Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(6):652-663. doi:10.7150/ijbs.10250 This issue Cite
Research Paper
Role of the Renin-Angiotensin System, Renal Sympathetic Nerve System, and Oxidative Stress in Chronic Foot Shock-Induced Hypertension in Rats
Tao Dong1, *, Jing-Wei Chen2, *, Li-Li Tian1, *, Lin-Hui Wang1, Ren-Di Jiang1, Zhe Zhang1, Jian-Bing Xu1, Xiao-Dong Zhao2, Wei Zhu3, Guo-Qing Wang1, Wan-Ping Sun4, ![]() , Guo-Xing Zhang1,
, Guo-Xing Zhang1, ![]()
1. Department of Physiology and Neuroscience, Medical College of Soochow University, Suzhou 215123, P.R. China
2. Department of Internal Medicine, the Affiliated Suzhou Chinese Traditional Medicine Hospital, Nanjing University of Chinese Medicine, Suzhou 215003, P.R. China
3. Department of Internal Medicine, the Second Affiliated Hospital, High-tech zone hospital, Soochow University, Suzhou 215151, P.R. China
4. Laboratory of Molecular Diagnostics, Medical College of Soochow University, Suzhou 215123, P.R. China
* These authors contributed equally to this work.
Received 2014-8-2; Accepted 2015-3-14; Published 2015-4-26
Abstract

Objective: The renin-angiotensin system (RAS) and renal sympathetic nerve system (RSNS) are involved in the development of hypertension. The present study is designed to explore the possible roles of the RAS and the RSNS in foot shock-induced hypertension.
Methods: Male Sprague-Dawley rats were divided into six groups: control, foot shock, RSNS denervation, denervation plus foot shock, Captopril (angiotensin I converting enzyme inhibitor, ACE inhibitor) plus foot shock, and Tempol (superoxide dismutase mimetic) plus foot shock. Rats received foot shock for 14 days. We measured the quantity of thiobarbituric acid reactive substances (TBARS), corticosterone, renin, and angiotensin II (Ang II) in plasma, the activities of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px), and renal noradrenaline content. RAS component mRNA and protein levels were quantified in the cerebral cortex and hypothalamus.
Results: The two week foot shock treatment significantly increased systolic blood pressure, which was accompanied by an increase in angiotensinogen, renin, ACE1, and AT1a mRNA and protein expression in the cerebral cortex and hypothalamus, an increase of the plasma concentrations of renin, Ang II, corticosterone, and TBARS, as well as a decrease in plasma SOD and GSH-Px activities. Systolic blood pressure increase was suppressed by denervation of the RSNS or treatment with Captopril or Tempol. Interestingly, denervation or Tempol treatment both decreased main RAS components not only in the circulatory system, but also in the central nervous system. In addition, decreased antioxidant levels and increased TBARS and corticosterone levels were also partially restored by denervation or treatment with Tempol or Captopril.
Conclusions: RAS, RSNS and oxidative stress reciprocally potentiate to play important roles in the development of foot shock-induced hypertension.
Keywords: renal sympathetic nerve denervation, foot shock, hypertension, renin-angiotensin system (RAS), oxidative stress
Introduction
Hypertension is a major risk factor for many diseases, including coronary heart disease, cerebrovascular accidents (stroke), cardiac insufficiency and renal insufficiency [1, 2]. Over half a century ago, Imhof et al. observed that psychological stress leads to an increase in blood pressure [3]. Data gathered since then show that chronic psychological stress is one of the major causes of sustained elevated blood pressure [4, 5]. Psychological stress has been shown to activate the hypothalamus-pituitary-adrenal cortex system in humans [6, 7] and in animals [8], which may be one of the mechanisms by which it affects blood pressure , which was mainly mediated by release of catecholamine, cortisol, vasopressin, endorphins and aldosterone [9, 10]. Another putative mechanism is through the activation of the sympathetic-adrenal medullar system [9, 11-13]. However, the precise mechanisms by which psychological stress induces hypertension are not clearly defined.
Clinical studies have shown the benefits of renal sympathetic denervation in patients with refractory hypertension [14, 15]. Percutaneous renal sympathetic denervation has been promising in clinical trials in reducing blood pressure in a safe and sustained manner. The procedure has been accepted as a novel, minimally invasive, device-based therapy that specifically targets and ablates the renal artery nerve with radiofrequency waves without the need for permanent implantation [16, 17]. Besides its effects on reducing blood pressure, additional beneficial effects from renal sympathetic denervation have also been reported in diseases such as diabetes [18], renal dysfunction [15], cardiac hypertrophy [14], heart failure [19], and other diseases [20]. However, an international expert consensus statement has pointed out the limitation of application in patients due to lack of the observation of the long-term physiological effects of renal sympathetic nerve [18]. Therefore, long-term observation in clinical trials should be carried out to ensure the safety, reliability and efficacy of renal sympathetic denervation. Furthermore, extensive basic research must also be performed to provide more solid evidence for the use of this procedure in clinical patients.
The renin-angiotensin system (RAS) has been shown to be a main regulator of blood pressure and fluid homeostasis [21]. Recent studies have shown the existence of local tissue RAS products: angiotensin II, III, IV and Ang 1-7, each of which has its own function in the tissue it is generated. These tissues and organs include the brain, bone marrow, adipose tissue, epididymis, carotid body, liver, and pancreas [22]. Similar to the critical role of RAS in regulating blood pressure in the circulatory system, several studies have probed the role of RAS in regulating blood pressure in the central nervous system. Currently, all known components of the RAS have been identified in the brain, including the precursor of RAS and the enzymes required for the production and metabolism of angiotensin peptides and specific angiotensin type 1 (AT1) and AT2 receptors, [23]. Ang II has been reported to be involved in blood-pressure control and body fluid and electrolyte balance through its interaction with AT1 receptors in the brain [24]. In the foot shock stress-induced high blood pressure model, Ang II has been shown to regulate blood pressure via activating hypothalamic vasopressin synthesis and release [3, 10]. Our recent report also demonstrated that RAS potentiate hypothalamic vasopression expression in foot shock stress-induced high blood pressure [25].
