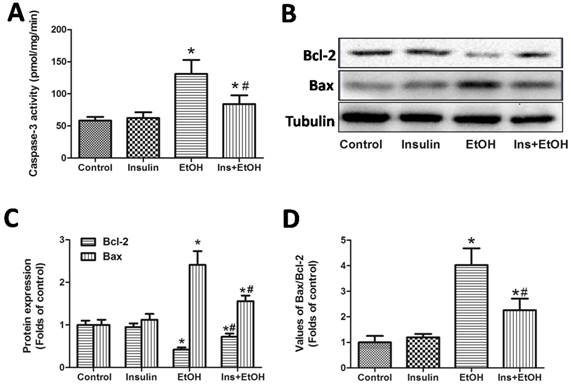
To investigate whether insulin pre-administration could alleviate apoptosis in liver of mice exposed to ethanol, we next examined the hepatic caspases-3 activity, protein expressions of hepatic Bcl-2 and Bax. As shown in Fig.2A, obvious increase of hepatic caspases-3 activity was observed in ethanol-treated mice compared with that of control group, while pre-treatment with insulin significantly reduced caspases-3 activity. To confirm the alleviative effects of insulin pre-administration on apoptosis in liver, we then examined the alteration of Bcl-2 and Bax expressions. As illustrated in Fig.2B-D, insulin administration alone had no effects of Bcl-2 and Bax expressions, but insulin pre-administration could effectively blunt the ethanol-induced the down-regulation of Bcl-2 expression, up-regulation of Bax expression and the increase of value of Bax/Bcl-2. These results indicated that insulin pre-administration could protect liver cells from apoptosis in mice suffering ethanol insult, and inhibition of mitochondrial apoptosis pathway might contribute to the insulin pre-administration-induced protective actin in apoptosis.
Insulin pre-administration ameliorated acute ethanol exposure-induced apoptosis in liver of mice. (A) Hepatic Caspase-3 activity; (B) Bcl-2, Bax protein expressions in liver determinate by Western blotting; (C) Values of the Bcl-2, Bax protein expressions expressed as the folds of control; (D) Values of Bax/Bcl-2 expressed as the folds of control. Normalizations of Western blots were ensured by Tubulin. Data of A were expressed as mean ± SD of eight mice per group (n = 8). Data of C and D were expressed as the mean ± SD of three independent experiments (n = 3). *P< 0.05, compared to control group; #P < 0.05, compared to EtOH group.
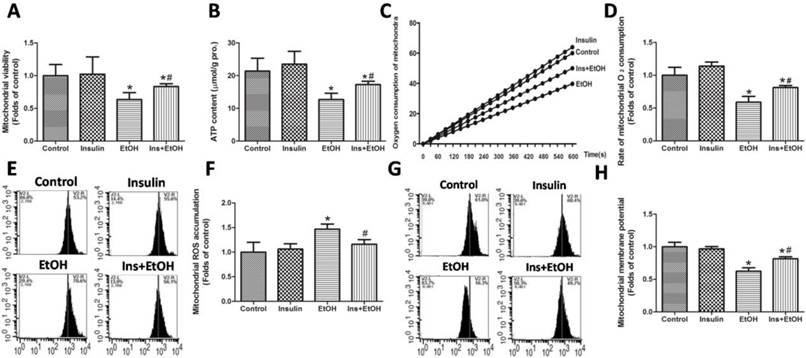
To address whether insulin pre-administration could alleviate ethanol exposure-induced mitochondrial dysfunction in liver, we next investigated the mitochondrial function by measuring mitochondrial viability, ATP level, Rate of mitochondrial oxygen consumption, mitochondrial ROS accumulation and MMP in mice receiving different treatments. As shown in Fig.3A, treatment with ethanol resulted in significant decrease in mitochondrial viability, which was markedly prevented by pre-administration with insulin. As noted in Fig.3B, the liver from ethanol-treated mice showed a marked decrease in ATP level compared with control group, but mice pre-administration with insulin exhibited a higher ATP level compared with the ethanol-treated ones, indicating that insulin improved the energy metabolism in mitochondria. Cellular respiration process occurred in the mitochondria needs oxygen supplement. As illustrated in Fig.3C and D, results using Clark electrode showed that there was remarkable inhibition of mitochondrial oxygen consumption rate in mice receiving ethanol, while insulin pre-administration effectively alleviated the inhibition. As shown in Fig.3E and F, we confirmed that there was significant increase in mitochondrial ROS accumulation in ethanol-treated mice, and insulin pre-administration almost completely abolished the increase. As illustrated in Fig.3G and H, ethanol exposure notably decreased MMP compared with the control group, however, a significantly increase of MMP in liver cells of mice pre-administration with insulin was observed compared to that of ethanol treatment mice. Taking together with the data about apoptosis in Fig.2, above results indicated that the inhibition of mitochondrial dysfunction might be responsible for the anti-apoptotic and protective effects exerted by insulin pre-administration against ethanol toxicity.
Insulin pre-administration ameliorated acute ethanol exposure-induced mitochondrial dysfunction in liver of mice. (A) Mitochondrial viability measured by MTT; (B) Hepatic ATP level; (C) Mitochondrial oxygen consumption in a thermostatically controlled oxygen apparatus; (D) Rate of mitochondrial oxygen consumption expressed as the folds of control; (E) Mitochondrial ROS accumulation analyzed by MitoSOX staining using flow cytometer; (F) Values of mitochondrial ROS accumulation calculated by MitoSOX fluorescence intensity expressed as the folds of control; (G) MMP analyzed by rhodamine 123 staining using flow cytometer; (H) Values of MMP calculated by rhodamine 123 fluorescence intensity expressed as the folds of control. Data of A and B were expressed as mean ± SD (n = 8). Data of D, F and H were expressed as the mean ± SD of three independent experiments (n = 3). *P< 0.05, compared to control group; #P < 0.05, compared to EtOH group.
