Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(8):868-878. doi:10.7150/ijbs.12045 This issue Cite
Research Paper
Regulation of β-Adrenergic Receptor Trafficking and Lung Microvascular Endothelial Cell Permeability by Rab5 GTPase
Junjun Yang*, 1, Huan Sun*, 1, #, Jihang Zhang2, Mingdong Hu1, Jianchun Wang1 ![]() , Guangyu Wu3, Guansong Wang1
, Guangyu Wu3, Guansong Wang1 ![]()
1. Institute of Respiratory Diseases, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, China
2. Department of Cardiology, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, China
3. Department of Pharmacology and Toxicology, Medical College of Georgia, Georgia Regents University, Augusta 30912, USA
* These authors contributed equally to this work.
# Current address: Intensive Care Unit, Mianyang Central Hospital, Mianyang 621000, China
Received 2015-3-4; Accepted 2015-5-5; Published 2015-6-1
Abstract

Rab5 GTPase modulates the trafficking of the cell surface receptors, including G protein-coupled β-adrenergic receptors (β-ARs). Here, we have determined the role of Rab5 in regulating the internalization of β-ARs in lung microvascular endothelial cells (LMECs) and in maintaining the integrity and permeability of endothelial cell barrier. Our data demonstrate that lipopolysaccharide (LPS) treatment disrupts LMEC barrier function and reduces the cell surface expression of β-ARs. Furthermore, the activation of β-ARs, particularly β2-AR, is able to protect the LMEC permeability from LPS injury. Moreover, siRNA-mediated knockdown of Rab5 inhibits both the basal and agonist-provoked internalization of β-ARs, therefore, enhancing the cell surface expression of the receptors and receptor-mediated ERK1/2 activation. Importantly, knockdown of Rab5 not only inhibits the LPS-induced effects on β-ARs but also protects the LMEC monolayer permeability. All together, these data provide strong evidence indicating a crucial role of Rab5-mediated internalization of β-ARs in functional regulation of LMECs.
Keywords: lung, microvascular endothelial cell, Rab5, β-adrenergic receptor, internalization, trafficking, permeability, lipopolysaccharide, small interfering RNA
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are within the same disease spectrum characterized by acute lung inflammation together with increased vascular permeability and hypoxemic respiratory insufficiency resulted from pulmonary edema. Patients suffering from ALI and ARDS exhibit low ratio of arterial oxygen partial pressure to fractional inspired oxygen (PaO2/FiO2), usually less than 300 and 200 mmHg, respectively. The mortality of ARDS at one year is substantially higher (41%) [1, 2]. Although both ALI and ARDS have been extensively investigated over the past years, their therapeutic improvement remains slow and insufficient. Currently, the only approved ALI/ARDS therapy involves low volume mechanical ventilation [2]. However, the mortality is unacceptably high.
It has been well defined that lung microvascular endothelial cells (LMECs) play an important role in both ALI and ARDS. The enhanced permeability of LMECs, which occurs due to ALI/ARDS, can be induced by several soluble factors such as bacterial lipopolysaccharide (LPS) and endogenous pro-inflammatory cytokines [3]. The LMEC injury and barrier dysfunction result in the protein-rich edema fluid leakage and the circulation of neutrophils into the pulmonary interstitium and alveolar space [3]. Therefore, pulmonary alveolar-capillary membrane injury and barrier dysfunction are the key pathophysiological characters of ALI/ARDS [1, 2].
β-Adrenergic receptors (β-ARs) are prototypic G protein-coupled receptors and they are known to be involved in maintaining the barrier integrity and function of vascular endothelial cells (ECs) and LMECs [4]. There are three β-AR subtypes, namely β1-AR, β2-AR and β3-AR. β2-AR is broadly expressed while β1-AR is primarily expressed in the heart and the brain and β3-AR in the adipose tissue [5, 6]. Furthermore, β-ARs are highly expressed in vascular ECs [7]. A previous study using human MECs has shown that cAMP production in response to stimulation of a non-selective β-AR agonist isoproterenol can be antagonized by the β-AR blocker propranolol [8]. Another study has shown that norepinephrine-induced IL-6 release in human dermal MECs is primarily mediated through β2-AR which contributes to the immune and inflammatory responses [9]. Our previous studies suggest that β2-AR may play a role in the LPS-induced cell hyperpermeability [10].
Rab5 is a Ras-like small GTPase that localizes to the early endosomes and orchestrates the initial steps of endocytotic trafficking of cell surface receptors [11, 12]. In particular, Rab5 has been shown to modulate the internalization of G protein-coupled receptors, including β-ARs, from the plasma membrane to endosomes [13-15]. Interestingly, globotriaosylceramide treatment increased Rab5 expression in the mouse aortic ECs and human umbilical vein ECs and reducing Rab5 expression could enhance KCa3.1 expression and endothelium-dependent relaxation [16]. However, little is known regarding the function of Rab5 in LMECs. In this study, we have investigated whether trafficking of endogenous β-ARs is regulated by Rab5 GTPase in primary LMECs. We have also determined the role of Rab5 and β-ARs in maintaining the integrity and permeability of LMECs. Our results have demonstrated that Rab5-regulated β-AR trafficking likely plays a crucial role in controlling the endothelial permeability of LMECs.
Materials and methods
Reagents and materials
[3H]-CGP12177 (specific activity 30-60 Ci/mmol, PerkinElmer, USA). Isoproterenol (ISO), atenolol, alprenolol, ICI 118,551 and lipopolysaccharide (LPS, from E. coli0111:B4) (Sigma, St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM) and trypsin (HyClone, Thermo Scientific, Beijing, China), and fetal bovine serum (FBS, Gibco, Invitrogen, Carlsbad, USA). Dynabeads® Sheep anti-Mouse IgG (Invitrogen, Carlsbad, USA). Antibodies against Rab5a (Abcam Hong Kong Ltd, China). Antibodies against phospho-ERK1/2, total ERK1/2 and CD31 (Santa Cruz Biotechnology, Inc., CA). Biotin conjugated BSA (biotin-BSA, Thermo Scientific Pierce Biotechnology, IL, USA).