In the circulatory system, renal sympathetic nerves and RAS have been reported to interact via two mechanisms [26]. First, Ang II has been shown to increase sympathetic nerve activity and oxidative stress [27-29]. Indeed, our previous studies have supported the hypothesis that Ang II induces hypertension through an increase in oxidative stress [30-32]. Second, the activation of the sympathetic nerve system increases RAS expression, synthesis or release into the circulatory system [33, 34]. These two mechanisms exacerbate one another, resulting in the development of hypertension and renal injury [34]. However, the relationship between renal sympathetic nerves, central RAS and oxidative stress is still unclear in the context of stress induced hypertension.
Our present study was designed to use the foot shock-induced hypertension model in rats to elucidate the interactions between the renal sympathetic nerve system (RSNS), RAS, and oxidative stress. We investigated possible mechanisms involved in foot shock-induced hypertension using renal sympathetic denervation, administration of angiotensin converting enzyme inhibitor, or an antioxidant. Our data show that RAS, RSNS and oxidative stress interact reciprocally, and all are involved in foot shock-induced hypertension.
Methods
1. Animal preparation
Ten-week-old male Sprague-Dawley rats were used in this study. The animals were obtained from Shanghai Laboratory Animal Center and maintained at a controlled temperature of 25 °C with a 12/12 light/dark cycle. Rats were divided into eight groups: control, foot shock, RSNS denervation, denervation plus foot shock, angiotensin converting enzyme inhibitor (ACEI), (ACEI) plus foot shock, antioxidant, and antioxidant plus foot shock. Fifteen animals were used in each group. Rats exposed to foot shock were individually placed into a foot shock stress box. In this device, rats received electrical foot shocks (0.15 mA, shocks of 5 sec every 30 sec) for 4 hr each session through an electrified grid floor. Body weight and systolic blood pressure were monitored for 30 minutes every day 2 hours after foot shock during the test period by tail-cuff method. Rats in the RSNS denervation group were surgically denervated under anesthesia (10% chloral hydrate) as previously reported [16]. In brief, rats were received bilateral flank incision, and RSNS denervation was initiated by stripping nervous and connective tissues from the renal arteries and veins under light microscope, followed by coating these vessels with s solution of 10% phenol in ethanol. Control-operated rats received a similar surgical incision, but the renal nerves were left intact. Rats were allowed to recover for one week before beginning the foot shock procedure. Rats receiving Angiotensin I-converting enzyme inhibitor (Captopril, 100 mg/kg/day) or superoxide dismutase (SOD) mimetic (Tempol, 10 mg/kg/day) were treated by intraperitoneal (ip) injection from the first day after the start of stress to the end of stress [35-37]. On day 14, rats were sacrificed under 10% chloral hydrate anesthesia (350 mg/kg ip). Plasma, cerebral cortex, hypothalamus, and and kidney tissue were collected and stored at -80 °C for further analysis. The present study conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All procedures were approved by the Animal Care and Use Committee of the University of Szeged.
2. Real-time PCR
Total RNA was isolated from frozen tissues by guanidinium isothiocyanate-acid phenol extraction and quantified by measuring the absorbance at 260 nm. One microgram of total RNA was used for reverse transcription. Rat angiotensinogen (AGT), ACE1, ACE2, and AT1a mRNAs were quantified by real time PCR (prism7000; Applied Biosystems, Foster, California). The primer pairs for AGT cDNA were 5′- CAG GTT TGT GCA GGC TGT GA -3′ (forward) and 5′- GAG CAT GGG CAC AGA CAC TGA -3′ (reverse). The primer pairs for ACE1 cDNA were 5′- TGC CTC AGC CTG GGA CTT CTA -3′ (forward) and 5′- CCC ATT TCG TGG TGG GCT A -3′ (reverse). The primer pairs for ACE2 cDNA were 5′- AAT CGT AGG CTC TGG GCT TGG-3′ (forward) and 5′- TTC GAT CAA CTG GTT TCG GTT GTA-3′ (reverse). The primer pairs for AT1a cDNA were 5′-AGT CCT GTT CCA CCC GAT CAC-3′ (forward) and 5′-TCA GAC ACT ATT CGA AAT CCA CTT G -3′ (reverse). The primer pairs for β-actin cDNA were 5′- GGA GAT TAC TGC CCT GGC TCC TA-3′ (forward) and 5′- GAC TCA TCG TAC TCC TGC TTG CTG-3′ (reverse). AGT, ACE1, ACE2 and AT1a mRNAs were normalized to β-Actin mRNA.
3. Western blot of cerebral RAS components
Expressions of cerebral RAS component AGT, ACE1, ACE2, and AT1awere measured by western blotting, as previous described [30, 35]. Briefly, cortex or hypothalamus tissue was homogenized in cold lysis buffer (Bio-Rad). Equal amount of protein (80 ug) from each homogenate or lysate was resolved by 10% SDS-PAGE. Proteins were transferred to PVDF membranes (Hybond TM-ECL; Amersham Pharmacia Biotech, Inc.). The membranes were blocked for 2 h at room temperature with 5% skimmed milk in PBS and 0.1% Tween 20. The blots were incubated overnight with 1:1000 diluted primary antibodies: monoclonal anti-AGT, monoclonal anti-ACE1, anti-ACE2, anti-AT1a, and anti-GAPDH (Santa Cruz Biotech, Inc.), followed by incubation for 1 h with a secondary antibody (HRP-conjugated anti-rabbit IgG; 1:2000). Immunoreactive bands were visualized using enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech) and analyzed by NIH image software. Data were normalized by GAPDH.
4. Quantification of effector molecules and activities
Renal tissue were homogenized to measure noradrenaline concentration, plasma concentrations of corticosterone, renin, Ang II, lipid peroxidation marker (malondialchehyche, MDA), and SOD (U.S.A. TSZ biological Trade Co., Ltd. USA, noradrenaline: R6413, corticosterone: R6410, renin: R6774, Ang II: R6430, MDA: R6734, SOD: R6733) and GSH-Px (Westang Biotechonology Inc. Shanghai, China, F15626) activities, and were measured using a commercially available enzyme-linked immunosorbent assay (ELISA) kits. All steps were performed according to the manufacturer's instructions.
5. Statistical analysis
All data are presented as the mean ± S.E.M. Statistical significance between more than two groups was tested using two way ANOVA followed by the Newman-Keel test or an unpaired two tail Student's t-test. P values < 0.05 are considered statistically significant.
Results
1. Effects of shock, denervation, ACEI and Tempol on weight gain
Rat body weights were tracked during the entire experimental procedure. Our data show no increase in body weight in all groups receiving foot shock, when compared with the control group, (Figure 1). These results are different from our recent observations about restrain stress model reduced body weight gain in mice [38].