Insulin pre-administration reduced ethanol-mediated oxidative stress and enhanced antioxidant defense system in liver of mice
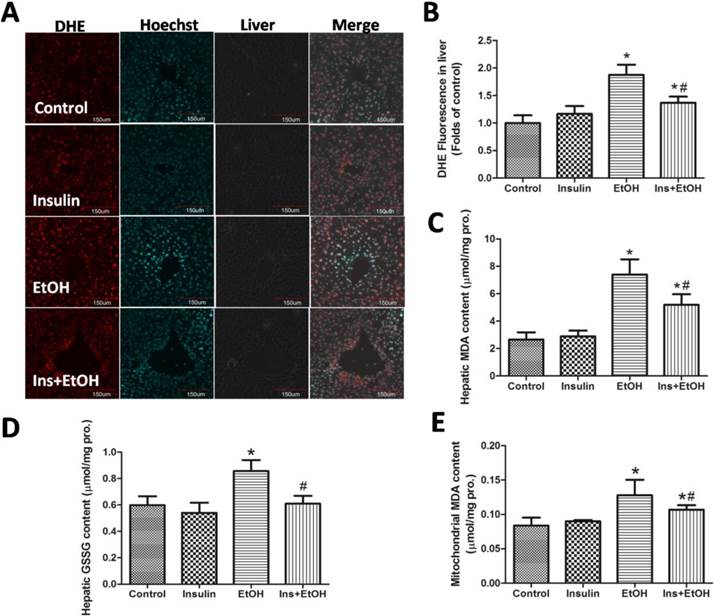
To examine whether insulin pre-administration reduced ethanol-mediated oxidative stress, we investigated the hepatic ROS accumulation, hepatic MDA level, hepatic GSSG level and mitochondrial MDA level. As the results determinate by DHE staining using confocal microscopy shown in Fig.4 A and B, there was no significant difference in the changes of hepatic ROS accumulation between insulin administration mice and control mice, and ethanol exposure induced a notable increase of hepatic ROS accumulation, which was significantly alleviated by insulin pre-administration. As noted in Fig.4 C, hepatic MDA level increased significantly in ethanol group compared to the control group, insulin pre-administration reduced MDA to a certain extent toward normal. As illustrated in Fig.4 D, insulin pre-administration markedly inhibited the increase of hepatic GSSG level in ethanol treatment mice. Mitochondrial oxidative damage also played an important role in mitochondrial dysfunction (18). As illustrated in Fig.4 E, the mitochondria from liver of mice treated with ethanol showed increased MDA level compared to control, indicating that ethanol insult resulted in oxidative injury in mitochondria. However, the lower level of mitochondrial MDA in insulin pre-administration mice was detected, which suggested that insulin could notably alleviate the oxidative stress in hepatocytes mitochondria in mice treated with ethanol. These data demonstrated that pre-administration with insulin could effectively reduce ethanol-mediated oxidative stress.
Insulin pre-administration reduced ethanol-mediated oxidative stress in liver of mice. (A) Hepatic ROS accumulation in cryosections analyzed by DHE staining using confocal microscopy; (B) Values of hepatic ROS accumulation calculated by DHE fluorescence intensity expressed as the folds of control; (C) Hepatic MDA level; (D) Hepatic GSSG level; (E) Mitochondrial MDA level. Data of B were expressed as the mean ± SD of three independent experiments (n = 3). Data of C-E were expressed as mean ± SD (n = 8). *P< 0.05, compared of control group; #P < 0.05, compared to EtOH group.
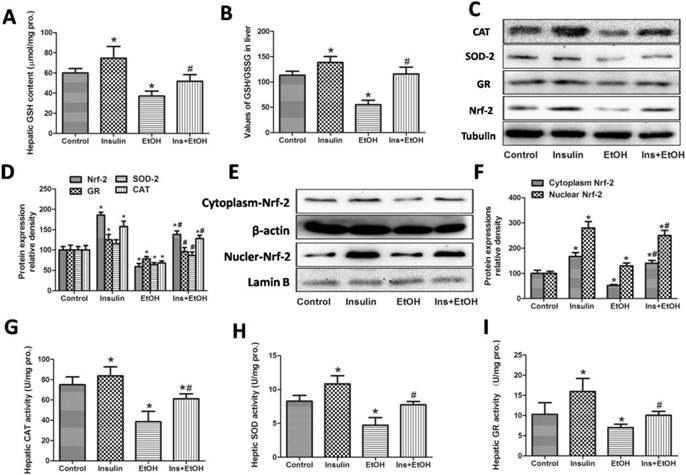
The mechanisms underlying the insulin-induced antioxidative protective effects against ethanol is not entirely clear until now. To further investigate the mechanisms of the antioxidative actions exerted by insulin pre-administration, we measured the antioxidant defense system such as antioxidant GSH, the value of GSH/GSSG, the protein expressions of Nrf-2 and related antioxidant enzymes, and the activities of SOD, CAT and GR. As shown in Fig.5 A and B, in ethanol-treated mice there was significant decrease both in hepatic GSH level and GSH/GSSG ratio compared with those in control group. However, insulin pre-administration notability suppressed the decrease of the level of GSH and the value of GSH/GSSG induced by ethanol exposure. Results in Fig.5 C and D showed that an increase of the protein expressions of GR, CAT and total Nrf-2 in insulin-treated mice and a decrease of the protein expressions of GR, SOD-2, CAT and total Nrf-2 in ethanol-treated mice were observed. As expect, in the insulin pre-administration mice the protein expressions of GR, SOD-2, CAT and total Nrf-2 were significantly up-regulated compared with these of mice treated with ethanol. For further and precisely investigate the role of Nrf-2 in the antioxidative actions against ethanol, we next examined the expressions of Nrf-2 in cytoplasm and nuclear extract. As demonstrated in Fig.5 E and F, insulin administration alone markedly increased and ethanol exposure slightly increased the nuclear Nrf-2 protein expressions, indicating both of insulin and ethanol treatment could promote the nuclear translocation of Nrf-2. Insulin pre-administration showed a notably increase of nuclear Nrf-2 expression compared with ethanol group, suggesting insulin significantly promoted the nuclear translocation of Nrf-2 induced by ethanol. We then determined the activities of antioxidant enzymes including SOD, CAT and GR to evaluate the antioxidative effects of insulin pre-administration. As illustrated in Fig.5 G-I, insulin administration alone increased and ethanol treatment decrease the activities of SOD, CAT and GR, and insulin pre-administration significantly suppressed the decline of these antioxidant enzymes activities mediated by ethanol exposure. Together with the oxidative data, these results indicated that insulin pre-administration reduced oxidative stress and enhanced antioxidant system in mice in mice subjected to ethanol exposure might through the activation of Nrf-2.
