Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(8):923-934. doi:10.7150/ijbs.10896 This issue Cite
Research Paper
Sensitization of Chemo-Resistant Human Chronic Myeloid Leukemia Stem-Like Cells to Hsp90 Inhibitor by SIRT1 Inhibition
Hak-Bong Kim1, Su-Hoon Lee1, Jee-Hyun Um2, Mi-Ju Kim1, Suh-Kyung Hyun1, Eun-Ji Gong1, Won Keun Oh3, Chi-Dug Kang1,* ![]() , Sun-Hee Kim1,*
, Sun-Hee Kim1,* ![]()
1. Department of Biochemistry, Pusan National University School of Medicine, Yangsan 626-870, Korea
2. Korea Mouse Metabolic Phenotyping Center, Lee Gil Ya Cancer and Diabetes Institute, Gachon University, Incheon 406-840, Korea
3. College of Pharmacy, Seoul National University, Seoul 151-818, Korea
* These authors contributed equally to this study as corresponding authors.
Received 2014-10-24; Accepted 2015-5-15; Published 2015-6-11
Abstract

Development of effective therapeutic strategies to eliminate cancer stem-like cells (CSCs), which play a major role in drug resistance and disease recurrence, is critical to improve cancer treatment outcomes. The current investigation was undertaken to examine the effectiveness of the combination treatment of Hsp90 inhibitor and SIRT1 inhibitor in inhibiting the growth of chemo-resistant stem-like cells isolated from human chronic myeloid leukemia K562 cells. Inhibition of SIRT1 by use of SIRT1 siRNA or SIRT1 inhibitors (amurensin G and EX527) effectively potentiated sensitivity of Hsp90 inhibitors (17-AAG and AUY922) in CD44high K562 stem-like cells expressing high levels of CSC-related molecules including Oct4, CD34, β-catenin, c-Myc, mutant p53 (mut p53), BCRP and P-glycoprotein (P-gp) as well as CD44. SIRT1 depletion caused significant down-regulation of heat shock factor 1 (HSF1)/heat shock proteins (Hsps) as well as these CSC-related molecules, which led to the sensitization of CD44high K562 cells to Hsp90 inhibitor by SIRT1 inhibitor. Moreover, 17-AAG-mediated activation of HSF1/Hsps and P-gp-mediated efflux, major causes of Hsp90 inhibitor resistance, was suppressed by SIRT1 inhibitor in K562-CD44high cells. Our data suggest that combined treatment with Hsp90 inhibitor and SIRT1 inhibitor could be an effective therapeutic approach to target CSCs that are resistant to current therapies.
Keywords: Hsp90 inhibitor, K562, CD44, SIRT1 inhibitor, multidrug resistance, cancer stem-like cells
Introduction
Many cancers contain small population of highly tumorigenic and intrinsically drug-resistant cancer stem cells (CSCs). CSCs with elevated expression of the CSC-related molecules such as CD44, Oct4, Nanog, β-catenin and several ATP-binding cassette (ABC) transporters, have the ability to self-renew and high tumorigenic potential, and have been associated with resistance to chemo- and radio-therapy [1, 2]. Therefore, therapeutic strategies that specifically target CSCs are likely to be effective in eradicating tumors and in reducing the risk of relapse and metastasis.
Heat shock protein 90 (Hsp90) has emerged as a major pharmaceutical target in cancer therapy, since Hsp90 is known to bind and stabilize a wide range of proteins with activity in the cell cycle, signal transduction and transcription, many of which are widely implicated in cancers [3]. Indeed, drugs aimed at inhibiting Hsp90 have been shown to possess multiple intracellular targeting capacities and could potentially be active against many types of cancer through degradation of a large number of client proteins [4]. It has been known that Hsp90 is required for the activity of the pluripotency transcription factors such as Oct4, Nanog and c-Myc, which are essential for maintaining stemness, and prevents them from degradation by the ubiquitin proteasome pathway [5, 6], and for stabilization of mutant p53 (mut p53) in cancer cells as well [7], suggesting a fundamental requirement of Hsp90 for CSC phenotype. In addition, Hsp90 has an approximately 100-fold greater affinity for its inhibitors in cancer cells than in normal cells due to multi-chaperone complexes of Hsp90 in an activated, high-affinity conformation, enabling selective targeting of Hsp90 of cancer cells [8, 9]. However, direct inhibition of Hsp90 leads to disruption of complexes of Hsp90 with heat shock factor 1 (HSF1), thereby causing HSF1-mediated induction of cytoprotective heat shock proteins (Hsps) such as Hsp70 and Hsp27, which contribute to resistance to Hsp90 inhibitors [10]. Moreover, HSF1 activation increases expression of multidrug resistance 1 (ABCB1, MDR1)/P-glycoprotein (P-gp) [11], and subsequently P-gp-mediated efflux of Hsp90 inhibitor [10]. Therefore, prevention of the Hsp90 inhibitor-mediated rebound induction of cytoprotective Hsps and P-gp is required to use Hsp90 inhibitor as an anti-cancer drug.