Rat body weights in each group. Rat body weights were measured on days 0, 3, 7, 10, and 14. Black circles represent the control group (n = 15), Black squares represent the stress group (n = 15), black triangles represent the denervation group (n = 15), inversed black triangles represent the denervation plus stress group (n = 15), black diamonds represent the Captorpril group (n = 15), white circles represent the Captopril plus stress group (n = 15), white squares represent Tempol group (n = 15), and white triangles represent Tempol plus stress group (n = 15). Data are represented as mean ± SEM.
2. Effects of shock, denervation, ACEI and Tempol on systolic blood pressure (SBP) and heart rate
SBP was measured on days 0, 3, 7, 10 and 14. The SBP was significantly increased when compared with the control group on day 7 (119.8 ± 3.1 vs. 95.0 ± 4.7 mmHg, p<0.05), day 10 (135.5 ± 3.9 vs. 99.5 ± 4.7 mmHg, p<0.05), and day 14 (134.2 ± 8.7 vs. 101.7 ± 4.7 mmHg, p<0.05) (Figure 2A). The group receiving denervation plus shock had SBP values of 97.8 ± 2.6, 105.3 ± 2.0, and 100.6 ± 1.8 mmHg on days 7, 10, and 14, respectively, which were markedly lower than SBP values measured in the foot shock group (p<0.05). Similarly, SBP values of the ACEI treatment group were 106.0 ± 1.2, 97.5 ± 2.7, 86.5 ± 4.0 mmHg on days 7, 10, and 14, which were markedly lower than SBP values measured in the foot shock group (p<0.05) (Figure 2A). SBP values in the Tempol treatment group were 109.0 ± 3.4, 103.9 ± 3.8, 100.3 ± 3.7 mmHg on days 7, 10, and 14, respectively, also markedly lower than the foot shock group (p<0.05) (Figure 2A). Denervation of renal sympathetic nerve did not markedly affect the SBP when compared to the control group. ACEI alone slightly decreased SBP, and Tempol alone had no effect on SBP (Figure 2A)
Hemodynamic changes. (A) SBP changes in each experimental group. (B) Heart rates of each test group. Black circles represent the control group (n = 15), black squares represent the stress group (n = 15), black triangles represent the denervation group (n =15), inversed black triangles represent the denervation plus stress group (n = 15), black diamonds represent the Captorpril group (n = 15), white circles represent the Captorpril; plus stress group (n = 15), white squares represent Tempol group (n = 15), and white triangles represent Tempol plus stress group (n = 15). Data are represented as mean ± SEM. * p< 0.05 compared with control group on the corresponding day. † p<0.05 compared with stress group on the corresponding day.
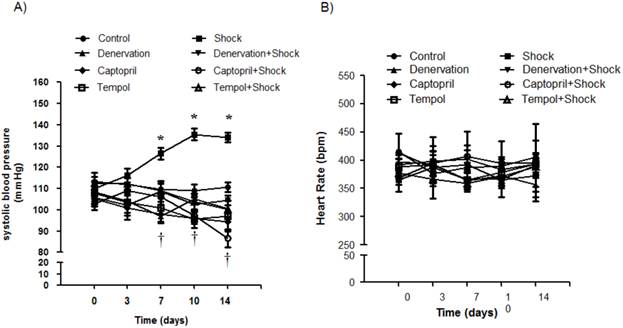
Heart rates were also monitored on days 0, 3, 7, 10 and 14. The measured heart rates were not markedly different throughout the whole experimental period between all groups (Figure 2B). These results are concordant with previously published observations [10, 39].
These results indicate that RSNS, RAS and oxidative stress play important role in the foot shock-induced hypertension.
3. Effects of shock, denervation, ACEI and Tempol treatments on plasma concentration of renin and Ang II
In the foot shock group, the renin concentration was 90.36 ± 9.13 μg/L, which was markedly higher than that the concentration in the control group (63.45 ± 7.63 μg/L, p<0.05, Figure 3A). The plasma concentration of renin in the denervation plus shock, ACEI, and Tempol groups was 66.96 ± 6.83, 70.64 ± 9.17 and 72.18 ± 10.92 μg/L, respectively, which was significantly lower than levels measured in the foot shock group (p<0.05, Figure 3A). Denervation alone did not affect plasma renin concentration. ACEI alone increased plasma renin concentration compared with control group (78.45 ± 6.54 μg/L, p<0.05, Figure 3A), but Tempol alone had no effect on plasma renin concentration (60.78 ± 5.92 μg/L).
Plasma concentrations of renin and Ang II. Concentrations of renin (A) and Ang II (B) in plasma from each test group after a two week stress period. Data of each group (n = 12) were presented as mean ± SEM. * p<0.05 compared with the control group. † p<0.05 compared with the stress group.
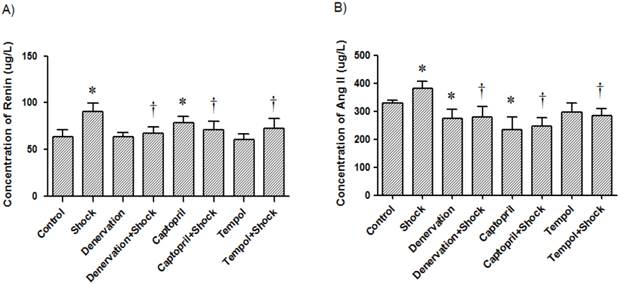
The Ang II concentration in the foot shock group was 380.48 ± 25.80 μg/L, which was higher than the measured concentration of the control group, which was 328.53 ± 11.71 μg/L (p<0.05, Figure 3B). Denervation, ACEI, and Tempol treatments significantly reduced Ang II concentration to 279.36 ± 36.68, 246.73 ± 30.84, and 283.52 ± 24.47 μg/L, respectively. These concentrations are significantly lower than the concentration measured in the foot shock group (p<0.05, Figure 3B). Denervation alone also reduced plasma Ang II concentration to 272.95 ± 33.10 μg/L, a level markedly lower than measured in the control group (p<0.05, Figure 3B). ACEI alone decreased plasma Ang II concentration compared with control group (234.70 ± 45.30 μg/L, p<0.05, Figure 3A), but Tempol alone had no effect on plasma renin concentration (297.80 ± 31.70 μg/L).