Insulin pre-administration enhanced antioxidant defense system in liver of mice exposed to ethanol. (A) Hepatic GSH level; (B) The value of GSH/GSSG in liver; (C) CAT, SOD-2, GR and total Nrf-2 protein expressions in liver determinate by Western blotting; (D) Value of the CAT, SOD-2, GR and total Nrf-2 protein expressions expressed as the folds of control; (E) Protein expression of Nrf-2 in cytoplasm and nuclear fractions in liver determinate by Western blotting; (F) Value of the Nrf-2 protein expression in cytoplasm and nuclear fractions expressions expressed as the folds of control; (G) Hepatic CAT activity; (H) Hepatic SOD activity; (I) Hepatic GR activity. Normalizations of Western blots were ensured by Tubulin in total protein, β-actin in cytoplasm fraction and Lamin B in nuclear fraction. Data of A-B and G-I were expressed as mean ± SD (n = 8). Data of D and F were expressed as the mean ± SD of three independent experiments (n = 3). *P< 0.05, compared to control group; #P < 0.05, compared to EtOH group.
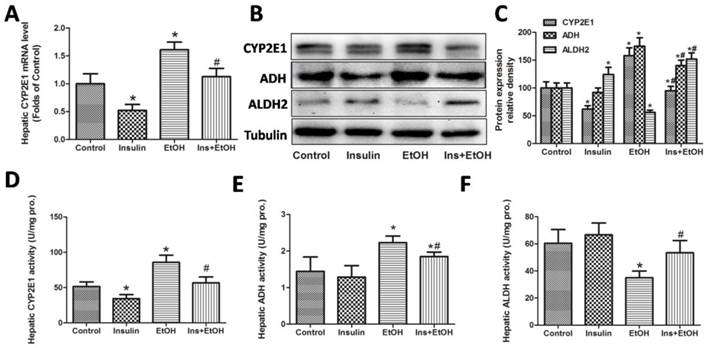
Insulin pre-administration modified ethanol metabolism pathway might be involved in the anti-oxidative and protective effects against acute ethanol exposure
Having demonstrated that insulin pre-administration reduced ethanol-induced liver injury, we further investigated whether modification of ethanol metabolism pathway is involved in this protective function. We first determined the mRNA expression of CYP2E1 in liver. As shown in Fig.6 A, mRNA expression of hepatic CYP2E1was significantly down-regulated in insulin group and significantly up-regulated in ethanol group. We also found that the up-regulation of CYP2E1 induced by ethanol was notably reversed by insulin pre-administration. We next investigated the protein expressions of hepatic CYP2E1, ADH and ALDH2 by Western blotting. As illustrated in Fig.6 B and C, in insulin administration group, hepatic CYP2E1 protein expression was also significantly down-regulated and hepatic ALDH2 protein expression was significantly up-regulated, but hepatic ADH protein expression had no significant change. However, insulin pre-administration effectively suppressed the increase of CYP2E1, ADH protein expressions and the decrease of ALDH2 protein expression in mice exposed to ethanol. To further study the role of these key ethanol metabolism enzymes in protection of insulin, we also determinate the activities of these enzymes. As demonstrated in Fig.6 D-F, our results revealed that ethanol markedly increased the activities of CYP2E1, ADH and decreased activity of ALDH compared with control group, and insulin pre-administration effectively alleviated these trends to normal control group, which is consistent with the results of protein expressions. Taken together, these data suggested that protective and antioxidative effects exerted by insulin pre-administration might involve in the modification of ethanol metabolism pathway.
Ethanol metabolism pathway modified by insulin pre-administration might be involved in the anti-oxidative and protective effects against acute ethanol exposure in mice. (A) Hepatic CYP2E1 mRNA level determinate by RT-PCR; (B) CYP2E1, ADH and ALDH2 protein expression in liver determinate by Western blotting; (C) Value of the CYP2E1, ADH and ALDH2 protein expression expressed as the folds of control; (D) Hepatic CYP2E1 activity; (E) Hepatic ADH activity; (F) Hepatic ALDH2 activity. Normalizations of Western blots were ensured by Tubulin. Data of A and C were expressed as the mean ± SD of three independent experiments (n = 3). Data of D-F were expressed as mean± SD (n = 8). *P< 0.05, compared to control group; #P < 0.05, compared to EtOH group.
