Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Molecular Basis of Autophagy...
Roles of Autophagy in Cardiac...
Autophagy as a Therapeutic...
Conclusions and Perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(6):672-678. doi:10.7150/ijbs.11883 This issue Cite
Review
Functions of Autophagy in Pathological Cardiac Hypertrophy
Zhenhua Li, Jian Wang ![]() , Xiao Yang
, Xiao Yang ![]()
State Key Laboratory of Proteomics, Collaborative Innovation Center for Cardiovascular Disorders, Genetic Laboratory of Development and Disease, Institute of Biotechnology, Beijing 100071, China.
Received 2015-2-13; Accepted 2015-3-13; Published 2015-4-27
Abstract

Pathological cardiac hypertrophy is the response of heart to various biomechanical and physiopathological stimuli, such as aging, myocardial ischemia and hypertension. However, a long-term exposure to the stress makes heart progress to heart failure. Autophagy is a dynamic self-degradative process necessary for the maintenance of cellular homeostasis. Accumulating evidence has revealed a tight link between cardiomyocyte autophagy and cardiac hypertrophy. Sophisticatedly regulated autophagy protects heart from various physiological and pathological stimuli by degradating and recycling of protein aggregates, lipid drops, or organelles. Here we review the recent progresses concerning the functions of autophagy in cardiac hypertrophy induced by various hypertrophic stimuli. Moreover, the therapeutic strategies targeting autophagy for cardiac hypertrophy will also be discussed.
Keywords: autophagy, cardiac hypertrophy, heart failure, therapeutic target
Introduction
Pathological cardiac hypertrophy refers to the enlargement of heart in a variety of diseases e.g. aging, myocardial ischemia and hypertension [1]. At cellular level, pathological cardiac hypertrophy is characterized by an increase in cardiomyocyte size, disarrangement of sarcomeric structure, enhanced protein synthesis and re-expression of fetal genes [2]. Although pathological cardiac hypertrophy initially compensates for the biomechanical and physiopathological stimuli, it progresses generally to detrimental cardiac remodeling and heart failure, which is a clinical risk factor for death [3, 4]. The mechanisms of pathological cardiac hypertrophy have been studied at multiple levels, such as genetic mutations in structural proteins, alterations of molecular signaling pathways, detrimental changes in subcellular organelles and communications among various cell types in hearts [5-8]. Further understanding the mechanisms underlying pathological cardiac hypertrophy would provide novel therapeutic strategies for the treatment of heart failure.
Autophagy is a conserved catabolic process by which cellular components are transported to and degraded in the lysosomes [9]. Autophagy occurs in response to various stimuli, such as nutrient limitation and cellular stress [10, 11]. Not only does autophagy play essential roles in tissue homeostasis by degradating long-lived proteins and unwanted organelles [12], it also acts as a recycling system to produce new energy and materials for the renovation of cells [13]. Constitutive autophagy is required for cell survival, while excessive autophagy triggers autophagic cell death, indicating dual roles of autophagy in pro- or anti-survival potential [14]. Particularly, studies by using autophagy-defective animal models have elucidated the significant physiopathological functions of autophagy at whole-organism or tissue-specific levels. Dysregulation of autophagy is associated with many clinical disorders, such as cancer, neurodegeneration, infection and metabolic syndromes [12, 15].
Recently, a number of studies have begun to show that autophagy plays important roles in cardiac hypertrophy under various pathological stimuli, thus indicating a possible link between autophagy dysregulation and cardiac hypertrophy. In this review, we focus on how autophagy is regulated and what are the effects of altered autophagy on cardiac hypertrophy. In addition, the possibility of autophagy as a therapeutic target for cardiac hypertrophy will also be highlighted.