It has been known that several markers associated with CSCs are involved in multidrug resistances. CD44 is a multifunctional transmembrane glycoprotein expressed in many cells and tissues [12]. It has been reported that CD44 is a key regulator of human acute myeloid leukemic stem cells to maintain their stem cell properties [13], and is required for the homing and engraftment of BCR-ABL-expressing leukemic stem cells [14]. ABC transporters, which are important determinants in acquiring drug resistance by exporting drugs out of cells, are often expressed highly in CSCs, greatly reducing effectiveness of many anti-cancer drugs [1, 15]. Among the ABC transporters, ABCG2 (breast cancer resistance protein, BCRP) and ABCB1(MDR1) are preferentially expressed in CSCs [16]. Hyaluronan-CD44 interaction activates Nanog-Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells [17]. The Wnt/β-catenin signaling pathway activation increases MDR1 expression through binding of β-catenin to the MDR1 promoter in chronic myeloid leukemia (CML) [18]. The promoter regions of ABC transporter genes involving MDR1 and ABCG2 are known to contain binding sites for the Oct4, which is also considered a marker of tumor stem cells [19], and it has been demonstrated that Oct4 overexpression enhanced whereas Oct4 knockdown reduced liver cancer cell resistance to chemotherapeutic drugs in vitro and in xenograft tumors [20]. The emerging notion that mutations in p53 play a major role in formation of CSCs is greatly supported by the correlation between tumors expressing mut p53 alleles and their undifferentiated phenotype. Indeed, mut p53 was shown to allow stem-like phenotype in breast and lung cancers [21] and transcriptionally induced the expression of MDR1 gene by stimulating its promoter [22], whereas wild-type p53 repressed the expression of CD44, Nanog and Oct4 [23, 24]. Therefore, many CSC-related molecules are considered to be responsible for acquisition of drug-resistance in CSCs.
In addition, there are some evidences showing that inhibition of histone deacetylase (HDAC) may be useful to inhibit CSCs. Indeed, SIRT1, a class III HDAC, directly bind to and deacetylates c-Myc and depletion/inhibition of SIRT1 reduces c-Myc stability [25]. Chemical inhibitors of HDAC are able to deplete Nanog with concomitant suppression of Oct4 and Sox2 [26]. In addition, inhibition of SIRT1 increases acetylation of mut p53 in p53-mutated human keratinocytes cell line [27], and acetylation of some mut p53 protein prevents its function, possibly through a conformational change [28]. Therefore, we speculated that inhibition of HDAC could augment the effectiveness of Hsp90 inhibitors through inactivation of the CSC-related molecules such as c-Myc, Nanog, Oct4 and mut p53, as well as ABC transporters.
Here, we provide the first line of evidence that combination of Hsp90 inhibitor and SIRT1 inhibitor would be a more effective therapeutic approach to target CML-stem like cells such as CD44high CML K562 cells exhibiting many CSC-related molecules.