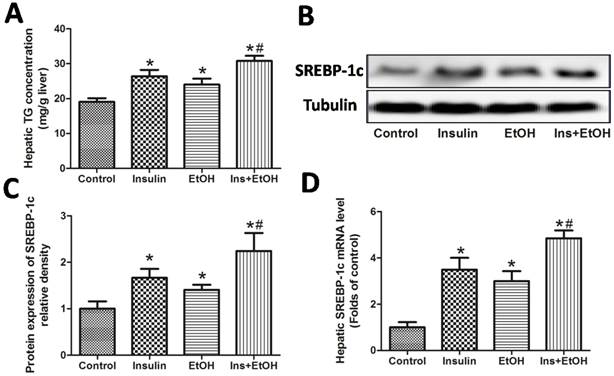
Insulin pre-administration could deteriorate hepatic steatosis in mice exposed to ethanol might be through SRBEP-1c activation
Hepatic steatosis (the accumulation of hepatic triglycerides) is the typical pathological changes and is considered to play a key role in the development of ALD (15). So we next examined the hepatic triglycerides level and the expression of SRBEP-1c, which is an important regulating factor for the process of fat metabolism. As shown in Fig.7 A, insulin administration alone and ethanol exposure could both increase the level of triglycerides in liver, and insulin pre-administration enhanced the increase of hepatic triglycerides level, suggesting insulin pre-administration could deteriorate ethanol-induced hepatic steatosis. As illustrated in Fig.7 B-D, our results revealed that protein and mRNA expressions of SREBP-1c were all up-regulated in insulin group and ethanol group, and as expect, insulin pre-administration further increased the up-regulations of SREBP-1c protein and mRNA expressions in mice exposed to ethanol, suggesting the activation of SREBP-1c might contribute the deteriorative effects of insulin on hepatic steatosis in mice exposed to ethanol.
Insulin pre-administration could deteriorate hepatic steatosis in mice exposed to ethanol might be through SRBEP-1c activation. (A) Hepatic TG level; (B) SREBP-1c protein expression in liver determinate by Western blotting; (C) Value of the SREBP-1c protein expression expressed as the folds of control; (D) Hepatic SREBP-1c mRNA level determinate by RT-PCR. Normalizations of Western blots and RT-PCR were ensured by Tubulin or β-actin. Data of A were expressed as mean ± SD (n = 8). Data of C and D were expressed as the mean ± SD of three independent experiments (n = 3). *P< 0.05, compared to control group; #P < 0.05, compared to EtOH group.
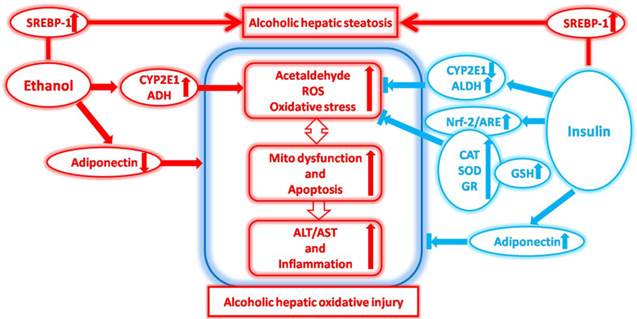
The effects of insulin pre-administration in mice exposed to ethanol: Alleviating hepatic oxidative injury through anti-oxidative, anti-apoptotic activities and deteriorating hepatic steatosis through SRBEP-1c activation.
Discussion
ALD is an important disease hazarded the health of crowd with drinking (36). Oxidative stress, inflammation, apoptosis are all involved in the pathogenesis of ALD (6). High dose of ethanol exposure can interference the normal insulin signal transduction and lead to the occurrence of insulin resistance in liver (37). Insulin is now increasingly considered to have multiple biological activities besides the regulation of blood glucose (34). Previous studies have found that insulin has anti-inflammatory (38), anti-apoptotic (39) and regulatory ability on redox balance (35). In the present study, using an acute high dose ethanol exposure model in vivo pre-treatment with insulin or not, we have made several novel findings. First, we found insulin pre-administration alleviated acute ethanol exposure-induced liver injury and inflammation reflected by decreasing serum activities of AST and ALT, the improvement of pathological alteration, and the inhibition of TNF-α and IL-6 expressions. Second, insulin pre-administration could significantly reduce apoptosis and ameliorate mitochondrial dysfunction in liver of mice exposed to ethanol, supporting by decreasing caspases-3 activities and the ratio of Bax/Bcl-2, increasing mitochondrial viability and mitochondrial oxygen consumption, inhibition of the decline of ATP levels and mitochondrial ROS accumulation. Third, insulin pre-administration prevented ethanol-mediated oxidative stress and enhance antioxidant system, which is evaluated by the decline of MDA levels and the rise of GSH/GSSG, the up-regulations of antioxidant enzymes CAT, SOD, GR through Nrf-2 dependent pathway. Forth, the modification of ethanol metabolism pathway such as the inhibition of CYP2E1and ADH, the activation of ALDH might be involved in the anti-oxidative and protective effects exerted by insulin pre-administration against acute ethanol exposure in mice. Finally, insulin pre-administration deteriorated hepatic steatosis in mice exposed to ethanol might be through SRBEP-1c activation. To the best of our knowledge, we first demonstrated that insulin pre-administration could effectively alleviate liver oxidative injury through anti-inflammatory, anti-oxidative and anti-apoptotic activities but also deteriorated hepatic steatosis through SRBEP-1c activation in mice exposed to ethanol in mice exposed to ethanol.
Previous studies have revealed that high ethanol exposure could induce significant the swelling of hepatocytes and the damage of arranged structure in liver (40). ALT and AST activities in serum are the most frequently indicators used for evaluation liver injury (41). Our previous studies have shown that insulin could protect human liver cells against tBHP-induced injury (35). In our study, insulin pre-administration significant alleviated the increase of liver weight/body weight ratio, liver damage in structure, and the increase of serum ALT and AST activities in mice exposed to ethanol (Table 1 and Fig.1). The results of our study first confirmed that that insulin pre-administration protected liver against ethanol-induced injury. It has come to light that the activation of inflammatory cells and the accumulation of inflammatory cytokines play a key role in the pathogenesis of ALD, especially alcoholic hepatitis (42). The release of inflammatory cytokines including TNF-α and IL-6 could directly lead to the hepatocytes apoptosis, but also led to the ROS accumulation with the results of oxidative stress (28). Previous researches have confirmed that insulin therapy exerts good anti-inflammatory activity in myocardial ischemia reperfusion (43), burn and sepsis (44) besides its metabolic regulatory activity. However, whether insulin pre-administration is beneficial to suppressing the inflammatory reaction in ALD is still not well understand, which might have a clinical significance. In our study, it was shown that in mice high dose of ethanol exposure increased the levels of hepatic MPO activity, hepatic mRNA expressions of TNF-α and IL-6, and serum TNF-α level, which were significantly inhibited by insulin pre-administration (Fig.1). These results suggested the inhibition of inflammatory response through inactivation of inflammatory cells and inflammatory cytokines might be involved in the protective effects of insulin pre-administration against alcoholic liver injury. In addition, Adiponectin as an important adipose-derived hormone has many beneficial biological and regulatory functions such as insulin-sensitizing, anti-inflammation, anti-oxidation and so on (31). The decrease of Adiponectin level is reported to be involved in the pathogenesis of ALD (32). Our study revealed that insulin administration alone could significantly increase serum Adiponectin level and insulin pre-administration effectively alleviated the decrease of serum Adiponectin level in mice exposed to ethanol, suggesting that the increase of Adiponectin level in the circulation system might also contribute to the protective effects of insulin pre-administration against ALD.