Cell culture and LPS challenge
According to the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and the procedure by the Ethical Committee of the Third Military Medical University, primary cultures of rat LMECs were performed using previously described [10, 17]. Primary cultures were isolated from the lung microvessels of male Sprague-Dawley rats (150-200 g) with an explant technique. Rat lungs were chopped into 1.5 mm2 pieces for a 25-cm2 flask. Then the pieces were cultured in medium by supplement with 20% FBS, heparin (90 µg/ml), and L-glutamine (4 mM) at 37 °C in an incubator containing 5% CO2 and 95% air. Following the tissue was removed at 60 h, and the remaining adherent cells were kept in medium containing 20% FBS for another 72 h. The cultures were digested with a solution containing 0.25% trypsin and 0.02% EDTA, and bound to the Dynabeads coated by anti-rat CD31 antibodies (Dynabeads® Sheep anti-Mouse IgG) at 4 °C (on ice) for 30 min. The Dynabeads were washed, and the cells were cultured in medium containing 10% FBS and 2% endothelial cell growth supplement (ECGS). Then, the cells were confirmed to be LMECs based on their morphology and the expression of a proprietary marker (CD31, FITC-labeled Bandeiraea simplicifolia isolectin B4, and factor VIII). The cells used in the experiments were between passages 4 and 10. The LMECs were grown as a monolayer, serum-starved (0.5% serum) for 6 h and then exposed to LPS at the indicated concentration for the selected period.
Confocal microscopy
Confocal microscopy was performed in a Leica DMRA2 epifluorescence microscope as described previously [10, 18, 19] . After transfection, LMECs were fixed with 4% paraformaldehyde and washed in phosphate-buffered saline (pH = 7.4). The nuclei were stained with diamidino-phenyl-indole (DAPI).
Western blotting
The protein concentrations of the cell extracts were measured using a NanoDrop 2000/2000c Spectrophotometer (Thermo Scientific, USA). Western blotting and densitometric analysis were performed using QuantiScan software [10].
siRNA-mediated depletion of Rab5
siRNA targeting rat Rab5a (CGCCAUAGUUGUGUAUGAUTT and AUCAUACACAACUAUGGCGTT) and a control non-silencing siRNA were purchased from Invitrogen (Valencia, CA, USA). The LMECs were cultured on 25-cm2 flasks at a density of 2×105 cells/ml for 24 h prior to transfection. siRNA were delivered into the LMECs using X-tremeGENE siRNA Transfection Reagent according to the manufacturer's instructions. Briefly, X-tremeGENE siRNA Transfection Reagent (20 μl) and the siRNA (10 μg) were diluted in 200 μl of Opti-MEM medium in separate tubes. These tubes were combined within 5 min and incubated for additional 20 min. Finally, the transfection mixture was added into the culture dishes.
Plasmid transfection
The LMECs were cultured on 24-well dishes at a density of 4×104 cells/well and transfection was performed when the cells reached 80% confluence. The LEMCs were co-transfected with the GFP-tagged β2-AR, GFP-Rab5a-WT or siRNA using X-tremeGENE HP DNA Transfection Reagent according to the manufacturer's instructions. Briefly, X-tremeGENE HP DNA Transfection Reagent (2.5 μl), GFP-tagged β2-AR (0.5 μg) and GFP-Rab5a-WT (0.5 μg), or siRNA (0.25 μg) were diluted in 50 μl of Opti-MEM medium in separate tubes. After the mixtures were combined and incubated for 30 min; then added to the culture dishes. After 48 h, the cells were processed for fluorescence microscopy as described above.
Measurement of the cell surface β-AR expression
The cell surface expression of β-ARs was determined by ligand binding of intact live cells as described previously [10, 20, 21]. Briefly, the LMECs were cultured on 12-well plates and transfected for 48 h. The cells were washed once with binding buffer and incubated for 2 h at room temperature in 6 nM [3H]-CGP12177. The levels of each β-AR subtype were measured by pre-incubating the cells with atenolol, a β1-AR-selective antagonist, or ICI 118,551, a β2-AR-selective antagonist, at a concentration of 1 µM for 30 min. The cells were washed three times with ice-cold phosphate-buffered saline (pH=7.4) and lysed in 500 µl of 1 M NaOH. Then, the radioactivity of the samples was measured by liquid scintillation spectrometry. Nonspecific binding was defined in the presence of alprenolol (10 μM).
To measure the internalization of β-ARs, the cells were cultured in 12-well plates and treated with ISO at a concentration of 1 µM at 37 °C for the indicated period. The cells were washed twice with cold phosphate-buffered saline (pH=7.4) and the cell surface receptor levels were determined by intact cell ligand binding assays as described above.
Measurement of ERK1/2 activation
The activation of ERK1/2 was measured as described previously [10, 18]. The LMECs were cultured and transfected with Rab5a siRNA and Rab5a plasmids for 48 h. The cells were stimulated with ISO (1 µM) for 15 min with or without pretreatment with ICI 118,551 or atenolol (100 nM) for 30 min. The stimulation was terminated by adding 1× SDS gel-loading buffer. Immunoblotting was used to determine the activation of ERK1/2 by measuring the levels of ERK1/2 phosphorylation with phospho-ERK1/2 antibodies.
Monitoring endothelial barrier function
The cell monolayer barrier function was determined using the iCELLigence System (Roche Applied Science, Germany). This is a highly sensitive method for obtaining real-time variations in electrical impedance, representing the global cellular status such as barrier function. The system includes an analyzer, a 16-well E-plate and a device station [22]. The assays were performed according to the manufacturer's instructions and the cell index (CI) and the normalized CI were defined as described previously [22]. Briefly, the plates were filled with 200 µl of DMEM containing 10% FBS and connected to the iCELLigence System to produce a time zero control for each plate and well. After the cells were seeded on the plates for 30 min, the plates were returned to the iCELLigence system and the CI was monitored using the installed software. The cells were allowed to grow to reach plateau (typically 3 days) and then exposed to various conditions as described in each experiment. The CI was measured every 15 min by the installed software for the duration of the experiments.
Measurement of the endothelial cell monolayer permeability
Permeability assays were determined based on the pervasion of biotinylated bovine serum albumin (biotin-BSA) into the lower chamber as described previously [10]. Briefly, the cells were seeded on 12-well dishes (Costar, Cambridge, MA) lined with polycarbonate filters (pore size 0.4 µM) and transfected with control or Rab5a siRNA for 2 days. The cells were treated with ISO (1 µM), ISO plus ICI 118,551 (1 µM) or ISO plus atenolol (1 µM) for 30 min before LPS treatment (10 µg/ml) for 6 h. The upper chamber wells were incubated with 500 µg/ml biotin-BSA and 100 µl of the lower chamber media aspirated after 30 min. The biotin-BSA concentration in the media was measured by enzyme-linked immunosorbent assays.
Statistical analysis
Significant differences were calculated via one-way analysis of variance (ANOVA) and a value of P < 0.05 was considered to be statistically significant. The data are expressed as the means ± S.E.