Molecular Basis of Autophagy Process
Autophagy could be either a non-specific degradation of cytoplasmic cargo (general autophagy) or a specific removal of unwanted organelles (selective autophagy), such as the clearance of mitochondria (mitophagy), peroxisomes (pexophagy), and lipid droplets (lipophagy) [12]. In both processes, intracellular components are first surrounded by double membrane-bound autophagic vesicles which then fuse with lysosomes to form autolysosomes for the degradation [16]. Autophagy process is governed by distinct functional complexes of autophagy-related gene (ATG) products. The induction of autophagy is relied on the unc-51 like kinase (ULK) complex stimulated by upstream nutrient-sensing kinases. Starvation or growth factor deprivation leads to the activation of ULK1/2 kinase, thus promoting autophagy induction [17-19]. After induction, the class III PI3K-Beclin1-VPS34 complex is activated by ULK complex [20] and recruited to the mitochondria, endoplasmic reticulum (ER), ER-mitochondria contact sites, Golgi apparatus, recycling endosome or plasma membrane to form an isolated membrane, which is called phagophore nucleation [21-25]. Apparently, the phagophore originates differently in different cell types and different physiologic or stress-related conditions. The phagophore elongation and formation of complete autophagosomes require two ubiquitin-like protein conjugation systems: the one is the ATG12-ATG5 conjugate which functions as a dimeric complex together with ATG16L1; the other is phosphatidylethanolamine (PE)-conjugated LC3, which is cleaved by ATG4 and subsquencely activated by ATG7 and E2-like enzyme ATG3. The mature autophagosome then fuse with lysosome, which is facilitated by the lysosomal membrane protein LAMP2 and the small GTPase RAB7. After fusion, lysosomal acid hydrolases, including Cathepsin-B, Cathepsin-D and Cathepsin-L, participate in the degradation of its contents [14, 26, 27].
Autophagy usually occurs at low basal levels to maintain the cellular homeostasis, and could be stimulated in response to various cellular stresses, such as nutrient limitation, oxidative stress and ER stress. Nutrient limitation exists in many pathological conditions and serves as one of the most typical triggers of autophagy. In mammalian cells, there exist two kinds of energy sensors, the short-term kinases and long-term transcriptional factors, to make accurate autophagic responses according to the status of energy metabolism. The AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) are two major short-term energy sensors for the rapid regulation of autophagy. AMPK is activated in response to low ATP/AMP ratio and promotes the induction of autophagy [28, 29]. mTORC1 is another important energy sensor to both amino acids and glucose, and acts as a central mediator of multiple autophagy-regulating signals. Nutrient inadequacy promotes autophagy by the inhibition of mTORC1 [29, 30]. In addition, autophagy is also regulated by long-term transcriptional factors. The nuclear-sensing receptors peroxisome proliferator-activated receptor-α (PPARα), cAMP response element-binding protein (CREB) and farnesoid X receptor (FXR), share common binding sites in many autophagic gene promoters and collaborate to regulate autophagy as key physiological switches [31, 32].
Oxidative stress, caused by overproduction of reactive oxygen species (ROS) excess to cellular antioxidant ability, plays vital roles in activating autophagy [33]. On one hand, ROS inhibits ATG4 activity via oxidation of an essential cysteine residue, which blocks the cleavage of PE from PE-conjugated LC3 and promotes autophagy [34]. On the other hand, the increased ROS levels result in the loss of membrane potential and the subsequent activation of Parkin-dependent mitophagy [35, 36].
ER stress is another trigger of autophagy in mammalian cells. ER stress happens upon a wide variety of physiopathological stimuli which disrupt protein folding capacity of ER and cause subsequent accumulation of unfolded or misfolded proteins [37]. ER stress promotes autophagy by two means in accordance with its dual roles in regulating unfolded protein response (UPR) and Ca2+ handling. UPR activates autophagy via regulation of autophagy- related kinases or by ATF4-dependent transcriptional regulation [37, 38]. Ca2+ release from ER promotes autophagy via activation of several Ca2+-dependent signaling pathways [39].