These results indicate that RSNS and oxidative stress are involved in foot shock-induced increases of circulatory RAS.
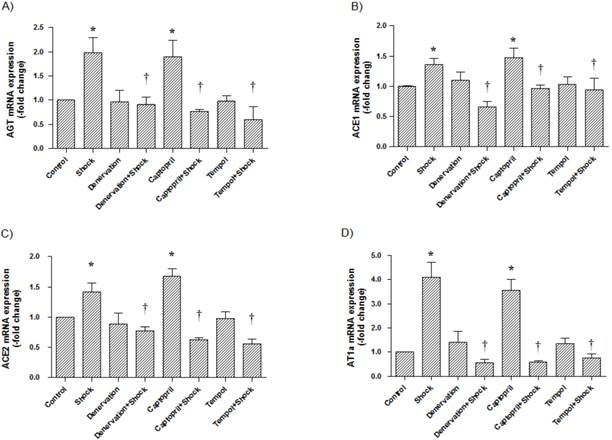
4. Effects of shock, denervation, ACEI and Tempol treatments AGT, ACE1, ACE2 and AT1a mRNA expression in the cerebral cortex and hypothalamus
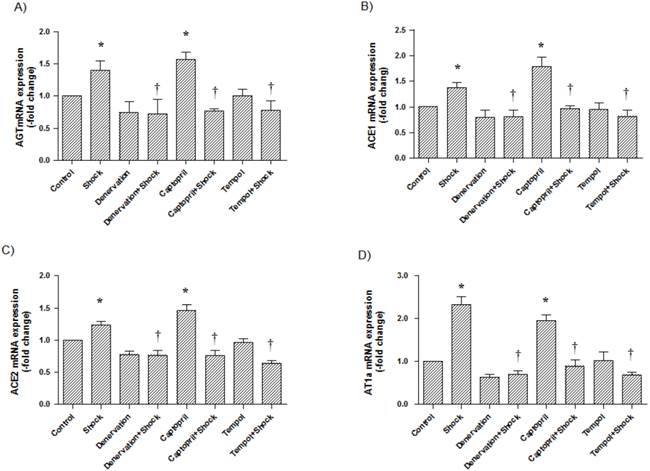
In the cerebral cortex, AGT, ACE1, ACE2, and AT1a mRNA expression (1.97 ± 0.31, 1.36 ± 0.10, 1.41 ± 0.15, 4.09 ± 0.64-fold of control, respectively) was significantly increased in the foot shock group when compared with the control group (p<0.05 vs. control, Figure 4A, 4B, 4C, 4D). Denervation, ACEI, and Tempol treatments significantly attenuated the foot shock-induced increases in AGT, ACE1, ACE2, and AT1a mRNA expression (p<0.05 vs. foot shock group, Figure 4A, 4B, 4C, 4D). Denervation itself and Tempol alone did not affect AGT, ACE1, ACE2 and AT1a mRNA expression in the cerebral cortex. ACEI alone markedly increased mRNA expressions of AGT, ACE1, ACE2 and AT1a in the cerebral cortex.
AGT, ACE1, ACE2, and AT1a mRNA expression in the cerebral cortex. (A) AGT mRNA expression from each test group, (B) ACE1 mRNA expression from each test group, (C) ACE2 mRNA expression from each test group, (D) AT1a mRNA expression from each test group. All data were normalized to β-actin. Control group data (n = 15) are expressed as 1. Data of stress group (n = 15), denervation group (n = 15), denervation plus stress group (n = 15), Captorpril plus stress group (n = 15) and Tempol plus stress group (n = 15) were expressed as fold of control group. Data were presented as mean ± SEM. * p< 0.05 compared with control group. † p<0.05 compared with stress group.
In the hypothalamus, AGT, ACE1, ACE2, and AT1a mRNA expression (1.39 ± 0.15, 1.38 ± 0.10, 1.23 ± 0.06, 2.33 ± 0.18-fold of control, respectively) was significantly increased in the foot shock group when compared with the control group (p<0.05 vs. control, Figure 5A, 5B, 5C, 5D). Denervation, Captopril, and Tempol treatments significantly inhibited foot shock-induced increases in AGT, ACE1, ACE2, and AT1a mRNA expression (p<0.05 vs. foot shock group, Figure 5A, 5B, 5C, 5D). Denervation itself and Tempol alone did not affect AGT, ACE1, ACE2, and AT1a expression in hypothalamus. ACEI alone markedly increased mRNA expressions of AGT, ACE1, ACE2 and AT1a in the cerebral hypothalamus.
AGT, ACE1, ACE2 and AT1a mRNA expression in the hypothalamus. (A) AGT mRNA expression in each test group, (B) ACE1 mRNA expression in each group, (C) ACE2 mRNA expression in each group, (D) AT1a mRNA expression in each group. All data were normalized to β-actin. Control group data (n = 15) were expressed as 1. Data of stress group (n = 15), denervation group (n = 15), denervation plus stress group (n = 15), Captopril group (n = 15), Captorpril plus stress group (n = 15), Tempol group (n = 15) and Tempol plus stress group (n = 15) were expressed as fold of control group. Data were presented as mean ± SEM. * p<0.05 compared with control group. † p<0.05 compared with stress group.
5. Effects of shock, denervation, ACEI and Tempol treatments AGT, ACE1, ACE2 and AT1a protein expression in the cerebral cortex and hypothalamus
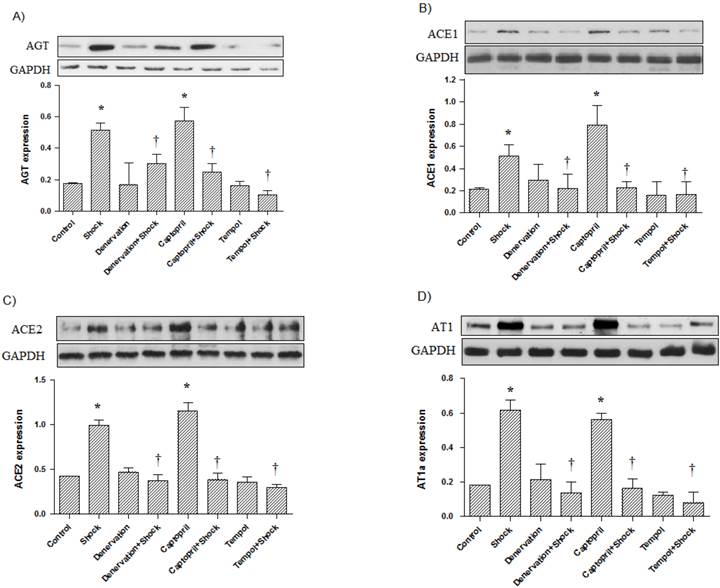
In the cerebral cortex, AGT, ACE1, ACE2, and AT1a protein expression was significantly increased in the foot shock group when compared with the control group (p<0.05 vs. control, Figure 6A, 6B, 6C, 6D). Denervation, ACEI, and Tempol treatments significantly attenuated the foot shock-induced increases in AGT, ACE1, ACE2, and AT1a protein expression (p<0.05 vs. foot shock group, Figure 6A, 6B, 6C, 6D). Denervation itself and Tempol alone did not affect AGT, ACE1, ACE2 and AT1a protein expression in the cerebral cortex. ACEI alone markedly increased protein expression of AGT, ACE1, ACE2 and AT1a in the cerebral cortex.