The morphological evidence of apoptosis has been obviously detected in the pathogenesis of ALD regardless of in crowd with drinking and animal experiments (21). Although apoptosis is always believed to be one kind of protective mechanisms for maintaining cellular homeostasis, but if the intensity of apoptosis in liver is beyond the adaptive range, apoptosis will become pathological and could cause liver damage (45). More and more evidence revealed that apoptosis as a key event may play an important role in the pathogenesis of ALD (21). A lot of previous studies have shown that insulin could exert a significant anti-apoptosis effect in exogenous chemical injuries or diseases such as myocardial infarction (34). In the following experiments, we detected the changes of apoptosis index. Caspase-3 is the key implementation molecular in the process of apoptosis (46). The ratio of pro-apoptotic protein Bax and anti-apoptotic protein Bcl-2 is usually used as the indicator of apoptosis and is involved in the mitochondrial pathway (47). As expect, insulin pre-administration ameliorated acute ethanol exposure-induced apoptosis (as measured by the decrease of caspase-3 activity and Bax/Bcl-2 ratio in Fig.2).
Mitochondrial dysfunction is believed to play an essential role in apoptosis (18). Mitochondrial dysfunction is always accompanied with the electronic leakage in the mitochondria electron transport chain and the damnification of mitochondria membrane, which could result in the mitochondrial ROS accumulation and the decline of MMP (19). Many studies have highlighted that mitochondrial dysfunction is associated with excessive ROS accumulation and plays a decisive role in the apoptosis (19). ATP is synthesized in the mitochondria and is the direct cellular energy source, and the level of ATP reflects the function of mitochondrial energy metabolism (20). The mitochondrial membrane potential (MMP) plays important roles in the normal mitochondrial function and controls the opening of the mitochondrial permeability transition pore (mPTP). The decline of MMP will induce the activation of caspases pathway and initiation the apoptotic process (48). The involvement of energy metabolism disorder like ATP depletion has attracted intense interest in the pathogenesis in ALD (49). In the next experiment we confirmed that insulin pre-administration also ameliorated acute ethanol exposure-induced mitochondrial dysfunction (as measured by the increase of mitochondrial viability, ATP level, rate of mitochondrial oxygen consumption, MMP and the decrease of mitochondrial ROS accumulation in Fig.3). These results indicated that the restoration of mitochondria homeostasis might be a key mechanism involved in the protective effects of insulin against ethanol cytotoxicity.
Excess ROS accumulation during ethanol metabolism results in oxidative stress, which plays a fundamental and decisive role in the pathogenesis of alcoholic liver disease (12). ROS can directly lead to the oxidative modifications on intracellular biological active molecules such as proteins, lipid and nucleic acids, it could also abnormal cell signal conduction, and the above damage caused mitochondrial dysfunction and apoptosis (11). We speculated that the protective effects of insulin against oxidative stress might be due to its inhibitive ability of ROS accumulation. MDA is an important indicator for measurement of lipid peroxidation (50). And GSSG as the oxidized form of GSH is also an indicator for oxidative damage (51). We then examined whether the protective effects of insulin were related to the inhibition of oxidative stress through determination of hepatic ROS accumulation in cryosections, hepatic MDA level, hepatic GSSG level and mitochondrial MDA level. And we found that insulin pre-administration was able to significantly inhibit the oxidative stress in liver of mice exposed to ethanol, suggesting that the antioxidative activity and the enhancing of antioxidant system might be involved in the protective actions of insulin.