Roles of Autophagy in Cardiac Hypertrophy
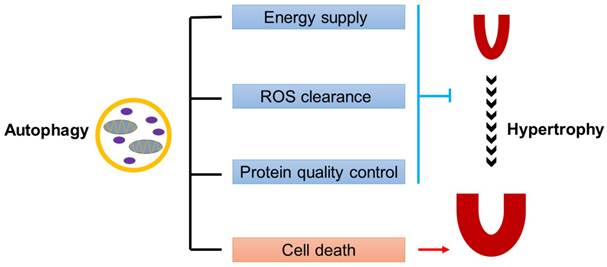
Autophagy plays complicated but indispensable roles in the maintenance of cardiac homeostasis [40]. On one hand, autophagy can act as a pro-survival factor. Firstly, autophagy promotes the degradation and recycling of long-lived proteins, lipid drops, or even damaged organelles, demonstrating a survival response to energy deprivation during cardiac hypertrophy. Secondly, the autophagy-lysosome system collaborates with ubiquitin-proteasome system in the protein quality control, which is essential for preventing cells from cytotoxicity caused by misfolded protein aggregates. Thirdly, autophagy participates in the clearance of damaged mitochondria accumulated in hypertrophic hearts, thus eliminating the ill effects of mitochondria-derived ROS on cardiomyocytes. On the other hand, autophagy can trigger type-II programmed cell death, which makes it a pro-death factor (Fig.1).
Dual roles of autophagy in cardiac hypertrophy. Baseline autophagy maintains cardiac structure and function by energy supply, ROS clearance and protein quality control, while excess autophagy is detrimental to the heart by enhancing cell death.
Pathological cardiac hypertrophy is accomplished by dysfunction of numerous hypertrophic related signaling pathways, many of which are also broadly involved in the regulation of cardiomyocyte autophagy [41-46]. Ca2+/Calcineurin signaling pathway, which promotes cardiac hypertrophy by synergistically activating hypertrophic gene transcription, has been shown to inhibit cardiomyocyte autophagy in an AMPK-depedent manner [41, 42]. PI3K/AKT pathway are involved in cardiac hypertrophy by two well-established downstream targets, glycogen synthase kinase-3 (GSK-3) and mTOR, both of which are main regulators of cardiomyocyte autophagy [43-45]. Mitogen-activated protein kinases (MAPKs) singaling, another significant hypertrophic signaling pathway, is also implicated in cardiomyocyte autophagy and mitophagy [46]. These studies suggest the link between cardiomyocyte autophagy and cardiac hypertrophy.
Furthermore, the experiments of disruption of basal autophagy in mouse heart establish the direct link between autophagy and cardiac hypertrophy [47-49]. Deletion of Atg5 specifically in adult hearts causes severe cardiac hypertrophy and dysfunction [47]. Similar results are obtained by ablation of Vps34 in the hearts, which show increased left ventricular wall thickness and mass, and decreased cardiac contractility with lower ejection fraction and fractional shortening [48]. In addition, loss of Mcl1, an anti-apoptotic BCL2 protein, inhibits autophagy and leads to rapid contractile dysfunction, cardiac hypertrophy and early mortality accompanied by cardiac fibrosis and inflammation [49].
Autophagy in aging-related cardiac hypertrophy
Autophagy acts as a defense mechanism to protect cardiomyocytes from aging, and thus plays pivotal roles in the amelioration of aging-related cardiac hypertrophy (Fig.2). The heart develops cardiac hypertrophy and diastolic dysfunction with aging when the autophagic activity is inhibited [50, 51]. Growing evidence suggests that the mechanisms involve signaling controlling cardiac aging, including NAD+-dependent class III histone deacetylases Sirtuins (SIRT), and endothelin-1 (ET-1) [52, 53]. The impaired SIRT1 activity might be an important cause for the inhibition of autophagy in aging hearts, as SIRT1-regulated AKT signaling and ATG protein activity are all necessary for the normal autophagy maintenace [52, 54-56]. In addition, the increased plasma level of endothelin-1 (ET-1) with aging also triggers a decrease in autophagy [53].