Results
LPS injury decreases the cell index and the cell surface expression of β-ARs in LMECs
Rat LMECs initially grew as capillary-like structures and assumed the typical EC-like cobblestone morphology at confluence (Supplementary Fig. 1). Positive expression of factor VIII-related antigen (Supplementary Fig. 1A) and CD31 antigen (Supplementary Fig. 1B and 1C) in the cells confirmed their endothelial origin. Furthermore, these cells were able to bind to FITC-labeled BSI-B4 and displayed positive staining with lectin BSI-B4 (Supplementary Fig. 1A), which are principal characteristics of microvascular ECs.
We then determined the effect of LPS treatment on the barrier permeability of LMECs by measuring the CI. Lung EC barrier dysfunction is characterized by increased permeation of fluid and macromolecules into interstitial and alveolar tissues, and is associated with a decreased CI [23]. To determine whether LPS treatment affects the EC barrier function, we challenged confluent cell monolayers with different doses of LPS (0, 5, 10, 20, or 50 µg/ml) for 9 h. We found that the CI decreased between 0 h and 9 h in a concentration-dependent manner. At 5 µg/ml, LPS did not significantly affect the normalized CI after 4 h (CI = 0.9656; P>0.05 compared to baseline). However, by 6 and 8 h, 5 µg/ml LPS significantly reduced the CI (0.8945 and 0.8794, respectively; P<0.05 compared to baseline). At 10 µg/ml, LPS significantly decreased the normalized CI at 6 and 8 h (0.7567 and 0.7407, respectively; P<0.05 compared to either baseline) (Fig. 1A). Similar results were obtained when 20 µg/ml of LPS were used. The normalized CI markedly decreased when the cells were treated with 50 µg/ml LPS for 9 h as compared with control group (P<0.05, Fig. 1A). The normalized CI values in all LPS treatment groups were significantly decreased compared to the control group. These data demonstrated that the response to LPS treatment developed gradually and peaked after 6 h administration.
Effects of LPS on the cell permeability and the cell surface expression of β-ARs in LMECs. (A) LPS treatment decreased the CI of LMECs in a dose- and time-dependent manner. The cells were cultured and the CI was measured in the iCELLigence System as described in “Materials and methods”. The images shown are representative CI traces after treatment with LPS at concentrations of 0 (black), 5 μg/ml (blue), 10 μg/ml (red), 20 μg/ml (green) and 50 μg/ml (pink), and were taken from one of four independent measurements. (B) Quantification of total cell surface β-ARs after LPS treatment. The mean values of specific [3H]-CGP12177 binding were 1859 ± 221, 1605 ± 176, 1467 ± 158, 1015 ± 146, and 997 ± 128 cpm at 0, 2, 4, 6, and 8 h groups, respectively. The data are expressed as the means ± S. E. (n=4). *P< 0.05 versus the 0 h group and ^P< 0.05 versus the 4 h group.
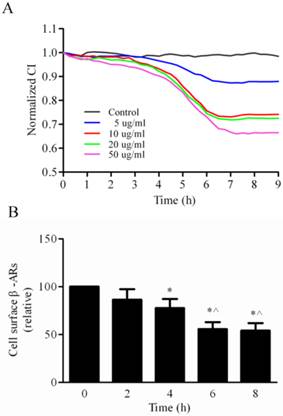
We next measured the effect of LPS treatment on the cell surface expression of total β-ARs in the LMECs. The cells were treated with 10 µg/ml LPS and the cell surface β-ARs measured by [3H]-CGP12177 ligand binding assays. The levels of β-ARs were significantly reduced by 22.4% after LPS treatment for 4 h (P<0.05, Fig. 1B). The receptor levels were obviously lower after 6 or 8 h treatment than 4 h treatment (P<0.05, Fig. 1B). These data demonstrate that LPS stimulation reduces the expression of endogenous β-ARs at the cell surface in LMECs.
β-AR activation suppresses LPS-induced hyperpermeability in LMECs
We first determined the effect of β-AR activation on the barrier function of LMECs under the normal condition. The activation of β-ARs by ISO (1 µM) significantly increased the normalized CI of LMECs (Fig. 2A and 2B), whereas the treatment of ICI118,551, a β2-AR-selective antagonist, produced an opposite effect. In contrast, the treatment with atenolol, a β1-AR antagonist, did not clearly influence the CI of LMECs. Consistently, the pretreatment with ICI118,551, but not atenolol, blocked the effect of ISO-mediated activation of β-ARs on the CI (Fig. 2A, 2B). These data suggest that the activation of β2-AR, but not β1-AR, is an important event in regulating the permeability of LMECs.
β-AR activation attenuates LPS-induced disruption of the cell permeability. (A) Effect of β-AR activation on the normalized CI values in LMECs. The LMECs were cultured and the barrier function was measured in real-time with the iCELLigence System. ISO was used to stimulate the cells with or without pretreatment of atenolol or ICI118,551 for 0.5 h. The data are expressed as the means ± S.E. of three separate experiments. (B) Quantitative data of the normalized CI after ISO stimulation at 1 h and 6.5 h. The data are expressed as the means ± S.E. (n=3). * p < 0.05 versus control group and ^ p < 0.05 versus ISO group. (C) β-AR activation suppressed LPS-induced CI decrease. The cells were treated with atenolol or ICI118,551 for 1 h, ISO, atenolol plus ISO or ICI118,551 plus ISO for 0.5 h, and then stimulated with LPS (10 μg/ml). The data are expressed as the means ± S.E. of three separate experiments. (D) Quantitative CI values after treatment with 10 μg/ml LPS for 6 h (n=4). *P< 0.05 versus control group and ^P< 0.05 versus LPS group.
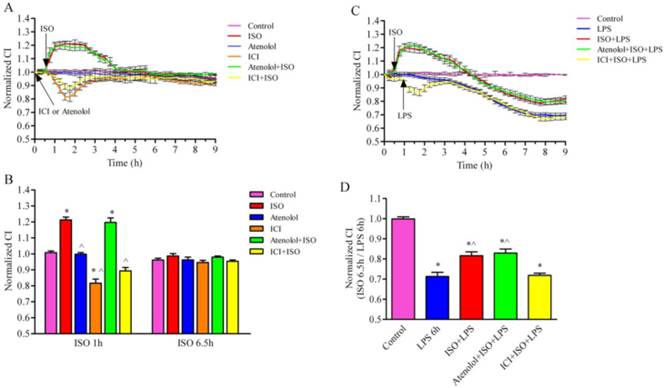
We then determined the effect of β-AR activation on the LPS-induced hyperpermeability. LPS treatment (10 µg/ml) decreases the CI (normalized CI = 0.713) which was inhibited by pre-treatment with ISO alone (CI = 0.816) or ISO plus atenolol (CI = 0.829) at 7 h (Fig. 2C and 2D). In contrast, the pretreatment with 1 µM ISO plus ICI118,551 did not produce significant effects compared with the LPS alone group (Fig. 2C and 2D). These data indicate that the activation of β2-AR is able to protect the cell barrier function induced by LPS.