AGT, ACE1, ACE2, and AT1a protein expression in the cerebral cortex (n = 10). (A) AGT protein expression from each test group, (B) ACE1 protein expression from each test group, (C) ACE2 protein expression from each test group, (D) AT1a protein expression from each test group. All data were normalized to GAPDH protein expression. Data were presented as mean ± SEM. * p< 0.05 compared with control group. † p<0.05 compared with stress group.

In the hypothalamus, AGT, ACE1, ACE2, and AT1a protein expression was also significantly increased in the foot shock group when compared with the control group (p<0.05 vs. control, Figure 7A, 7B, 7C, 7D). Denervation, Captopril, and Tempol treatments significantly inhibited foot shock-induced increases in AGT, ACE1, ACE2, and AT1a protein expression (p<0.05 vs. foot shock group, Figure 7A, 7B, 7C, 7D). Denervation itself and Tempol alone did not affect AGT, ACE1, ACE2, and AT1a expression in hypothalamus. ACEI alone markedly increased protein expression of AGT, ACE1, ACE2 and AT1a in the cerebral hypothalamus.
AGT, ACE1, ACE2, and AT1a protein expression in the cerebral hypothalamus (n = 10). (A) AGT protein expression from each test group, (B) ACE1 protein expression from each test group, (C) ACE2 protein expression from each test group, (D) AT1a protein expression from each test group. All data were normalized to GAPDH protein expression. Data were presented as mean ± SEM. * p< 0.05 compared with control group. † p<0.05 compared with stress group.
These results suggest that RSNS and oxidative stress play a role in foot shock-induced increases of cerebral RAS expression.
6. Effects of shock, denervation, ACEI, and Tempol treatments on plasma SOD, activity of GSH-Px, and TBARS levels
The activity of SOD in plasma in the foot shock group was 55.30 ± 5.78 U/L, which was markedly higher than the activity level found in the control group, which was 137.48 ± 7.38 U/L (p<0.05, Figure 8A). The plasma SOD activity in the denervation plus shock, ACEI, and Tempol groups was 84.56 ± 4.24, 126.35 ± 5.48, and 102.00 ± 4.00 U/L, respectively. These levels were markedly higher than levels measured in the foot shock group (p<0.05, Figure 8A). However, denervation alone and Tempol also significantly affected plasma SOD activity when compared with the control group (Figure 8A). ACEI alone had no effect on plasma SOD activity.
Plasma SOD and GSH-Px activities and TBARS levels. Plasma SOD activity (A), GSH-Px activity (B), and TBARS levels (C) in each test group after two week stress period were measured. Data from each group (n = 15) are presented as mean ± SEM. * p<0.05 compared control group. † p<0.05 compared with stress group.

The activity of GSH-Px in plasma from the stress group was 104.08 ± 11.16 U/L, which was reduced when compared with the control group (141.56 ± 8.56 U/L, p<0.05, Figure 8B). The activity of GSH-Px in the denervation plus shock, ACEI, and Tempol treatment groups was 133.52 ± 11.52, 128.50 ± 13.14, and 120.44 ± 5.28 U/L, respectively. These activity levels were markedly higher than level measured in the foot shock group (p<0.05, Figure 8B). Denervation, ACEI and Tempol alone did not affect plasma GSH-Px activity (Figure 8B).
Foot shock markedly increased plasma TBARS levels when compared with the control group (131.18 ± 0.91 vs. 108.20 ± 4.37 mmol/L, p<0.05). Plasma TBARS level in the denervation plus shock, ACEI, and Tempol treatment groups was 116.93 ± 9.38, 111.11 ± 10.38 and 107.18 ± 6.65 mmol/L, respectively. These levels were markedly lower than those measured in the foot shock group (p<0.05, Figure 8C). Denervation and ACEI alone did not affect plasma TBARS levels (Figure 8C). While Tempol alone markedly reduced plasma TBARS levels.
These results reveal that RSNS and RAS play a role in foot shock-induced increase of oxidative stress.
7. Effects of shock, denervation, ACEI, and Tempol on renal noradrenaline concentration and plasma concentration of corticosterone
The renal noradrenaline concentration in the foot shock group was 76.60 ± 7.38 ng/mg protein, which was significantly higher than the concentration in control group, which was 58.81 ± 4.14 ng/mg protein. Interestingly, denervation, ACEI and Tempol reduced renal noradrenaline levels to 34.32 ± 3.06, 53.60 ± 6.16, and 47.60 ± 5.16 ng/mg protein, respectively (p<0.05 vs. foot shock group, Figure 9A). However, denervation, ACEI and Tempol alone had no effect on renal noradrenaline levels. These results indicate that foot shook do increase renal sympathetic nerve system, and confirm the success of renal sympathetic nerve denervation.
Renal concentration of noradrenaline and plasma concentrations of corticosterone. The concentration of noradrenaline in renal tissue (A) and the concentration of corticosterone in plasma (B) in each test group were measured after the two week stress period. Data from each group (n = 15) are presented as mean ± SEM. * p<0.05 compared control group. † p<0.05 compared with stress group.