Materials and methods
Cell culture and reagents
Human K562 CML cell line was obtained from American Type Culture Collection (Manassas, VA). CD44high K562 cells were established during isolation of imatinib-resistant K562 cells after treatment with increasing concentrations of imatinib, and were stable in complete medium without imatinib. The profile of cell surface CD44 expression was carried out on both cells and gated using mouse anti-human CD44-FITC (BD Biosciences, San Jose, CA, USA).We also used CD44+ HCT-15 cells with high CD44 expression and CD44- HCT-15 cells with low CD44 expression isolated from human colon cancer cell line HCT-15 [29]. Cells were maintained in RPMI medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% (v⁄v) heat-inactivated FBS (Gibco BRL, Life Technologies, Carlsbad, CA, USA), 100 U⁄ml penicillin and 100 mg⁄ml streptomycin (Sigma-Aldrich, St.Louis, MO, USA) in a 5% CO2 humidified incubator at 37°C. 17-AAG and AUY922 were purchased from Enzo Life Sciences Inc. (Farmingdale, New York, USA) and Selleck Chemicals (Houston, TX, USA), respectively. EX527 was purchased from BioVision Inc. (Milpitas, CA, USA). Amurensin G, a natural SIRT1 inhibitor, was supplied Dr. Oh (Seoul National University, Seoul, Korea) as described previously [30].
Cell proliferation assay
Cell proliferation was measured by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Exponentially growing cells (2x104 cells/well) were plated in plated in a 96-well plate and incubated in growth medium containing the indicated concentrations of 17-AAG (or AUY922) and/or amurensin G (or EX527) at 37°C for 96 h. Inhibition of cell proliferation was expressed as percentages of untreated control cell growth. At least two separate experiments were performed in triplicate.
Real-time reverse transcription PCR (RT-PCR)
Total RNA was isolated by using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. The RNA from each sample was transcribed to cDNA TOPscript TM RT -DryMIX(Enzynomics, Daejeon, South Korea). During real time PCR, 6-carboxyl fluorescein was used as the fluorescent reporter dye to detect amplified cDNA. Real time PCR was performed by the SYBR Green real time PCR method using 2X Power SYBR® Green PCR Master Mix (Applied Biosystems, CA, USA) using Rotor-Gene Q system (QIAGEN, Hilden, Germany) to subject the samples to denaturation at 95°C for 15 min followed by cycles of denaturation at 95°C for 20 s, annealing at 60°C for 30 s and elongation at 72°C for 30 s. Each sample was tested in triplicate to ensure statistical significance. Quantification of the relative gene expression was performed using the comparative CT method. In all experiments, Actin was used as the endogenous control. ABCG2 (forward) 5'-GACAGTTTCCAATGACCTGA-3' and (reverse) 5'-TTACATTTGAAATTGGCAGG-3'; ABCB1 (forward) 5'-GGAAGCCAATGCCTATGACTTTA-3' and (reverse) 5'-GAACCACTGCTTCGCTTTCTG-3'; Hsp70 (forward) 5'-GGAAGATCCCTCGAGATCCA-3' and (reverse) 5'-AGCATGGCAGTCACTTGCTC-3'; Hsp27 (forward) 5'-ATGGCGTGGTGGAGATCACC-3' and (reverse) 5'-CAAAAGAACCACCAGGTGGC-3'; Beta-actin (forward) 5'-ACCAACTGGGACGACATGGA-3' and (reverse) 5'-GTGAGGATCTTCATGAGGTA-3'.
Western blot analysis
Protein samples were separated by SDS-PAGE and blotted to nitrocellulose membrane (Hybond-ECL, GE Healthcare). The membrane was incubated with antibody as specified, followed by secondary antibody conjugated with horseradish peroxidase. Specific antigen-antibody complexes were detected by enhanced chemiluminescence (PerkinElmer, Life science, USA). Western blot analysis was performed with the following antibodies: CD44, Oct4, β-catenin, SIRT1 and phospho Lyn (Cell Signaling Technology, MA, USA), Hsp27, c-Myc, CD34 (Epitomics, CA, USA), p53, PARP, Bcl-2, Bax, HSF1, P-gp, Bcr-Abl, NF-κB and STAT5 (Santa Cruz Biotechnology, CA, USA), Hsp70, Hsp90 (Enzo Life Sciences, Inc. NY, USA), BCRP (Signet Laboratories, Inc., Dedham, MA, USA) and β-actin (Sigma-Aldrich, St. Louis, MO, USA).
siRNA Transfection
The siRNAs used for the targeted silencing of SIRT1 (5′-CUAAUCUAGACCAAAGA AUdTdT-3′), and nonspecific scrambled control (5′-CUUCCCGAAAACUUGAGACdTdT-3′) were purchased from Bioneer (Daejeon, Republic of Korea). In brief, CD44high K562 cells (2x105 cells/ml) were seeded on 6-well plates and transfected with indicated concentration of SIRT1siRNA or control siRNA using oligofectamine reagent. After 48 h, cells were treated with 17-AAG for an additional 24 h and collected for western blotting to determine the levels of indicated proteins.