Rab5 regulates β2-AR trafficking
Our previous studies have demonstrated that Rab1 regulates the export of endogenous β2-AR from the ER to the cell surface and Rab1 significantly increases the cell surface expression of total β-ARs and β2-AR [10]. Here, we studied the role of Rab5 in β2-AR trafficking in LMECs. The expression levels of Rab5 were manipulated by the siRNA-mediated depletion and overexpression strategies. Cy3-tagged Rab5a siRNA (Fig. 3A) and GFP-tagged Rab5a (Fig. 3B) were distributed throughout the transfected cells with the transfection efficiencies of 95% and 83%, respectively. Western blot analysis revealed that Rab5a expression was significantly decreased by 75% after Rab5a siRNA transfection for 48 h (Supplementary Fig. 2). These data confirmed the successful manipulation of the Rab5 levels in the primary LMEC cultures.
Effect of Rab5a on the subcellular localization of β2-AR in LMECs. (A) Transfection efficiency of Rab5a siRNA. The LMECs were cultured on coverslips and transfected with Cy3-tagged Rab5a siRNA for 12 h using X-tremeGENE siRNA Transfection Reagent. Rab5a siRNA was detected by fluorescence microscopy. Blue, DNA staining with DAPI (nuclear); red, Cy3. Scale bar, 50 μm. (B) Transfection efficiency of Rab5a WT-GFP. The LMECs were transfected with GFP-tagged Rab5a WT for 48 h using X-tremeGENE HP DNA Transfection Reagent. DNA staining with DAPI (nuclear, Blue); GFP (Green). Scale bar, 25 μm. (C) Rab5a knockdown modified the subcellular localization of β2-AR. The LMECs were transiently transfected with GFP-tagged β2-AR alone or together with Rab5a siRNA or Rab5a WT for 48 h. The cells were treated with ISO (1 μM) for 1 h and the subcellular distribution of β2-AR was revealed by confocal microscopy. The data are representative images of three separate experiments. Green, GFP-tagged receptor; blue, DNA stained with DAPI (nuclei). Scale bars, 10 μm.
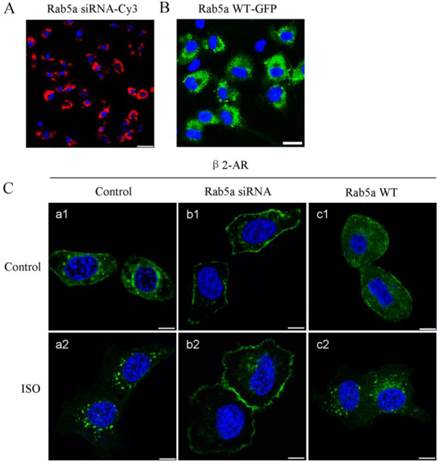
In the absence of agonists, as expected, β2-AR-GFP was clearly localized to the cell surface in LMECs. Interestingly, it is also apparent that β2-AR-GFP was expressed inside the cell (Fig. 3C, a1). In the Rab5a knockdown cells, β2-AR-GFP was primarily localized to the cell surface and its intracellular expression was decreased (b1), whereas overexpression of Rab5a did not affect β2-AR subcellular distribution under the baseline conditions (c1). These data suggest that, although β2-AR is mainly expressed at the cell surface, it may undergo constitutive internalization in the LMECs which can be blocked by depleting Rab5.
Next, we examined the effect of ISO stimulation on β2-AR trafficking. Upon ISO stimulation, β2-AR-GFP primarily localized inside the cells and the expression of β2-AR at the cell surface was clearly reduced (a2). These data suggest that ISO stimulation promotes β2-AR internalization from the plasma membrane to endosomes in the LEMCs. Similar results were observed in the Rab5a-overexpressing cells (c2). In the Rab5a siRNA-transfected cells, β2-AR remained at the cell surface and was not affected by ISO treatment (b2), indicating that Rab5 knockdown blocks β2-AR internalization. Altogether, these data suggest that Rab5 regulates β2-AR trafficking in the presence and absence of receptor agonists.
Rab5 modulates the internalization of β-ARs
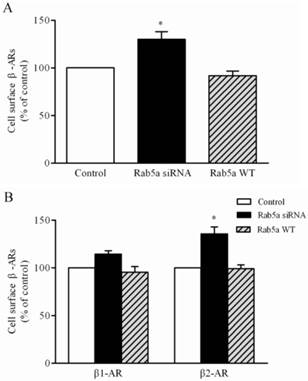
To further define the role of Rab5 in regulating the trafficking of β-ARs, we quantitatively measured the cell surface expression of the total β-ARs, β1-AR and β2-AR by intact cell ligand binding assays. Total β-AR expression was increased by 29.4% in the LMECs after Rab5a siRNA transfection (Fig. 4A). A pronounced effect of Rab5 knockdown on β2-AR expression was observed by 35.6% in the LMECs. These increases were not due to the effects of Rab5a knockdown on the total LMEC numbers (Supplementary Fig. 3B). These data indicate that Rab5a modulates the cell surface expression of endogenous β-ARs, especially β2-AR, in the LMECs. In contrast, the expression of Rab5a WT did not alter the total and the cell surface expression of β-ARs (Fig. 4B); suggesting that the expression level of endogenous Rab5a is not a rate-limiting factor for the trafficking of β-ARs.
Effect of Rab5a on the cell surface expression of β-ARs. (A) Quantitative analysis of total β-ARs on the cell surface of LMECs after Rab5a siRNA and Rab5a WT transfection. The LMECs were transfected with control siRNA, Rab5a siRNA or Rab5a WT for 48 h. The expression of β-ARs at the cell membrane was determined by intact cell binding to the [3H]-CGP12177 ligand. The mean values for specific binding were 2034 ± 132, 2632 ± 135, and 1867 ± 168 cpm in control, Rab5a siRNA, and Rab5a WT group, respectively (n = 3, each in triplicate). (B) Quantitative analysis of the cell surface β1-AR and β2-AR in the LMECs. The mean values for specific [3H]-CGP12177 binding to β1-AR were 572 ± 582, 656 ± 100 and 548 ± 112 cpm and the binding values for β2-AR were 1456 ± 49, 1978 ± 252, and 1376 ± 174 cpm in control, Rab5a siRNA and Rab5a WT group, respectively (n = 3, each in triplicate). The data are expressed as the means ± S.E. (n=3). *p < 0.05 versus control group.