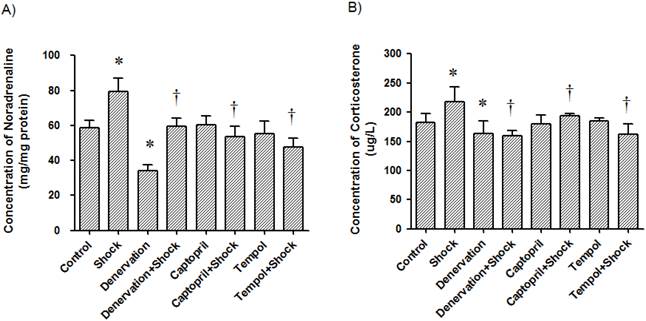
The plasma concentration of corticosterone in the foot shock group was 218.51 ± 24.95 μg/L, which was significantly higher than the concentration in the control group, which was 182.99 ± 14.58 μg/L. Similarly, denervation, ACEI, and Tempol reduced plasma corticosterone levels to 160.24 ± 8.27, 194.19 ± 3.32 and 162.51 ± 17.83 μg/L, respectively (p<0.05 vs. foot shock group, Figure 9B). However, denervation, ACEI and Tempol alone had no effect on plasma corticosterone levels. These results confirm that foot shock induce rat under stress condition, and this stress condition could be partially alleviated by renal sympathetic denervation, ACEI or antioxidant.
Discussion
In the present study, we observed that renal sympathetic denervation, ACEI and antioxidant treatment attenuated foot shock-induced increases of SBP, cerebral and circulatory RAS components, lipid peroxidation, and stress hormone. The treatments also attenuated the foot shock-induced decreases in antioxidant levels, suggesting important roles for RAS and oxidative stress in regulating blood pressure in response to chronic stress. These results also indicate an interaction between central RAS, the peripheral nervous system, and oxidative stress.
Previous evidence has shown that stress is an independent factor in cardiovascular events, possibly by maintaining high blood pressure [6, 9, 40-42]. It was widely accepted that stress increases blood pressure via the hypothalamus-pituitary-adrenal cortex [6, 7] and sympathetic-adrenal systems [9, 11-13]. Our present study demonstrates the increase of plasma corticosterone and renal tissue noradrenaline contents in response to foot-shock, indicating the activation of adrenal system. Renal denervation reduced RAS expression and oxidative stress markers suggest that activation of adrenal system is responsible for the activation of RAS and the increase of oxidative stress.
Since the discovery of RAS, its role in the regulation of blood pressure has been widely investigated, not only in the peripheral circulatory system, but also in the central nervous system [10, 43, 44]. It was reported that Ang II participates in stress-induced high blood pressure by stimulating the synthesis and release of hypothalamic vasopressin [10]. Other RAS components have also been demonstrated to be involved in the regulation of blood pressure in the nucleus tractus solitarii [44], paraventricular nucleus and rostral ventrolateral medulla [45]. Our present study also demonstrates the increase of RAS component mRNA levels in the central nervous system (cerebral cortex and hypothalamus). Interestingly, we observed that the increase in mRNA expression of these components is suppressed by both ACEI and Tempol treatment and renal sympathetic nerve denervation. This data clearly suggest that one: oxidative stress play a role in increase of RAS expression, two: sympathetic nerve activation feed-back to regulate central nervous system RAS component expression, which plays a role in the elevation of blood pressure in our foot shock stress model. Although we observed that ACEI alone had significant effects on central nervous RAS mRNA expressions and circulatory renin content due to negative feed-back effect, and also the inhibitory effects on circulatory Ang II, we could not find these effects in ACEI treated foot-shock group. Further study may be needed to explore this issue.
It should also be noted that brain ACE2 has been reported to exert antihypertensive effects in neurogenic hypertension [46]. However, our present paper shows that chronic foot shock stress also increases ACE2 mRNA expression in the cerebral cortex and hypothalamus, accompanied by other components such as AGT, ACE1 and AT1 and an increase in blood pressure. Our interpretation is that the increase may be due to auto-regulation in response to elevated blood pressure or ACE2 could not antagonize the hypertensive effects of other components such as Ang II, AT1. Alternatively, increases of ACE2 mRNA expression in the cerebral cortex and hypothalamus are maybe due to the different animal model applied in the our experiments and the previously published report.
Although RAS components increase their expressions in foot shock-induced hypertension, the mechanism by which they affect hypertension must still be elucidated. Our previous study demonstrated that, following the administration of Ang II, the increased blood pressure was sensitive to antioxidant treatment, which suggests that body antioxidant levels are important in maintaining high blood pressure [30-32]. In the present study, our data show that chronic foot shock reduces plasma antioxidant levels (GSH-Px and SOD activities) and increases lipid peroxidation. Antioxidant treatment not only reversed antioxidant levels and decreased lipid peroxidation, but it also attenuated the increase in blood pressure, suggesting that oxidative stress is involved in foot shock-induced hypertension. Furthermore, denervation and ACEI treatments also restored antioxidant levels and decreased lipid peroxidation, suggesting that sympathetic nerve activation and the subsequent RAS increase-induced hypertension are mediated by oxidative stress. More importantly, antioxidant treatment also reduced central RAS component expression, suggesting that central RAS has a complex mechanism of regulation.
Although it was reported that Ang II can influence the hypothalamus-pituitary-adrenal system by enhancing the synthesis and secretion of corticotrophin-releasing hormone, adrenocoticotrophic hormone, and corticosterone [47-49], how exactly Ang II and other RAS components influence these systems is still unclear. In the present study, we also observed a foot shock-induced increase in plasma corticosterone levels, suggesting that the hypothalamus-pituitary-adrenal system is activated in our model. However, our data also demonstrate that ACEI and antioxidant treatments both reduce plasma corticosterone levels, suggesting another mechanism by which the hypothalamus-pituitary-adrenal system may be activated.
Due to the high morbidity and mortality of cardiovascular diseases, more and more efforts are being made to explore the mechanisms behind their pathogenesis. Although new treatment strategies have been developed and widely applied, they have met with unsatisfactory outcomes in clinical patients. In our modern society, people inevitably encounter more and more stress, which directly affects their health. Therefore, it is increasingly important to elucidate the mechanism of stress induced hypertension and cardiovascular diseases.