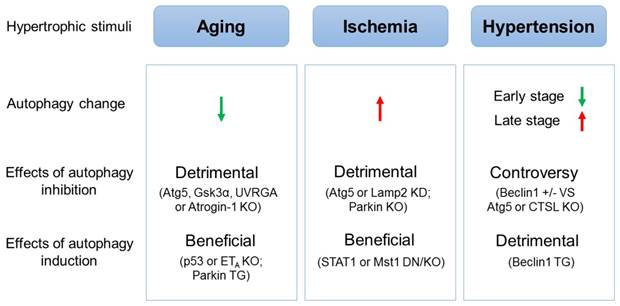
The effects of autophagic alteration on hearts under different hypertrophic stimuli. The autophagic activity is decreased in aging hearts but is increased in ischemic hearts. Inhibition of autophagy in mice is detrimental to, while induction of autophagy is beneficial for hearts suffering aging or ischemia. In the mouse model of hypertensive cardiac hypertrophy, autophagy is inhibited in the early stage but is induced in the late stage. Induction of autophagy is demonstrated to be detrimental to hypertensive cardiac hypertrophy, while contradict results concerning the effects of inhibited autophagy on hypertensive cardiac hypertrophy are reported. (↑: induction; ↓: inhibition; KO: knockout mice; TG: transgenic mice; KD: gene knockdown; DN: dominant negative transgenic mice)
Autophagy inhibition can induce aging-related cardiac hypertrophy [57-60]. Cardiac-specific Atg5-deficient mice show inhibited autophagy, shortened lifespan and aging-related cardiomyopathy, characterized by increased left ventricular dimension and decreased fractional shortening [57]. Gsk3α knockout mice demonstrate suppressed autophagy in an mTOR-dependent manner and develop cardiac hypertrophy and contractile dysfunction with age [58]. Deficiency of UVRGA, a protein which promotes autophagosome formation by binding directly to Beclin1-VPS34 complex and enhancing VPS34 activity, impairs autophagic flux and leads to age-related cardiomyopathy accompanied by enhanced inflammatory response [59]. In addition, loss of Atrogin-1 in mice fails to degrade CHMP2B, resulting in autophagy impairment and cardiomyopathy characterized by myocardial remodeling with reduced diastolic function and arrhythmias [60].
Induction of autophagy can reverse aging-associated cardiac hypertrophy and contractile dysfunction [53, 61, 62]. It has been shown that p53 can impair autophagy by disrupting Parkin translocation to damaged mitochondria and subsequent mitophagy. Parkin-overexpression or p53-deficiency ameliorates the functional decline in aging hearts accompanied by decreased senescence-associated β-galactosidase activity [61, 62]. Cardiomyocyte endothelin-1 receptor ETA deletion induces autophagy and alleviates aging-associated cardiac hypertrophy [53].
All these studies demonstrate the essential roles of autophagy in the maintenance of cardiac homeostasis during aging, suggesting the therapeutic potential of activation of autophagy for cardiac aging.
Autophagy in myocardial ischemia-induced cardiac hypertrophy
Autophagy contributes to cardiomyocyte survival and protects hearts from myocardial ischemia-induced cardiac pathological remodeling and dysfunction (Fig.2). Myocardial ischemia impairs cardiac function, which is due to inadequate nutrient and oxygen supply caused by the blockage of the coronary arteries. The ischemic hearts are exposed to starvation and hypoxia, which stimulate autophagy via AMPK activation and Bnip3 upregulation respectively [63-66]. Furthermore, the oxidative stress and ER stress, two significant triggers of autophagy mentioned aboved, are also induced by ischemia [67, 68].
Inhibition of autophagy could promote injury in myocardial ischemia [69, 70]. Knockdown of Atg5 or Lamp2 in vitro reduces ATP content and diminishes survival of cardiomyocytes subjected to anoxia [69]. Administration of autophagy inhibitor bafilomycin A1 significantly increases infarct size in a mouse model of acute myocardial infarction [69]. In addition, Parkin deficient mice are more sensitive to myocardial infarction and present decreased mitophagy, enlarged left ventricular and larger infarcts after the infarction [70].
Stimulation of autophagy plays protective roles in myocardial ischemia. STAT1 deficient hearts show increased levels of autophagy and significantly decreased infarct size following myocardial infarction, which is reversed by pre-treatment with the autophagy inhibitor [71]. Mst1 suppresses autophagy via phosphorylating Beclin1 and enhancing interaction between Beclin1 and BCL-2/BCL-XL. Activation of autophagy is found in Mst1 knockout and dominant negative transgenic mice, which reduces MI scar contraction, attenuates left ventricular enlargement and improves LV function [72, 73].