We next measured the effect of Rab5 on ISO-induced internalization of β-ARs (Fig. 5A). In the control group, the percentages of remaining cell surface β-ARs after ISO stimulation for 2 h, 6 h and 8 h were 83.9 ± 5.1%, 72.5 ± 5.8%, and 68.3 ± 4.3%, respectively. Rab5a overexpression did not significantly affect the surface levels of β-ARs at any time points examined as compared with control group. There was a higher percentage of remaining receptors in the Rab5a knockdown cells at all time points except for the 2 h time point, compared to the control group or the Rab5a WT group. These data demonstrate that agonist-induced β-AR internalization is suppressed by Rab5a knockdown in the LMECs.
Rab5 modulates β-AR internalization in LMECs. (A) Effect of Rab5a on the internalization of β-ARs after ISO stimulation. The cells were grown to 80% confluence and transfected with Rab5a WT or Rab5a siRNA. The cells were incubated with ISO (1 μM) at 37 °C for the indicated time periods. The levels of surface β-ARs were determined by intact cell ligand binding. The data are the means ± S.E. and represent three independent experiments. *p< 0.05 versus the time 0 in each group and ^p< 0.05 versus the same time point in the control group. (B) Effect of Rab5a on the cell surface expression of β-ARs after LPS treatment. The cells were transfected with Rab5a WT or Rab5a siRNA for 48 h and then subjected to LPS for 6 h. The mean values for specific [3H]-CGP12177 binding were 1542 ± 98, 887 ± 125, 2005± 98, 1470 ± 195, 1504 ± 222, 964 ± 43 cpm in control, LPS alone, Rab5a siRNA alone, Rab5a siRNA plus LPS, Rab5a alone, and Rab5a plus LPS groups, respectively (n = 3, each in triplicate). The data shown are the percentages of the mean values obtained from the LMECs transfected with the control plasmid and are presented as the means ± S.E. (n = 3, each in triplicate). *P< 0.05 versus control group and ^P <0.05 versus LPS group.
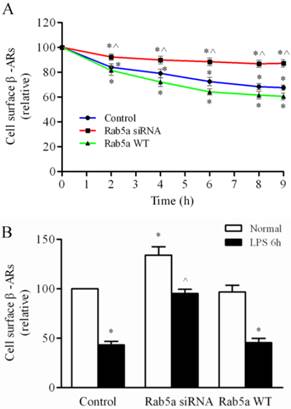
Then, we examined the effect of Rab5a on β-AR expression in the LPS-injured LMECs. The β-AR expression level was decreased to 43% by LPS treatment for 6 h and the level of β-AR expression was increased by 52% in cells transfected with Rab5a siRNA (Fig. 5B). There was no significant difference between the Rab5a WT and control groups (Fig. 5B). These data suggest that the knockdown of Rab5a reduces the effect of LPS injury on the expression of β-ARs in LMECs.
Regulation of β-AR-mediated ERK1/2 activation by Rab5 in LMECs
To determine whether or not Rab5a can regulate β-AR signaling through modifying receptor trafficking, we determined the effect of Rab5a on the β-AR-mediated activation of ERK1/2. As shown in Fig. 6A, the expression of Rab5a siRNA significantly enhanced ERK1/2 activation in response to stimulation with ISO in the LMECs, compared with control cells. However, Rab5a WT did not have obvious effects. The pretreatment with ICI118,551, but not atenolol, blocked the ISO-mediated activation of ERK1/2 (Fig. 6B-6D). These results demonstrate that Rab5a regulates not only β-AR trafficking but also receptor-mediated ERK1/2 activation.
Effect of Rab5a and LPS on the activation of ERK1/2 in LMECs. The cells were transfected with Rab5a siRNA, or Rab5a WT for 48 h and the activation of ERK1/2 was determined by Western blot analysis. Representative blots of pERK1/2 (upper panel), total ERK1/2 (middle panel) and GAPDH (bottom panel) are shown. (A) The LMECs were stimulated with ISO (1 μM) alone at 37 °C. (B) The cells were treated with ISO plus ICI 118,551 (1 μM). (C) The cells were treated with ISO plus atenolol (1 μM). (D) Quantitative data of ERK1/2 activation normalized to GAPDH expression. The data are expressed as the means ± S.E. (n=3). * p < 0.05 versus control group. (E) Effect of LPS on ERK1/2 activation. The LMECs were stimulated with LPS, ISO, LPS and then ISO, or ISO and then LPS. Bottom panel shows quantitative data of ERK1/2 activation normalized to GAPDH expression. The data are expressed as the means ± S.E. (n=3). * p < 0.05 versus control group and ^ p < 0.05 versus LPS alone group.
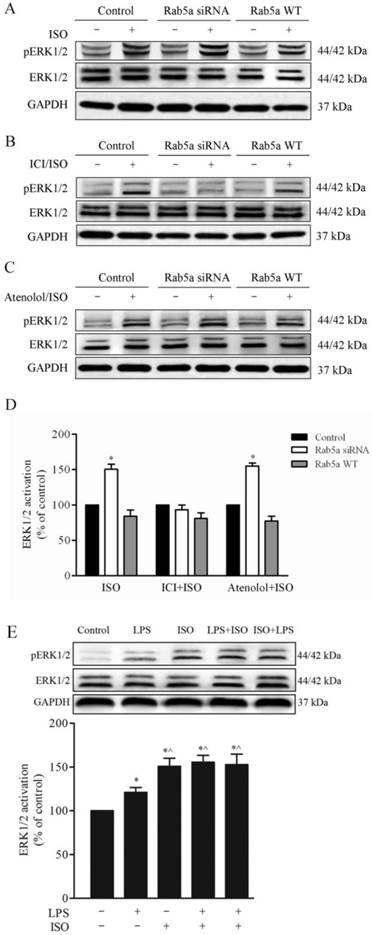
We next determined the effect of LPS on the β-AR-mediated activation of ERK1/2. As shown in Fig. 6E, the activation of ERK1/2 was modestly but significantly increased after LPS treatment compared with control group. Interestingly, LPS treatment did not influence ERK1/2 activation in cells stimulated with ISO.