Excessive ethanol exposure not only induces the accumulation of ROS, but also depletes antioxidant and impairs antioxidative enzymes to induce oxidative stress (1). Oxidative stress induced by ROS over-generation and mitochondrial dysfunction is considered to play a central role in the pathogenesis of ALD (10). Therefore the antioxidant therapy has become an important strategy for alcoholic liver injury. Although some antioxidants achieved good results in animal experiments and cell experiments (52), but in the actual process of clinical treatment antioxidants usually have not achieved the desired protective effects (53). We speculated that the activation of endogenous antioxidant system may be the key to prevention and treatment of oxidative damage induced by alcohol. Our previous study has showed that insulin treatment could effectively protect cells against oxidative insult by enhancing the battery of antioxidant system via the activation of Nrf-2 (35). In antioxidant system, GSH as an important non-enzymatic ROS scavenger plays a critical role in preventing liver oxidative injury. The value of GSH/GSSG reflects the homeostasis of GSH balance, which is essential for maintain of mitochondrial function (25). Deficiency of GSH could induce mitochondrial dysfunction and even cells death (54). In our study hepatic GSH and the ratio of GSH/GSSG after ethanol treatment was investigated to determine whether an intracellular redox change is involved in the protective effects of insulin, and we revealed that insulin pre-administration effectively suppressed the decrease of hepatic GSH and GSH/GSSG in mice exposed to ethanol, suggesting the restoring of GSH and the regulating the ratio of GSH/GSSG may be responsible for insulin's protective activity. And these results were in accordance with results of oxidative stress. Antioxidative enzymes play a core role in the maintaining of redox balance in the antioxidant defense system. Some of antioxidative enzymes like SOD and CAT can directly catalytic the remove of ROS, some of them like GR and GCLC also catalytic the synthetic of antioxidants to exert indirectly antioxidative activity (24). For example, SOD and CAT have the ability of scavenging superoxide anion and hydrogen peroxidase respectively, GR has the ability of forming reduced GSH (55). We then investigated the mechanisms of the antioxidative actions exerted by insulin pre-administration through determination of the protein expressions and activities of antioxidative enzymes. Insulin pre-administration up-regulated the protein expressions of GR, SOD-2, CAT and increased the activities of these enzymes. We concluded that insulin probably enhanced antioxidant system to exert its protective and anti-apoptotic effects. Activation of Nrf-2 is a crucial cellular antioxidative mechanism, which could interact with ARE to mediate the antioxidative enzymes expression (26). Nrf-2 is also important for maintaining GSH homeostasis through regulation of GSH synthesis relative enzymes (56). Our previous research has demonstrated that insulin can activate antioxidant enzymes via Nrf-2 pathway and plays a key role in redox balance (35). We hypothesized that alleviated effects of insulin on ethanol induced oxidative stress might be through Nrf-2 activation too. In our study, ethanol inhibited the expression of total Nrf-2 but promote the Nrf-2 nuclear translocation for adaptability. Interestingly, ethanol did not increase the protein expressions and activities of antioxidative enzymes such as GR, SOD, CAT, suggesting that ethanol exposure might inhibit the transcription activity of antioxidative enzymes. Insulin pretreatment significantly promotes the synthesis and nuclear translocation of Nrf-2 process in mice exposed to ethanol, which could finally increase the expressions and activities of antioxidase.
Ethanol metabolism pathway also plays a significant and pivotal role in the pathogenesis of ALD (40). In pathological condition, CYP2E1 as a significant ethanol catalyst enzyme has characteristic of forming large amount of ROS with the result of oxidative stress (9). ADH and ALDH can catalyze the production and removal of acetaldehyde respectively. Toxic acetaldehyde can lead to mitochondrial dysfunction and apoptosis, leading to seriously damage the liver function (57). Acetaldehyde plays an important role in the mechanism of alcoholic liver disease (13). Over-expression of ADH increases susceptibility to ethanol toxicity in myocardial cells (58). However, over-expression of ALDH has a protective effect on cardiac injury induced by alcohol (59). In the present study, we determined the protein expressions and activities of CYP2E1, ADH and ALDH. We found that insulin pre-administration could significantly suppress ethanol-induced CYP2E1 and ADH activation. We also observed that insulin pre-administration could significantly activate ALDH in mice exposed to ethanol. These results suggested that protective and antioxidative effects exerted by insulin pre-administration might be thought the modification of ethanol metabolism pathway via reducing ROS and acetaldehyde accumulation.
Because of the ethanol oxidative process, there is an increase of NADH/NAD+ value, which will induced the disorder of fat metabolism with the results of triglyceride accumulation in hepatocytes (60). And the hepatic steatosis is the early manifestation of ALD and is believed to be the foundational pathological change of other more severe alcoholic liver disease (16). Although we found that insulin pretreatment could exert a significant protective effects on oxidative damage and inflammatory reaction in liver against ethanol exposure, but we also observed that insulin pre-treatment significantly deteriorated hepatic steatosis in mice exposed to ethanol. Fatty acid synthesis in liver is mainly regulated by sterol regulatory element binding protein-1c (SREBP-1c), and a large number of studies show that ethanol exposure can significantly activate SREBP-1c, which is responsible for the formation of fatty liver (61). And insulin could also lead to the activation of SREBP-1c to increase the triglyceride in hepatocytes (62). In our present study, we confirmed that the expression of SREBP-1c were all up-regulated in insulin group and ethanol group, and insulin pre-administration could further increase the activation of SREBP-1c, suggesting SREBP-1c activation might contribute the deteriorative effects of insulin pre-administration on hepatic steatosis in mice exposed to ethanol. According to the “Two-hit” hypothesis for ALD, although steatosis is reversible, but it might be the basis of other serious liver disease and pathologies including steatohepatitis, fibrosis, cirrhosis, and even hepatocellular carcinoma (16). We think that insulin pretreatment might act as a double-edged sword in the pathogenesis of alcoholic liver injury, at least in the early stage: protections against oxidative stress and exacerbation of steatosis. And the exacerbation of steatosis mediated by insulin might increase susceptibility to later ethanol exposure or other oxidative insults, suggesting the diverse and complex effects of insulin on alcoholic liver injury.
In summary, our present study for the first time demonstrates that insulin pre-administration has multiple beneficial and side effects in the liver injury of mice exposed to ethanol. As shown in Fig.7, the protective activities of insulin pre-administration in liver against ethanol toxicity is involving in the suppressing of acetaldehyde and ROS accumulation through the activation of ALDH and the inhibition of CYP2E1, the increase of Adiponectin, the enhancing of antioxidant system through increasing GSH and activation antioxidative enzymes via Nrf-2 dependent pathway, and the inhibition of inflammatory response through inactivation of inflammatory cells and inflammatory factors (TNF-α and IL-6) in liver. These above protective abilities inhibited ethanol exposure-induced mitochondrial dysfunction and apoptosis, which can ultimately ameliorated alcoholic liver injury supporting by the improvement of H&E pathological alterations and the inhibition of intracytoplasmic enzymes release. However, we also found that insulin pre-administration deteriorated hepatic steatosis in mice exposed to ethanol might be through SRBEP-1c activation. This study gives us a new insight of the biological protective effects of insulin on the oxidative stress, apoptosis, mitochondrial dysfunction, and inflammation associated with ALD and also the harmful effects on steatosis associated with ALD. The pathogenesis of ALD is very complex, and the biological actin of insulin is also vary widely, the deeper investigations about the effects and underlying mechanisms of insulin and related signal transduction on ALD should be seriously considered in the further researches.