Flow-cytometric dye-efflux assay
Cell suspension (500 μl) of CD44high K562 cells treated with 17-AAG and/or amurensin G for 24 h incubated with 0.5 μg/ml rhodamine 123 (Rho123), a fluorescent substrate of P-gp, at 37°C for 30 min. After incubation, the cells were washed with ice-cold PBS and further incubated at 37°C for 3 h to allow P-gp-mediated drug efflux. Cells were pelleted by centrifugation at 500×g and resuspended in PBS. Cellular fluorescence was analyzed immediately by using a FACS flow cytometer (FACScalibur, BD Biosciences, San Jose, CA).
Statistical analysis
A Student's t-test was used to calculate the statistical significance of the experimental data and the level of significance was set as *P < 0.05, **P < 0.01 and ***p < 0.001.
Results
CD44high K562 cells exhibit higher expression of various CSC-related molecules and resistance to a variety of anti-cancer drugs in comparison to parental K562wt cells
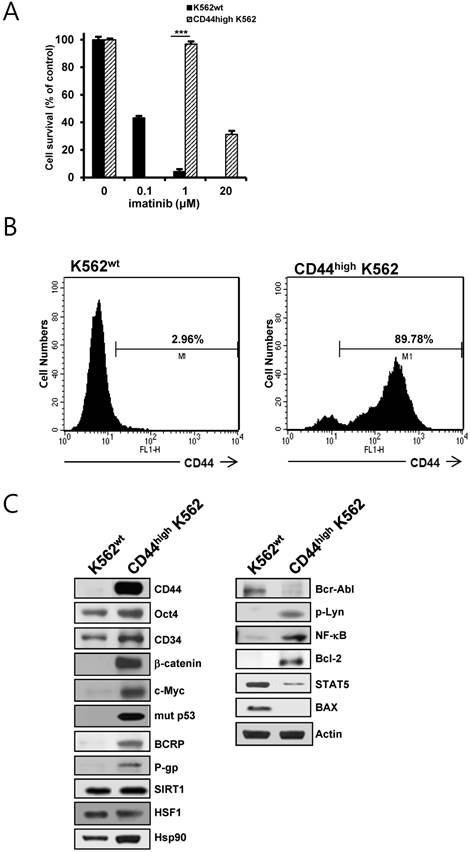
CD44high K562 cells, which exhibited higher resistance to imatinib, were obtained by continuous exposure of parental K562 (K562wt) cells to imatinib (Fig.1A). The phenotypes of K562wt and CD44high K562 leukemia cells were determined with flow-cytometric analysis (Fig. 1B), showing that CD44high K562 cells were highly enriched in CD44-positive population (89.98 %) as compared with K562wt cells (2.96 %). Interestingly, CD44high K562 cells showed higher protein levels of various CSC-related molecules (Oct4, CD34, β-catenin, c-Myc, mut p53), ABC transporter (BCRP and P-gp), Hsp90, SIRT1 and HSF1 as compared with those of K562wt cells (Fig. 1C, left panels). In addition, the expression level of Bcr-Abl was remarkably lower than that of K562wt cells. Since it has been demonstrated that some resistance to imatinib is associated with an acquired BCR-ABL-independent signaling characteristics [31, 32], we determined BCR-ABL-independent signaling molecules. The levels of phosphorylated Lyn, NK-κB and Bcl-2 were increased and the levels of STAT5 and Bax were decreased in CD44high K562 cells compared with K562wt cells (Fig. 1C, right panels). These results indicated that CD44high K562 CML cells might be leukemic CSCs with BCR-ABL-independent signaling characteristics.