Rab5 regulation of the LMEC permeability in response to LPS stimulation
Our previous data demonstrate that Rab5a knockdown inhibits agonist-induced β-AR internalization and the activation of β-AR protects the endothelial barrier from LPS injury. Based on these findings, we determined the effect of Rab5a on LPS-induced endothelial barrier dysfunction by monitoring the normalized CI using the iCELLigence System. The LPS-induced change of the normalized CI in the LPS alone group was similar to the LPS combined treatment with ISO plus ICI 118,551 pretreatment group after transfection with Rab5a siRNA and the control (NC) siRNA at 7 h. Under normal condition, the whole CI in the cells after transfection with Rab5a siRNA was similar to that after transfection with NC siRNA (Fig. 7A and Supplementary Fig. 3A). After 6 h of LPS treatment, the normalized CI was not significantly different in the Rab5a siRNA group compared to NC siRNA group. However, there was a significant difference in the normalized CI in the Rab5a siRNA cells pretreated with ISO or ISO plus atenolol compared to the corresponding control cells (P<0.05, Fig. 7B). ISO and ICI 118,551 pretreatment/LPS treatment produced no significant effect. Rab5a knockdown did not affect LMEC proliferation compared to NC siRNA (Supplementary Fig. 3B). Thus, Rab5a knockdown did enhance the protective effect of β2-AR activation.
Rab5a-mediated β-AR trafficking protects the LMEC permeability from LPS injury. (A) The LMEC barrier function (CI) was enhanced by the activation of β-ARs. After transfection with Rab5a siRNA for 48 h, the LMECs were pretreated with vehicle, atenolol or ICI118,551 for 1 h; with ISO, atenolol plus ISO or ICI118,551 plus ISO for 0.5 h, and then stimulated with or without LPS (10 μg/ml) at time 1 h. The responses of the LMECs were monitored with the iCELLigence System. The data are expressed as the means ± S.E. from three individual experiments. (B) Quantitative analysis of the normalized CI values of LMECs after the LPS challenge for 6 h. The data are expressed as the means ± S.E., n = 3. *p< 0.05 versus the LMEC control group; ^p< 0.05 versus the LMECs treated with LPS alone group. (C) Rab5a siRNA inhibited LPS-mediated the monolayer hyperpermeability of LMECs. The LMECs were grown to confluence on 0.4-μm polyester membranes in the upper chambers of coculture wells and transfected with control or Rab5a siRNA for 48 h. The cultures were pretreated with ISO, ISO plus atenolol, ISO plus ICI118,551 or vehicle, and then subjected to LPS (10 μg/ml) for 6 h. Biotin-BSA (500 μg/ml) was added to the upper chamber wells. Aliquots of the lower chamber media were aspirated at 0.5 h after treatment. The biotin-BSA concentrations in the media were determined via an enzyme-linked immunosorbent assay. The data are expressed as the means ± S.E., n = 4. *p< 0.05 versus the LMEC control group; ^p< 0.05 versus the LMECs treated with LPS alone group.
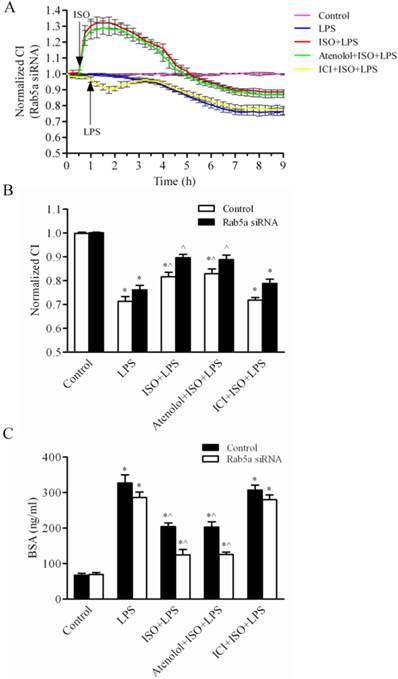
Next, we determined whether or not Rab5a affected the LMEC monolayer permeability. The cells were transfected with control or Rab5a siRNA, pretreated with ISO and then exposed to LPS for 6 h. The BSA diffusion coefficients in the LPS group transfected with control siRNA or Rab5a siRNA were 333.0 ± 30.9 and 286.3 ± 26.2 ng/ml, respectively (Fig. 7C). These values were significantly higher than those in the control group (78.1 ± 8.9 and 69.5 ± 9.6 ng/ml, respectively; P<0.01). The LPS-induced increase in the BSA diffusion coefficient was attenuated to 204.2 ± 17.8 and 202.9 ± 26.3 ng/ml by ISO and ISO plus atenolol pretreatment, respectively. In contrast, Rab5a siRNA attenuated the BSA diffusion coefficient to 124.8 ± 26.4 and 125.9 ± 11.3 ng/ml in cells pretreated with ISO or ISO plus atenolol, respectively (P<0.05). There was no difference between the cells transfected with control siRNA and Rab5a siRNA that were pretreated with ISO and ICI 118,551. These data suggest that, although Rab5a knockdown does not directly influence the LMEC permeability, it significantly decreases the LMEC permeability when combined with ISO pretreatment. These data are consistent with the effect of Rab5 on the trafficking and signaling of β-ARs. Taken together, these data indicate that Rab5a modulates endogenous β-AR trafficking in the LMECs, thereby regulating the barrier function and the monolayer permeability.
Discussion
LPS induces LMEC barrier dysfunction by disrupting β-ARs
Using the iCELLigence system, we have demonstrated that the LMEC barrier function is disrupted by LPS treatment in a dose- and time-dependent manner. Similar results were obtained in previous studies using impedance-based cell analysis - transendothelial electrical resistance (TER) or the [14C] BSA flux for endothelial cells [17, 24]. These data are also consistent with several reports showing that LPS induces endothelial barrier dysfunction both in vitro and in vivo [25-27]. We have also found that the LPS challenge to the LMECs markedly reduces the expression of β-ARs at the cell surface. These data suggest that the trafficking of β-ARs may play an important role in LPS-induced injury.
β-AR activation suppresses LPS-induced barrier disruption
In the LMECs, β-AR activation prevents LPS-induced damage to the cell barrier. Furthermore, β-ARs regulate other types of EC junctions that are directly involved in the endothelial barrier integrity of the brain, cardiovasculature and lung. β-AR stimulation has been shown to transactivate vascular endothelial growth factor receptor-2 (VEGFR2) signaling to further increase hemangioma-derived EC proliferation [28]. Both β-ARs and insulin inhibit retinal EC apoptosis under hyperglycemic conditions [29]. β-AR inhibition increases hydraulic conductivity, reduces the cAMP level and the MEC monolayer TER, and induces VE-cadherin protein fragmentation. β-AR signaling also contributes to endothelial barrier homeostasis under baseline conditions [30]. Here, we have demonstrated that β-AR activation partially alleviates the LPS-induced injury. Furthermore, we have identified that it is β2-AR, but not β1-AR, involved in the regulation of the LMEC permeability. Thus, β2-AR activation may enhance barrier function at least when subjected to the LPS stimulation.