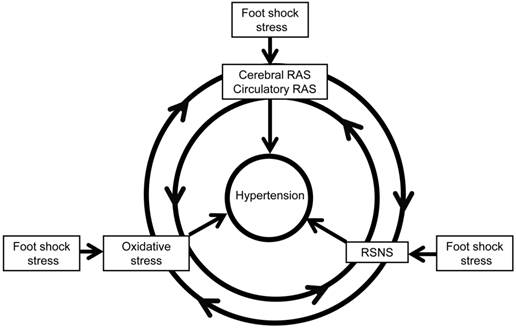
In conclusion, our present paper demonstrates the role of RAS and oxidative stress in chronic foot stress-induced hypertension, revealing a possible mechanism of chronic blood pressure regulation. This regulation may take place through the interactions between the sympathetic nerve, RAS, and oxidative stress (Figure 10), which is one factor could potentiate another factor to form a vicious circle to enhance the development of hypertension. Taken together, these data provide a better understanding of the pathogenesis of hypertension and present novel strategies in the prevention and treatment of stress-induced hypertension.
Possible mechanism of foot shock-induced hypertension. RAS indicates renin angiotensin system, and RSNS indicates renal sympathetic nerve system. RAS could increases oxidative stress and RSNS activity, oxidative stress could increase RAS expression and RSNS activity, also RSNS activation could increase oxidative stress and RAS expression. The reciprocal interaction of these factors contributes to stress-induced development of hypertension.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (81270316, 81271922, 81470563), the Research Program of Soochow University (Q413400111), Suzhou science and technology development projects (SYS201364, SYS201258, SYSD2012116).
Conflict of Interest
None declared.
References
1. Scolari F, Ravani P. Atheroembolic renal disease. Lancet. 2010;375(9726):1650-60
2. Dahlof B. Prevention of stroke in patients with hypertension. The American journal of cardiology. 2007;100(3A):17J-24J
3. Imhof P, Hurlimann A, Steinmann B. [Blood pressure increase caused by psychological stress]. Cardiologia. 1957;31(3):272-6
4. Sparrenberger F, Cichelero FT, Ascoli AM, Fonseca FP, Weiss G, Berwanger O. et al. Does psychosocial stress cause hypertension? A systematic review of observational studies. Journal of human hypertension. 2009;23(1):12-9
5. Landsbergis PA, Dobson M, Koutsouras G, Schnall P. Job strain and ambulatory blood pressure: a meta-analysis and systematic review. American journal of public health. 2013;103(3):e61-71
6. Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. Journal of hypertension. 1987;5(4):425-33
7. Whitworth JA, Saines D, Scoggins BA. Blood pressure and metabolic effects of cortisol and deoxycorticosterone in man. Clinical and experimental hypertension Part A, Theory and practice. 1984;6(4):795-809
8. Willard PW, Fuller RW. Functional significance of the sympathetic nervous system in production of hypertension. Nature. 1969;223(5204):417-8
9. Zimmerman RS, Frohlich ED. Stress and hypertension. Journal of hypertension Supplement: official journal of the International Society of Hypertension. 1990;8(4):S103-7
10. Lu LM, Wang J, Yao T. [Angiotensin II participates in stress-induced high blood pressure via stimulating hypothalamic vasopressin synthesis and release]. Sheng li xue bao: [Acta physiologica Sinica]. 2000;52(5):371-4
11. Anderson EA, Sinkey CA, Mark AL. Mental stress increases sympathetic nerve activity during sustained baroreceptor stimulation in humans. Hypertension. 1991;17(4 Suppl):III43-9
12. Wallin BG, Delius W, Hagbarth KE. Comparison of sympathetic nerve activity in normotensive and hypertensive subjects. Circulation research. 1973;33(1):9-21
13. Mustacchi P. Stress and hypertension. The Western journal of medicine. 1990;153(2):180-5
14. Leong KT, Walton A, Krum H. Renal sympathetic denervation for the treatment of refractory hypertension. Annual review of medicine. 2014;65:349-65
15. Aranda-Lara P, Martinez-Esteban MD, Munoz JJ, Hernandez-Marrero D. Renal sympathetic denervation: a new treatment strategy in the management of refractory arterial hypertension. Nefrologia: publicacion oficial de la Sociedad Espanola Nefrologia. 2012;32(5):555-7
16. Hu J, Ji M, Niu C, Aini A, Zhou Q, Zhang L. et al. Effects of renal sympathetic denervation on post-myocardial infarction cardiac remodeling in rats. PloS one. 2012;7(9):e45986
17. Krum H, Schlaich MP, Sobotka PA, Bohm M, Mahfoud F, Rocha-Singh K. et al. Percutaneous renal denervation in patients with treatment-resistant hypertension: final 3-year report of the Symplicity HTN-1 study. Lancet. 2014;383(9917):622-9
18. Schlaich MP, Schmieder RE, Bakris G, Blankestijn PJ, Bohm M, Campese VM. et al. International expert consensus statement: Percutaneous transluminal renal denervation for the treatment of resistant hypertension. Journal of the American College of Cardiology. 2013;62(22):2031-45
19. Schirmer SH, Sayed MM, Reil JC, Ukena C, Linz D, Kindermann M. et al. Improvements in left ventricular hypertrophy and diastolic function following renal denervation: effects beyond blood pressure and heart rate reduction. Journal of the American College of Cardiology. 2014;63(18):1916-23
20. Davies JE, Manisty CH, Petraco R, Barron AJ, Unsworth B, Mayet J. et al. First-in-man safety evaluation of renal denervation for chronic systolic heart failure: primary outcome from REACH-Pilot study. International journal of cardiology. 2013;162(3):189-92
21. Crowley SD, Coffman TM. Recent advances involving the renin-angiotensin system. Exp Cell Res. 2012;318(9):1049-56
22. Leung PS. The peptide hormone angiotensin II: its new functions in tissues and organs. Current protein & peptide science. 2004;5(4):267-73
23. Marc Y, Llorens-Cortes C. The role of the brain renin-angiotensin system in hypertension: implications for new treatment. Prog Neurobiol. 2011;95(2):89-103
24. Campos LA, Bader M, Baltatu OC. Brain Renin-Angiotensin system in hypertension, cardiac hypertrophy, and heart failure. Front Physiol. 2011;2:115
25. Wang LH, Dong T, Liu BB, Zhao XD, Chen JW, Murao K. et al. Contribution of the renin-angiotensin system in chronic foot-shock induced hypertension in rats. Life sciences. 2015;121:135-44