Identification of CD44high K562 cells and the difference in expression of CSC-related and other molecules between K562wt and CD44high K562 cells. A. Both cells were treated with various concentrations of imatinib for 96 h. Percentage of cell survival was determined after 96 h of incubation using MTT assay. Each bar represents the mean ± S.D. of triplicate experiments. ***p < 0.001. B: the cell surface expression of CD44 was quantified by flow cytometry after labeling both cells with anti-CD44 antibody. C: The protein levels of CD44, Oct4, CD34, β-catenin, c-Myc, mut p53, BCRP, P-gp, Bcl-2, Hsp90, Bcr-Abl, phospho-Lyn (p-Lyn), NF-κB, Bax and STAT5 in both cells were determined by western blot analysis. β-Actin (Actin) is used as a loading control of both cells.
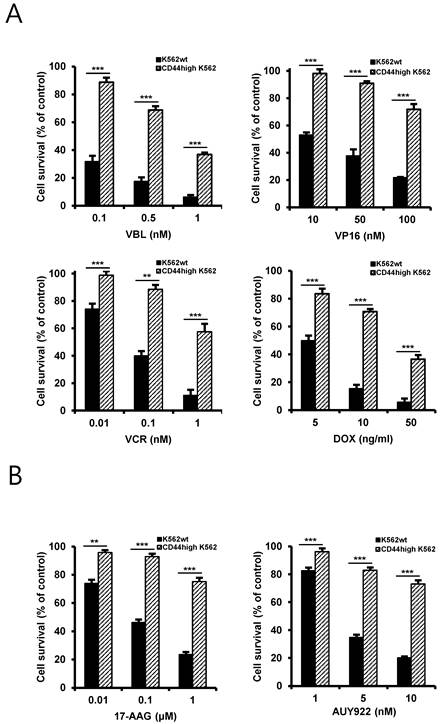
One characteristic of CSCs is believed to be responsible for resistance to chemotherapeutic drugs, possibly due to enrichment of ABC transporter proteins in CSCs and resultant rapid and effective efflux of drugs out of the cells [19]. Therefore, to determine the chemo-resistant property of CD44high K562 cells, the cells were treated with various chemotherapeutic drugs including vinblastine (VBL), etoposide (VP16), doxorubicin (DOX), vincristine (VCR) (Fig. 2A) and Hsp90 inhibitors including 17-allylamino-17-demethoxy-geldanamycin (17-AAG), an ansamycin Hsp90 inhibitor, and NVP-AUY922 (hereafter called AUY922), a non-ansamycin Hsp90 inhibitor (Fig. 2B). Our data showed that CD44high K562 cells exhibited resistance to Hsp90 inhibitors as well as chemotherapeutic drugs involving imatinib compared with K562wt cells, suggesting that CD44high K562 cells are enriched in CSCs with chemo-resistance phenotype.
Differential sensitivity of K562wt and CD44high K562 cells to chemotherapeutic drugs and Hsp90 inhibitors. Both cells were treated with various concentrations of vinblastine (VBL), etoposide (VP16), doxorubicin (DOX) and vincristine (VCR) (A), 17-AAG and AUY922 (B) for 96 h. Percentage of cell survival was determined after 96 h of incubation using MTT assay. Each bar represents the mean ± S.D. of triplicate experiments. **p < 0.01 and ***p < 0.001.