Rab5-mediated β-AR trafficking protects LMEC integrity from inflammatory damage
Rab5 has been well characterized to play a crucial role in regulating the internalization of cell surface receptors, including β-ARs, from the plasma membrane to endosomes [11-15]. Expression of constitutively activated Rab5a induces the generation of large endosomal structures beneath the plasma membrane that accumulate EPCR and FVIIa in CHO-EPCRs and HUVECs [31]. In addition, Rab5 knockdown inhibits early endosomes trafficking and fusion, which significantly blocks VEGF-induced VEGFR2 internalization in HUVECs [32]. Here, we have demonstrated that Rab5 is a crucial mediator in β2-AR internalization in the LMECs. More interestingly, β2-AR likely undergoes internalization in the absence of agonist stimulation and this constitutive trafficking process is mediated through a Rab5-dependent pathway. Therefore, it is most likely that Rab5 is involved in the endocytosis of β2-AR at both the basal level and in response to agonist stimulation. The most importantly, we have shown that Rab5-mediated β-AR trafficking protects the LMEC barrier from LPS-induced damage. It has been shown that β-AR-mediated signaling contributes to endothelial barrier maintenance under baseline conditions [33]. These findings suggest that β-ARs protect the MEC integrity under both the basal and pathological conditions.
There are many signal transduction pathways, such as cAMP production and the MAPK ERK1/2 activation, which can be activated by β-ARs [34-37]. Our results have shown that Rab5a siRNA significantly augmented β-AR-stimulated ERK1/2 activation which is in parallel with the reduction of the receptor at the cell surface in the LMECs. Therefore, Rab5 regulates not only β2-AR trafficking but also receptor-mediated signaling. However, it remains unknown whether or not the effect of Rab5 on β-AR-mediated ERK1/2 activation is directly linked to its function in regulating cell permeability in the LMECs. It is also interesting to note that LPS stimulation attenuates the cell surface expression of β-ARs but does not clearly influence ERK1/2 activation in response to ISO stimulation. One possible explanation for this is that LPS stimulation alone modestly activates ERK1/2.
Based on our results presented here, Rab5a and β2-AR are potential therapeutic targets for ALI. Indeed, it has been demonstrated that, in some animal models and clinical studies, the β-AR agonist salbutamol may stimulate epithelial repair - a potential pharmacological first in ARDS [38]. Matrix metalloproteinase-9 (MMP-9) is specifically upregulated by salbutamol treatment in the ARDS patients. MMP-9 may play a beneficial role in reducing pulmonary edema in ARDS by improving alveolar epithelial healing [39]. Although the epithelial injury biomarkers are not decreased, salmeterol reduces pneumonia and some alveolar inflammation [40]. ISO stimulation augments vascular volume expansion and eliminates extra-vascular volume, which may be due to enhanced endothelial barrier function in healthy volunteers [41]. Therefore, normal β-AR trafficking may be more important than β-AR activation. Based on our findings and others, further studies, particularly clinical trials for the action of β-AR trafficking, are necessary.
In conclusion, the present data have demonstrated that Rab5 GTPase regulates β-AR internalization, which in turn modulates the LMEC permeability. In addition, β-AR activation protects the endothelial barrier from LPS-induced injury and Rab5-coordinated homeostasis of β-ARs contributes to maintaining the cell barrier permeability.
Supplementary Material
Supplementary Figures 1-3.
Abbreviations
β-AR: beta-adrenergic receptor; β1-AR: beta1-adrenergic receptor; β2-AR: beta2-adrenergic receptor; LMEC: lung microvascular endothelial cell; LPS: lipopolysaccharide; Rab5a WT: wild-type Rab5a; ALI: acute lung injury; ARDS: acute respiratory distress syndrome; cAMP: cyclic AMP; GFP: green fluorescent protein; biotin-BSA: biotinylated bovine serum albumin; BSI-B4: Bandeiraea simplicifolia isolectin B4; CI: cell index; ISO: isoproterenol; HEK293: human embryonic kidney 293 cell line; VEGF: vascular endothelial growth factor; ERK1/2: extracellular signal-regulated kinase1/2; EC: endothelial cell; TER: transepithelial electrical resistance.
Acknowledgements
This work was supported by grants 81170066, and 8117067 (to G. Wang and J. Wang) from the National Science Foundation of China.
Authors' contributions
Concept and design, GW; acquisition of data, JY, HS, JZ, MH, JW, GW; analysis and interpretation, JY, HS, JW, GW, GW; drafting and editing of the manuscript, JY, HS, GW, GW. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
References
1. Wang CY, Calfee CS, Paul DW. et al. One-year mortality and predictors of death among hospital survivors of acute respiratory distress syndrome. Intensive Care Med. 2014;40:388-96
2. Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147-63
3. Gonzales JN, Gorshkov B, Varn MN. et al. Protective effect of adenosine receptors against lipopolysaccharide-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L497-507
4. Calvert JW, Lefer DJ. Role of beta-adrenergic receptors and nitric oxide signaling in exercise-mediated cardioprotection. Physiology (Bethesda). 2013;28:216-24
5. Roth Flach RJ, Matevossian A, Akie TE. et al. beta3-Adrenergic receptor stimulation induces E-selectin-mediated adipose tissue inflammation. J Biol Chem. 2013;288:2882-92
6. Yu JT, Wang ND, Ma T. et al. Roles of beta-adrenergic receptors in Alzheimer's disease: implications for novel therapeutics. Brain Res Bull. 2011;84:111-7
7. Guimaraes S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53:319-56
8. Namiecinska M, Wiktorowska-Owczarek A, Loboda A. et al. Cyclic AMP generating system in human microvascular endothelium is highly responsive to adrenaline. Pharmacol Rep. 2006;58:884-9
9. Stohl LL, Zang JB, Ding W. et al. Norepinephrine and adenosine-5'-triphosphate synergize in inducing IL-6 production by human dermal microvascular endothelial cells. Cytokine. 2013;64:605-12