Autophagy in hypertension-induced cardiac hypertrophy
There exist controversies concerning the function of autophagy in hypertension-induced cardiac hypertrophy (Fig.2). In clinic, hypertension is one of the most common causes of cardiac hypertrophy and heart failure. Hypertensive cardiac hypertrophy is due to not only the mechanical stress from elevated blood pressure but also the alternations of neurohormones, growth factors and cytokines. Autophagic level is changed during different periods of transverse aortic constriction (TAC)-induced cardiac hypertrophy [47]. Autophagy is suppressed in hypertrophied hearts after TAC for 1 week, and is upregulated in failing hearts after TAC for 4 weeks, revealed by alteration of LC3-II expression levels [47]. Renin-angiotensin-aldosterone system (RAAS) is a hormone system that regulates blood pressure and shows dual effects on autophagy via its two receptors, angiotensin II receptor, type 1 (AGTR1) and AGTR2 [74]. Moreover, complicated regulatory effects of numerous hypertrophic signalings might be responsible for the phase-dependent change of autophagy [41-46]. Disruption of autophagy-regulating genes is also associated with TAC-induced cardiac hypertrophy. Cardiac-specific deficiency of Atg5 early in cardiogenesis shows no pathological cardiac phenotypes at baseline, but displays severe cardiac dysfunction and left ventricular dilatation 1 week after TAC operation [47]. Cathepsin-L deficiency impairs autophagosomal content degradation and worsens ventricular remodeling and heart failure in response to TAC [75]. These results suggest the protective role of autophagy in pressure overload-induced cardiac hypertrophy. However, another study on Beclin1 draws an opposite conclusion. They find that TAC induces autophagic activity from 1 day till 3 weeks. Mice haploinsufficient for Beclin1 are resistant to TAC-induced pathological remodeling of the left ventricle. While cardiomyocyte-specific overexpression of Beclin1 promotes autophagy and pathological ventricular remodeling caused by TAC [76]. The study suggests that TAC-induced cardiac autophagy is a maladaptive response.
Autophagy as a Therapeutic Target for Cardiac Hypertrophy
Autophagy is a must for the maintenance of normal cardiac performance, and either inadequate or excessive autophagy in different cardiac pathological conditions is a maladaptive response. Efforts have been made to elucidate the possibility of tuning autophagy as a therapeutic target of cardiac hypertrophy.
Administration of regulators of autophagy related pathways has been shown to have the therapeutic potential for treating cardiac hypertrophy. A number of studies indicate that induction of autophagic activity is benefit for heart diseases. As the inhibitor of mTOR, rapamycin can prevent cardiac hypertrophy induced by TAC or thyroid hormone treatment, and reverse cardiac hypertrophy and dysfunction in aging mice [77-80]. Administration of AICAR or metformin, activators of AMPK, in a TAC mice model induces autophagy, inhibits cardiac hypertrophy and improves heart function [81]. Overexpression of Atg7 rescues the CryABR120G autophagic deficiency, and attenuates the accumulation of misfolded proteins and aggregates in cardiomyocytes [82]. Furthermore, Sustained Atg7-induced autophagy in the CryABR120G hearts decreases cardiac hypertrophy and interstitial fibrosis, ameliorates ventricular dysfunction [83]. Some studies show that inhibition of autophagy can also be benefit for heart diseases. Histone deacetylases inhibitor trichostatin A abolishes TAC-induced autophagic induction and blunts pressure overload-induced cardiac hypertrophy [84].
Recent studies have demonstrated that miRNAs play important roles in regulating cardiac autophagy [85-88]. Targeting autophagy-regulating miRNAs by chemically engineered oligonucleotides mimics/antagomirs has been used to treat cardiac hypertrophy. Intravenously injection of specific antagomirs against miR-132, an autophagy-inhibiting miRNA, rescues pressure overload-induced cardiac hypertrophy and heart failure in mice [87]. Intra-myocardial injection with miR-99a-overexpressing lentiviral vectors increases autophagy via inhibiting mTOR signaling, and improves cardiac function and survival after myocardial infarction [88]. These data suggest that autophagy-regulating miRNAs might be novel therapeutic targets for stress-triggered cardiac hypertrophy.