26. Moretti JL, Burke SL, Davern PJ, Evans RG, Lambert GW, Head GA. Renal sympathetic activation from long-term low-dose angiotensin II infusion in rabbits. Journal of hypertension. 2012;30(3):551-60
27. Huang C, Yoshimoto M, Miki K, Johns EJ. The contribution of brain angiotensin II to the baroreflex regulation of renal sympathetic nerve activity in conscious normotensive and hypertensive rats. The Journal of physiology. 2006;574(Pt 2):597-604
28. Campese VM, Shaohua Y, Huiquin Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension. 2005;46(3):533-9
29. Clayton SC, Haack KK, Zucker IH. Renal denervation modulates angiotensin receptor expression in the renal cortex of rabbits with chronic heart failure. American journal of physiology Renal physiology. 2011;300(1):F31-9
30. Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Abe Y. ROS during the acute phase of Ang II hypertension participates in cardiovascular MAPK activation but not vasoconstriction. Hypertension. 2004;43(1):117-24
31. Kimura S, Zhang GX, Abe Y. Malfunction of vascular control in lifestyle-related diseases: oxidative stress of angiotensin II-induced hypertension: mitogen-activated protein kinases and blood pressure regulation. Journal of pharmacological sciences. 2004;96(4):406-10
32. Kimura S, Zhang GX, Nagai Y, Miyata K, Nishiyama A, Shokoji T. et al. Time-dependent transition of tempol-sensitive reduction of blood pressure in angiotensin II-induced hypertension. Journal of hypertension. 2004;22(11):2161-8
33. Hocherl K, Kammerl M, Kees F, Kramer BK, Grobecker HF, Kurtz A. Role of renal nerves in stimulation of renin, COX-2, and nNOS in rat renal cortex during salt deficiency. American journal of physiology Renal physiology. 2002;282(3):F478-84
34. DiBona GF. Peripheral and central interactions between the renin-angiotensin system and the renal sympathetic nerves in control of renal function. Annals of the New York Academy of Sciences. 2001;940:395-406
35. Zhang GX, Ohmori K, Nagai Y, Fujisawa Y, Nishiyama A, Abe Y. et al. Role of AT1 receptor in isoproterenol-induced cardiac hypertrophy and oxidative stress in mice. Journal of molecular and cellular cardiology. 2007;42(4):804-11
36. Bolterman RJ, Manriquez MC, Ortiz Ruiz MC, Juncos LA, Romero JC. Effects of captopril on the renin angiotensin system, oxidative stress, and endothelin in normal and hypertensive rats. Hypertension. 2005;46(4):943-7
37. Yoshiyama M, Takeuchi K, Omura T, Kim S, Yamagishi H, Toda I. et al. Effects of candesartan and cilazapril on rats with myocardial infarction assessed by echocardiography. Hypertension. 1999;33(4):961-8
38. Liu N, Wang LH, Guo LL, Wang GQ, Zhou XP, Jiang Y. et al. Chronic restraint stress inhibits hair growth via substance P mediated by reactive oxygen species in mice. PloS one. 2013;8(4):e61574
39. Xia CM, Shao CH, Xin L, Wang YR, Ding CN, Wang J. et al. Effects of melatonin on blood pressure in stress-induced hypertension in rats. Clinical and experimental pharmacology & physiology. 2008;35(10):1258-64
40. Rosengren A, Hawken S, Ounpuu S, Sliwa K, Zubaid M, Almahmeed WA. et al. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):953-62
41. Lambert G, Schlaich M, Lambert E, Dawood T, Esler M. Stress reactivity and its association with increased cardiovascular risk: a role for the sympathetic nervous system? Hypertension. 2010;55(6):e20 author reply e1
42. Neelakantan S. Psychology: Mind over myocardium. Nature. 2013;493(7434):S16-7
43. von Bohlen und Halbach O, Albrecht D. The CNS renin-angiotensin system. Cell and tissue research. 2006;326(2):599-616
44. Cheng WH, Lu PJ, Hsiao M, Hsiao CH, Ho WY, Cheng PW. et al. Renin activates PI3K-Akt-eNOS signalling through the angiotensin AT(1) and Mas receptors to modulate central blood pressure control in the nucleus tractus solitarii. British journal of pharmacology. 2012;166(7):2024-35
45. Gabor A, Leenen FH. Central neuromodulatory pathways regulating sympathetic activity in hypertension. Journal of applied physiology. 2012;113(8):1294-303
46. Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res. 2013;113(9):1087-96
47. Jezova D, Ochedalski T, Kiss A, Aguilera G. Brain angiotensin II modulates sympathoadrenal and hypothalamic pituitary adrenocortical activation during stress. Journal of neuroendocrinology. 1998;10(1):67-72
48. Rivier C, Vale W. Effect of angiotensin II on ACTH release in vivo: role of corticotropin-releasing factor. Regulatory peptides. 1983;7(3):253-8
49. Sumitomo T, Suda T, Nakano Y, Tozawa F, Yamada M, Demura H. Angiotensin II increases the corticotropin-releasing factor messenger ribonucleic acid level in the rat hypothalamus. Endocrinology. 1991;128(5):2248-52
Author contact
![]() Corresponding authors: Guo-Xing Zhang, Department of Physiology, Medical College of Soochow University, 199 Ren-Ai Road, Dushu Lake Campus, Suzhou Industrial Park, Suzhou 215123, P.R. China. Tel: +86-512-65880127; Fax: +86-512-68222218; Email: zhangguoxingedu.cn. Co-corresponding author: Wan-Ping Sun, Laboratory of Molecular Diagnostics, College of Pharmaceutical Sciences, Soochow University, 199 Ren-Ai Road, Dushu Lake Campus, Suzhou Industrial Park, Suzhou 215123, P.R. China. Tel: +86-13584889256. Email: sunwanpingedu.cn
Corresponding authors: Guo-Xing Zhang, Department of Physiology, Medical College of Soochow University, 199 Ren-Ai Road, Dushu Lake Campus, Suzhou Industrial Park, Suzhou 215123, P.R. China. Tel: +86-512-65880127; Fax: +86-512-68222218; Email: zhangguoxingedu.cn. Co-corresponding author: Wan-Ping Sun, Laboratory of Molecular Diagnostics, College of Pharmaceutical Sciences, Soochow University, 199 Ren-Ai Road, Dushu Lake Campus, Suzhou Industrial Park, Suzhou 215123, P.R. China. Tel: +86-13584889256. Email: sunwanpingedu.cn