The up-regulated CSC-related molecules in CD44high K562 cells are suppressed by SIRT1 inhibition
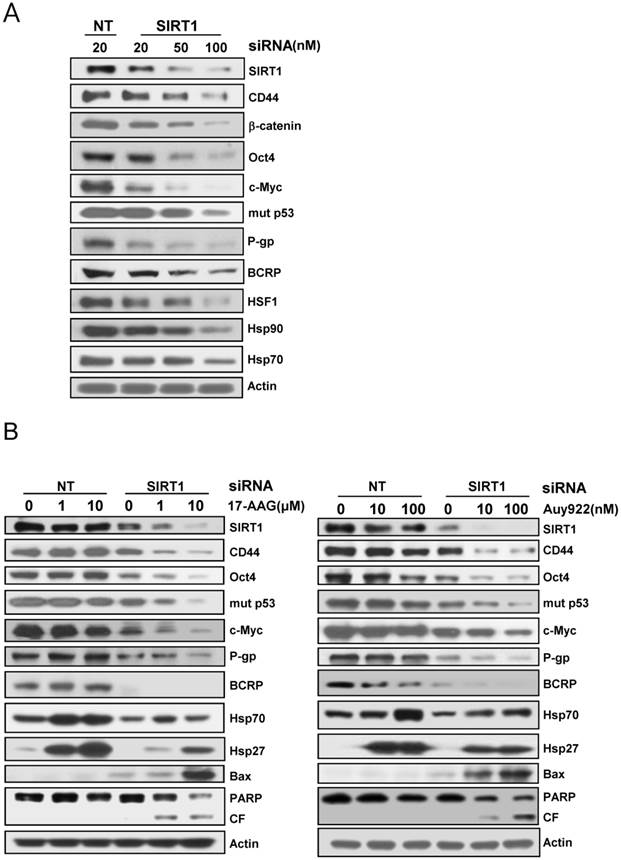
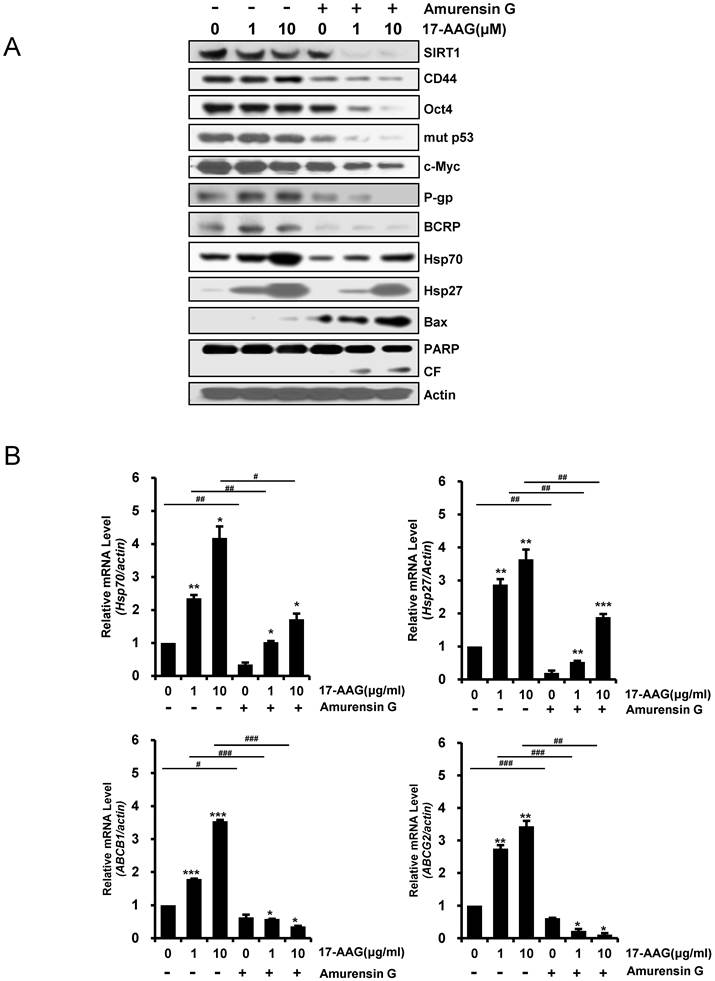
Since it has been showed that SIRT1 inhibition hinders BCR-ABL transformation of hematopoietic stem cells and CML disease development, suggesting that SIRT1 facilitates gain of function of CSC properties [33, 34], we determined if depletion of SIRT1 could modulate the up-regulated CSC-related molecules in CD44high K562 cells. When SIRT1 was knock-downed in CD44high K562 cells with increasing amounts of SIRT1 siRNA, the levels of CD44, Oct-4, β-catenin, c-Myc, mut p53, P-gp and BCRP were decreased dose-dependently (Fig. 3A). Since CD44 knockdown was associated with down-regulation of HSF1 [35], the levels of HSF1 and its target genes including Hsp 90 and Hsp70 were also determined. As expected, all of them were down-regulated after depletion of SIRT1 in CD44high K562 cells (Fig. 3A). Since it has been known that Hsp90 is required for the activity and stability of the pluripotency transcription factors such as Oct4, Nanog and c-Myc [5, 6], and for stabilization of mut p53 in cancer cells [7], the effect of 17-AAG on the expression of Hsps, CSC-related molecules and proapoptotic proteins in CD44high K562 cells was investigated in the presence or absence of SIRT1 inhibition with siRNA. The expression of CD44, Oct4, mut p53 and c-Myc was not significantly affected by treatment with 17-AAG alone, but was remarkably down-regulated by combined treatment of 17-AAG with SIRT1 siRNA. Moreover, the 17-AAG-stimulated induction of Hsp70/Hsp27 and BCRP/P-gp was suppressed by SIRT1 inhibition, which causes both up-regulation of Bax and cleavage of PARP (Fig. 3B). Similar results were obtained with combination treatment of AUY922 and SIRT1 siRNA (Fig. 3C), or 17-AAG and amurensin G, a potent natural SIRT1 inhibitor [30], in CD44high K562 cells (Fig. 4A). These results were followed by a significant suppression of 17-AAG-mediated mRNA induction of Hsp70, Hsp27, ABCB1 and ABCG2 genes by combined treatment with amurensin G in CD44high K562 cells (Fig. 4B). These results suggested a possibility that the effects of Hsp90 inhibitor can be potentiated by SIRT1 inhibition in CSCs.