Conclusions and Perspectives
Autophagy is a catabolic process essential for maintaining cardiac homeostasis in response to various stresses. Dysregulation of autophagy under normal or pathological conditions leads to severe cardiac hypertrophy and heart failure. More importantly, the potential of autophagy as a therapeutic target for cardiac hypertrophy has been demonstrated. Nevertheless, further study is needed to decipher the regulatory networks by which cardiac autophagy is sophisticatedly regulated. New regulators of cardiac autophagy should be explored. Long non-coding RNAs have been demonstrated to regulate heart development [89, 90] and cardiac hypertrophy [91], but their function in the regulation of cardiac autophagy needs to be further investigated. Hopefully, a better understanding of the molecular mechanisms controlling cardiac autophagy will eventually provide new promising therapeutic strategies for patients with cardiac hypertrophy.
Acknowledgements
This work was supported by the Chinese National Key Program on Basic Research (2012CB945103; 2011CB504202) and National Natural Science Foundation of China (31430057).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45-79
2. Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194:355-65
3. Lips DJ, deWindt LJ, van Kraaij DJ, Doevendans PA. Molecular determinants of myocardial hypertrophy and failure: alternative pathways for beneficial and maladaptive hypertrophy. Eur Heart J. 2003;24:883-96
4. Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2014;385:812-24
5. Adalsteinsdottir B, Teekakirikul P, Maron BJ, Burke MA, Gudbjartsson DF, Holm H. et al. Nationwide study on hypertrophic cardiomyopathy in Iceland: evidence of a MYBPC3 founder mutation. Circulation. 2014;130:1158-67
6. van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37-45
7. Zhou LY, Liu JP, Wang K, Gao J, Ding SL, Jiao JQ. et al. Mitochondrial function in cardiac hypertrophy. Int J Cardiol. 2013;167:1118-25
8. Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A. et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136-46
9. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717-21
10. Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517:302-10
11. Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45-59
12. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65-75
13. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-41
14. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741-52
15. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
16. Kaufmann A, Beier V, Franquelim HG, Wollert T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell. 2014;156:469-81
17. Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464-74
18. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y. et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981-91
19. Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci U S A. 2009;106:17049-54
20. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J. et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741-50
21. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK. et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656-67
22. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A. et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685-701
23. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N. et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389-93
24. van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2270-84
25. Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell. 2013;154:1285-99
26. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67-93
27. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84-91
28. Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W. et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290-303
29. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-41
30. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585-95
31. Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA. et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112-5
32. Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S. et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108-11
33. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181-90
34. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749-60
35. Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462-76
36. Chen Y, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471-5
37. Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol. 2010;2010:930509
38. Rzymski T, Milani M, Singleton DC, Harris AL. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle. 2009;8:3838-47
39. Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T. et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193-205
40. Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J, Hill JA. Cardiovascular autophagy: concepts, controversies, and perspectives. Autophagy. 2013;9:1455-66
41. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J. et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215-28
42. He H, Liu X, Lv L, Liang H, Leng B, Zhao D. et al. Calcineurin suppresses AMPK-dependent cytoprotective autophagy in cardiomyocytes under oxidative stress. Cell Death Dis. 2014;5:e997
43. Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220-3
44. Zhai P, Sadoshima J. Glycogen synthase kinase-3beta controls autophagy during myocardial ischemia and reperfusion. Autophagy. 2012;8:138-9
45. Zhang Y, Han X, Hu N, Huff AF, Gao F, Ren J. Akt2 knockout alleviates prolonged caloric restriction-induced change in cardiac contractile function through regulation of autophagy. J Mol Cell Cardiol. 2014;71:81-91
46. Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE. et al. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265
47. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M. et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619-24
48. Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM. et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003-8
49. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB. et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27:1365-77