Abbreviations
ALD, alcoholic liver disease; EtOH, ethanol; ROS, reactive oxygen species; ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; ATP, adenosine triphosphate; Rho123, rhodamine 123; DHE, dihydroethidium; MMP, mitochondrial membrane potential; MPTP, mitochondrial permeability transition pore; MPO, Myeloperoxidase; MTT, 4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide; DCFH-DA, dichlorofluorescein; DHE, dihydroethidium; LPO, lipid peroxidation; MDA, malondialdehyde; GR, glutathione reductase; SOD, superoxide dismutase; CAT, catalase; GSH, reduced glutathione; GSSG, oxidized glutathione; GCLC, glutamate-cysteine ligase catalytic subunit; HO-1, Heme oxygenase-1; ARE, antioxidant response element; Bax, B cell lymphoma 2-associated X protein; Bcl-2, B cell lymphoma 2; Nrf-2, NF-E2-related factor-2; CYP2E1,cytochrome P450 2E1; ADH, alcohol dehydrogenase; ALDH, acetaldehyde dehydrogenase; TNF-α, tumor necrosis factor-α; IL-6, interleukin- 6; PBS, phosphate buffered saline; SREBP-1, sterol regulatory element binding protein-1; TBS, Tris buffered saline; BSA, bovine serum albumin.
Acknowledgements
This work was supported by Program for Changjiang Scholars, Innovative Research Team in University (PCSIRT) and National Nature Scientific Foundation (NSFC-30872135 and NSFC-31400724). This work was also supported by Natural Science Foundation of Shaanxi Province (2014JQ4135).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hagymasi K, Blazovics A, Lengyel G. et al. Oxidative damage in alcoholic liver disease. Eur J Gastroenterol Hepatol. 2001;13:49-53
2. Shukla SD, Zakhari S. Epigenetics. New frontier for alcohol research. Alcohol Res. 2013;35:1-2
3. Shukla SD, Pruett SB, Szabo G. et al. Binge ethanol and liver: new molecular developments. Alcohol Clin Exp Res. 2013;37:550-557
4. Das SK, Vasudevan DM. Alcohol-induced oxidative stress. Life Sciences. 2007;81:177-187
5. Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev. 2010;3:178-185
6. Chae HB. Alcoholic liver disease. Korean J Gastroenterol. 2009;53:275-282
7. Stewart S, Jones D, Day CP. Alcoholic liver disease: new insights into mechanisms and preventative strategies. Trends Mol Med. 2001;7:408-413
8. Cunningham CC, Bailey SM. Ethanol consumption and liver mitochondria function. Biol Signals Recept. 2001;10:271-282
9. Cederbaum AI. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig Dis. 2010;28:802-811
10. Albano E. Alcohol, oxidative stress and free radical damage. Proc Nutr Soc. 2006;65:278-290
11. Bartosz G. Reactive oxygen species: destroyers or messengers? Biochem Pharmacol. 2009;77:1303-1315
12. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biology and Medicine. 2010;48:749-762
13. Barry RE. Role of acetaldehyde in the pathogenesis of alcoholic liver disease. Br J Addict. 1988;83:1381-1386
14. Yan M, Zhu KX, Meng FL. et al. Relationship between ALDH gene polymorphism and alcoholic liver diseases. Zhonghua Gan Zang Bing Za Zhi. 2003;11:654-656
15. Newton BW, Russell WK, Russell DH. et al. Proteomic methods for biomarker discovery in a rat model of alcohol steatosis. Methods Mol Biol. 2012;909:259-277
16. Mantena SK, King AL, Andringa KK. et al. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259-1272
17. Cadenas E, Davies KJA. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biology and Medicine. 2000;29:222-230
18. Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049-2063
19. Sastre J, Serviddio G, Pereda J. et al. Mitochondrial function in liver disease. Front Biosci. 2007;12:1200-1209
20. Saa A, Siqueira KM. Modeling the ATP production in mitochondria. Bull Math Biol. 2013;75:1636-1651
21. Higuchi H, Kurose I, Kato S. et al. Ethanol-induced apoptosis and oxidative stress in hepatocytes. Alcohol Clin Exp Res. 1996;20:340A-346A
22. Ishii H, Adachi M, Fernandez-Checa JC. et al. Role of apoptosis in alcoholic liver injury. Alcohol Clin Exp Res. 2003;27:1207-1212
23. Volkmann N, Marassi FM, Newmeyer DD. et al. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 2014;21:206-215
24. Pi J, Zhang Q, Fu J. et al. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicology and Applied Pharmacology Nrf2 in Toxicology and Pharmacology - Nrf2 in Toxicology and Pharmacology. 2010;244:77-83
25. Lu SC. Regulation of glutathione synthesis. Molecular Aspects of MedicineGlutathione in Health and Disease - Glutathione in Health. 2009;30:42-59
26. Niture SK, Kaspar JW, Shen J. et al. Nrf2 signaling and cell survival. Toxicology and Applied Pharmacology. 2010;244:37-42
27. Yan D, Dong J, Sulik KK. et al. Induction of the Nrf2-driven antioxidant response by tert-butylhydroquinone prevents ethanol-induced apoptosis in cranial neural crest cells. Biochem Pharmacol. 2010;80:144-149
28. Wang HJ, Gao B, Zakhari S. et al. Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012;32:343-368
29. Keegan A, Martini R, Batey R. Ethanol-related liver injury in the rat: a model of steatosis, inflammation and pericentral fibrosis. J Hepatol. 1995;23:591-600
30. Thiele GM, Freeman TL, Klassen LW. Immunologic mechanisms of alcoholic liver injury. Semin Liver Dis. 2004;24:273-287
31. Lee S, Kwak HB. Role of adiponectin in metabolic and cardiovascular disease. J Exerc Rehabil. 2014;10:54-59