10. Li Y, Wang G, Lin K. et al. Rab1 GTPase promotes expression of beta-adrenergic receptors in rat pulmonary microvascular endothelial cells. Int J Biochem Cell Biol. 2010;42:1201-9
11. Olkkonen VM, Stenmark H. Role of Rab GTPases in membrane traffic. Int Rev Cytol. 1997;176:1-85
12. Mizuno-Yamasaki E, Rivera-Molina F, Novick P. GTPase networks in membrane traffic. Annu Rev Biochem. 2012;81:637-59
13. Jiang X, Pan H, Nabhan JF. et al. A novel EST-derived RNAi screen reveals a critical role for farnesyl diphosphate synthase in beta2-adrenergic receptor internalization and down-regulation. FASEB J. 2012;26:1995-2007
14. Seachrist JL, Anborgh PH, Ferguson SS. beta 2-adrenergic receptor internalization, endosomal sorting, and plasma membrane recycling are regulated by rab GTPases. J Biol Chem. 2000;275:27221-8
15. Wang G, Wu G. Small GTPase regulation of GPCR anterograde trafficking. Trends Pharmacol Sci. 2012;33:28-34
16. Choi S, Kim JA, Na HY. et al. Globotriaosylceramide induces lysosomal degradation of endothelial KCa3.1 in fabry disease. Arterioscler Thromb Vasc Biol. 2014;34:81-9
17. Dudek SM, Munoz NM, Desai A. et al. Group V phospholipase A2 mediates barrier disruption of human pulmonary endothelial cells caused by LPS in vitro. Am J Respir Cell Mol Biol. 2011;44:361-8
18. Wu G, Zhao G, He Y. Distinct pathways for the trafficking of angiotensin II and adrenergic receptors from the endoplasmic reticulum to the cell surface: Rab1-independent transport of a G protein-coupled receptor. J Biol Chem. 2003;278:47062-9
19. Li C, Fan Y, Lan TH. et al. Rab26 Modulates the Cell Surface Transport of alpha2-Adrenergic Receptors from the Golgi. J Biol Chem. 2012;287:42784-94
20. Duvernay MT, Dong C, Zhang X. et al. Anterograde trafficking of G protein-coupled receptors: function of the C-terminal F(X)6LL motif in export from the endoplasmic reticulum. Mol Pharmacol. 2009;75:751-61
21. Duvernay MT, Dong C, Zhang X. et al. A single conserved leucine residue on the first intracellular loop regulates ER export of G protein-coupled receptors. Traffic. 2009;10:552-66
22. Pan T, Huang B, Zhang W. et al. Cytotoxicity assessment based on the AUC50 using multi-concentration time-dependent cellular response curves. Anal Chim Acta. 2013;764:44-52
23. Slanina H, Konig A, Claus H. et al. Real-time impedance analysis of host cell response to meningococcal infection. J Microbiol Methods. 2011;84:101-8
24. Gong P, Angelini DJ, Yang S. et al. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J Biol Chem. 2008;283:13437-49
25. Sun C, Beard RS Jr, McLean DL. et al. ADAM15 deficiency attenuates pulmonary hyperpermeability and acute lung injury in lipopolysaccharide-treated mice. Am J Physiol Lung Cell Mol Physiol. 2013;304:L135-42
26. Rafikov R, Dimitropoulou C, Aggarwal S. et al. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J Biol Chem. 2014;289:4710-22
27. Grinnell KL, Chichger H, Braza J. et al. Protection against LPS-induced pulmonary edema through the attenuation of protein tyrosine phosphatase-1B oxidation. Am J Respir Cell Mol Biol. 2012;46:623-32
28. Ji Y, Chen S, Li K. et al. The role of beta-adrenergic receptor signaling in the proliferation of hemangioma-derived endothelial cells. Cell Div. 2013;8:1
29. Panjala SR, Steinle JJ. Insulin and beta-adrenergic receptors inhibit retinal endothelial cell apoptosis through independent pathways. Neurochem Res. 2011;36:604-12
30. Unwalla HJ, Horvath G, Roth FD. et al. Albuterol modulates its own transepithelial flux via changes in paracellular permeability. Am J Respir Cell Mol Biol. 2012;46:551-8
31. Nayak RC, Keshava S, Esmon CT. et al. Rab GTPases regulate endothelial cell protein C receptor-mediated endocytosis and trafficking of factor VIIa. PLoS One. 2013;8:e59304
32. Park SY, Shi X, Pang J. et al. Thioredoxin-interacting protein mediates sustained VEGFR2 signaling in endothelial cells required for angiogenesis. Arterioscler Thromb Vasc Biol. 2013;33:737-43
33. Spindler V, Waschke J. Beta-adrenergic stimulation contributes to maintenance of endothelial barrier functions under baseline conditions. Microcirculation. 2011;18:118-27
34. Iaccarino G, Ciccarelli M, Sorriento D. et al. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res. 2005;97:1182-9
35. Evans BA, Hutchinson DS, Summers RJ. beta2-Adrenoceptor-mediated regulation of glucose uptake in skeletal muscle--ligand-directed signalling or a reflection of system complexity? Naunyn Schmiedebergs Arch Pharmacol. 2013;386:757-60
36. Dong C, Yang L, Zhang X. et al. Rab8 interacts with distinct motifs in {alpha}2B- and {beta}2-adrenergic receptors and differentially modulates their transport. J Biol Chem. 2010;285:20369-80
37. Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639-50
38. Sandri C, Caccavari F, Valdembri D. et al. The R-Ras/RIN2/Rab5 complex controls endothelial cell adhesion and morphogenesis via active integrin endocytosis and Rac signaling. Cell Res. 2012;22:1479-501
39. O'Kane CM, McKeown SW, Perkins GD. et al. Salbutamol up-regulates matrix metalloproteinase-9 in the alveolar space in the acute respiratory distress syndrome. Crit Care Med. 2009;37:2242-9
40. Perkins GD, Gates S, Park D. et al. The beta agonist lung injury trial prevention. A randomized controlled trial. Am J Respir Crit Care Med. 2014;189:674-83
41. Asmussen S, Salter M, Prough DS. et al. Isoproternenol increases vascular volume expansion and urinary output after a large crystalloid bolus in healthy volunteers. Shock. 2014;42:407-14
Author contact
![]() Corresponding author: Dr. Guansong Wang, Professor of Medicine, Institute of Respiratory Diseases and Critical Care, Xinqiao Hospital of Third Military Medical University, 183 Xinqiao Street, Chongqing 400037, China, Email: wanggs2003com, wanggsedu.cn; Tel: +8623 6545 9972; Fax: +8623 6521 1653
Corresponding author: Dr. Guansong Wang, Professor of Medicine, Institute of Respiratory Diseases and Critical Care, Xinqiao Hospital of Third Military Medical University, 183 Xinqiao Street, Chongqing 400037, China, Email: wanggs2003com, wanggsedu.cn; Tel: +8623 6545 9972; Fax: +8623 6521 1653