50. Ferrari AU, Radaelli A, Centola M. Invited review: aging and the cardiovascular system. J Appl Physiol. 2003;95:2591-7
51. Linton PJ, Gurney M, Sengstock D, Mentzer RM Jr, Gottlieb RA. This old heart: Cardiac aging and autophagy. J Mol Cell Cardiol. 2014 [Epub ahead of print]
52. Tong C, Morrison A, Mattison S, Qian S, Bryniarski M, Rankin B. et al. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. FASEB J. 2013;27:4332-42
53. Ceylan-Isik AF, Dong M, Zhang Y, Dong F, Turdi S, Nair S. et al. Cardiomyocyte-specific deletion of endothelin receptor A rescues aging-associated cardiac hypertrophy and contractile dysfunction: role of autophagy. Basic Res Cardiol. 2013;108:335
54. Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V. et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4:ra46
55. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374-9
56. Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114:368-78
57. Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I. et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600-6
58. Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E. et al. GSK-3alpha is a central regulator of age-related pathologies in mice. J Clin Invest. 2013;123:1821-32
59. Song Z, An L, Ye Y, Wu J, Zou Y, He L. et al. Essential role for UVRAG in autophagy and maintenance of cardiac function. Cardiovasc Res. 2014;101:48-56
60. Zaglia T, Milan G, Ruhs A, Franzoso M, Bertaggia E, Pianca N. et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest. 2014;124:2410-24
61. Kubli DA, Quinsay MN, Gustafsson AB. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol. 2013;6:e24511
62. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T. et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308
63. Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH. et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807-12
64. Takagi H, Matsui Y, Hirotani S, Sakoda H, Asano T, Sadoshima J. AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy. 2007;3:405-7
65. Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci U S A. 2002;99:12825-30
66. Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA. et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146-57
67. Dai DF, Rabinovitch P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy. 2011;7:917-8
68. Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275-82
69. Kanamori H, Takemura G, Goto K, Maruyama R, Ono K, Nagao K. et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol. 2011;300:H2261-71
70. Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK. et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915-26
71. McCormick J, Suleman N, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. STAT1 deficiency in the heart protects against myocardial infarction by enhancing autophagy. J Cell Mol Med. 2012;16:386-93
72. Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A. et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478-88
73. Odashima M, Usui S, Takagi H, Hong C, Liu J, Yokota M. et al. Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ Res. 2007;100:1344-52
74. Porrello ER, D'Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG. et al. Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension. 2009;53:1032-40
75. Sun M, Ouzounian M, de Couto G, Chen M, Yan R, Fukuoka M. et al. Cathepsin-L ameliorates cardiac hypertrophy through activation of the autophagy-lysosomal dependent protein processing pathways. J Am Heart Assoc. 2013;2:e000191
76. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA. et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782-93
77. Gao XM, Wong G, Wang B, Kiriazis H, Moore XL, Su YD. et al. Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:1663-70
78. Kuzman JA, O'Connell TD, Gerdes AM. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology. 2007;148:3477-84
79. Flynn JM, O'Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ. et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013;12:851-62
80. Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ. et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell. 2014;13:529-39
81. Li Y, Chen C, Yao F, Su Q, Liu D, Xue R. et al. AMPK inhibits cardiac hypertrophy by promoting autophagy via mTORC1. Arch Biochem Biophys. 2014;558:79-86
82. Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151-60
83. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM. et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284-97
84. Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y. et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123-8
85. Bo L, Su-Ling D, Fang L, Lu-Yu Z, Tao A, Stefan D. et al. Autophagic program is regulated by miR-325. Cell Death Differ. 2014;21:967-77
86. Su M, Wang J, Wang C, Wang X, Dong W, Qiu W. et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2014 [Epub ahead of print]
87. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S. et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078
88. Li Q, Xie J, Li R, Shi J, Sun J, Gu R. et al. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J Cell Mol Med. 2014;18:919-28
89. Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML. et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570-83
90. Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A. et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013;24:206-14
91. Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST. et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102-6
Author contact
![]() Corresponding authors: Xiao Yang and Jian Wang. Xiao Yang, Ph.D. Institute of Biotechnology, 20 Dongdajie, Beijing 100071, China. Tel/Fax: 86-10-63895937(O); E-mail: yangxac.cn. Jian Wang, Ph.D. Institute of Biotechnology, 20 Dongdajie, Beijing 100071, China. Tel/Fax: 86-10-63895937(O); E-mail: wangjian7773com
Corresponding authors: Xiao Yang and Jian Wang. Xiao Yang, Ph.D. Institute of Biotechnology, 20 Dongdajie, Beijing 100071, China. Tel/Fax: 86-10-63895937(O); E-mail: yangxac.cn. Jian Wang, Ph.D. Institute of Biotechnology, 20 Dongdajie, Beijing 100071, China. Tel/Fax: 86-10-63895937(O); E-mail: wangjian7773com