32. Tang H, Sebastian BM, Axhemi A. et al. Ethanol-induced oxidative stress via the CYP2E1 pathway disrupts adiponectin secretion from adipocytes. Alcohol Clin Exp Res. 2012;36:214-222
33. Monte S, Derdak Z, Wands JR. Alcohol, insulin resistance and the liver-brain axis. J Gastroenterol Hepatol. 2012;27(Suppl 2):33-41
34. Wang X, Tao L, Hai CX. Redox-regulating role of insulin: the essence of insulin effect. Mol Cell Endocrinol. 2012;349:111-127
35. Wang X, Wu H, Chen H. et al. Does insulin bolster antioxidant defenses via the extracellular signal-regulated kinases-protein kinase B-nuclear factor erythroid 2 p45-related factor 2 pathway? Antioxid Redox Signal. 2012;16:1061-1070
36. Bacio GA, Lunny KF, Webb JN. et al. Alcohol Use Following an Alcohol Challenge and a Brief Intervention among Alcohol-Dependent Individuals. Am J Addict. 2014;23:96-101
37. Van de Wiel A. Alcohol and insulin sensitivity. The Netherlands Journal of Medicine. 1998;52:91-94
38. Dandona P, Ghanim H, Bandyopadhyay A. et al. Insulin suppresses endotoxin-induced oxidative, nitrosative, and inflammatory stress in humans. Diabetes Care. 2010;33:2416-2423
39. Kang S, Song J, Kang H. et al. Insulin can block apoptosis by decreasing oxidative stress via phosphatidylinositol 3-kinase- and extracellular signal-regulated protein kinase-dependent signaling pathways in HepG2 cells. Eur J Endocrinol. 2003;148:147-155
40. Zima T, Kalousova M. Oxidative stress and signal transduction pathways in alcoholic liver disease. Alcohol Clin Exp Res. 2005;29:110S-115S
41. Reitman S, Frankel S. A calorimetric method for the de- termination of serum glutamate. Am. J. Clin. Pathol. 1957:56-63
42. Arai M, Nakano S, Okuno F. et al. Endotoxin-induced hypercoagulability: a possible aggravating factor of alcoholic liver disease. Hepatology. 1989;9:846-851
43. Sun Q, Li J, Gao F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J Diabetes. 2014;5:89-96
44. Chen Q, Yu W, Shi J. et al. Insulin alleviates the inflammatory response and oxidative stress injury in cerebral tissues in septic rats. J Inflamm (Lond). 2014;11:18
45. Jaeschke H, Gujral JS, Bajt ML. Apoptosis and necrosis in liver disease. Liver Int. 2004;24:85-89
46. Alvarez S, Blanco A, Fresno M. et al. TNF-alpha contributes to caspase-3 independent apoptosis in neuroblastoma cells: role of NFAT. PLoS One. 2011;6:e16100
47. Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92-101
48. Liu JX, Zhang JH, Li HH. et al. Emodin induces Panc-1 cell apoptosis via declining the mitochondrial membrane potential. Oncol Rep. 2012;28:1991-1996
49. Young TA, Bailey SM, Van Horn CG. et al. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol. 2006;41:254-260
50. Vendemiale G, Altomare E, Grattagliano I. et al. Increased plasma levels of glutathione and malondialdehyde after acute ethanol ingestion in humans. J Hepatol. 1989;9:359-365
51. Koch OR, Martino E, Boveris A. et al. Increased biliary glutathione disulfide (GSSG) release in chronically ethanol-treated rats. Adv Exp Med Biol. 1980;132:619-625
52. Cohen-Kerem R, Koren G. Antioxidants and fetal protection against ethanol teratogenicity. I. Review of the experimental data and implications to humans. Neurotoxicol Teratol. 2003;25:1-9
53. Cruz A, Muntane J. Alcoholic liver disease, oxidative stress, and antioxidants. Rev Esp Enferm Dig. 2011;103:393-395
54. Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. 2009;30:29-41
55. Laloi C, Apel K, Danon A. Reactive oxygen signalling: the latest news. Current Opinion in Plant Biology. 2004;7:323-328
56. Harvey CJ, Thimmulappa RK, Singh A. et al. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radical Biology and Medicine. 2009;46:443-453
57. Barry RE, McGivan JD. Acetaldehyde alone may initiate hepatocellular damage in acute alcoholic liver disease. Gut. 1985;26:1065-1069
58. Duan J, McFadden GE, Borgerding AJ. et al. Overexpression of alcohol dehydrogenase exacerbates ethanol-induced contractile defect in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2002;282:1216-1222
59. Li SY, Li Q, Shen JJ. et al. Attenuation of acetaldehyde-induced cell injury by overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene in human cardiac myocytes: role of MAP kinase signaling. J Mol Cell Cardiol. 2006;40:283-294
60. Donohue TJ. Alcohol-induced steatosis in liver cells. World J Gastroenterol. 2007;13:4974-4978
61. You M, Fischer M, Deeg MA. et al. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem. 2002;277:29342-29347
62. Shimomura I, Bashmakov Y, Ikemoto S. et al. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci. 1999;96:13656-13661
Author contact
![]() Corresponding author: Department of Toxicology, the Ministry of Education Key Lab of Hazard Assessment and Control in Special Operational Environment, Shaanxi Provincial Key Lab of Free radical biology and medicine, School of Public Health, The Fourth Military Medical University, Xi'an, 710032, Shaanxi, P. R. China. Tel: +86 02983374879. Fax: +86 02984774879. Email: cx-haiedu.cn.
Corresponding author: Department of Toxicology, the Ministry of Education Key Lab of Hazard Assessment and Control in Special Operational Environment, Shaanxi Provincial Key Lab of Free radical biology and medicine, School of Public Health, The Fourth Military Medical University, Xi'an, 710032, Shaanxi, P. R. China. Tel: +86 02983374879. Fax: +86 02984774879. Email: cx-haiedu.cn.