Effect of SIRT1 depletion on the expression of CSC-related molecules and HSF1/Hsps and on Hsp90 inhibitor-mediated regulation of these molecules in CD44high K562 cells. A: Cells treated were transfected with the indicated dose of 20 nM non targeted control siRNA (NT) or 20~100 nM SIRT1 siRNA (SIRT1) for 48 h, and western blot analysis was performed to monitor indicated protein levels. B: Cells were transfected with of 20 nM NT or SIRT1 siRNA for 48 h were additionally treated with indicated doses of 17-AAG or AUY922 for 24 h, and whole cell lysates of each transfectant were analyzed by western blotting to measure changed expression of indicated proteins.
Inhibition of protein levels of CSC-related molecules and Hsps, and activation of pro-apoptotic cascade by amurensin G, and suppression of mRNA levels of 17-AAG-mediated induction of Hsp70/Hsp27 and ABCB1/ABCG2 genes by amurensin G in CD44high K562 cells. A: Cells were pretreated with amurensin G (5 μg/ml) for 6 h, and treated with 17-AAG (1 or 10 μM) for additional 24 h, and the indicated proteins were detected by western blot analysis. CF; cleavage fragments. B: Relative mRNA levels of Hsp70/Hsp27 and ABCB1/ABCG2 genes in the cells treated with 17-AAG alone and co-treated with 17-AAG and amurensin G were determined by real time RT-PCR. Each bar represented the mean ± S.D. of triplicate experiments. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control, #p < 0.05, ##p < 0.01 and ###p < 0.001, 17-AAG alone vs. combination with amurensin G.
SIRT1 inhibitor blocks P-gp (or BCRP)-mediated efflux activity of Hsp90 inhibitor in CD44high K562 cells
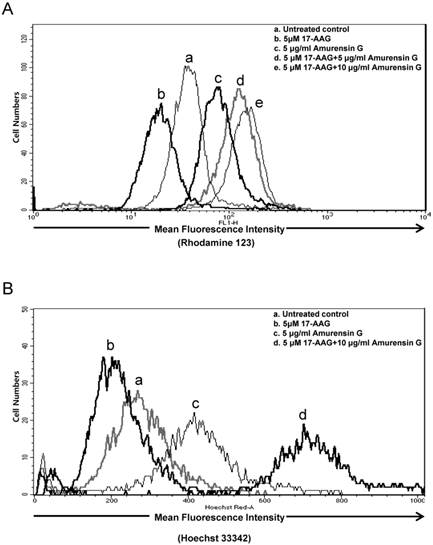
Since hyaluronan receptor CD44 forms complexes with multidrug transporters, BCRP and P-gp in the plasma membrane, and disassembly of CD44-transporter complexes with hyaluronan oligomers induces internalization of CD44, BCRP, and P-gp, and consequently, suppresses drug transporter activity and increases sensitivity to anticancer drug [36], we examined whether SIRT1 inhibition blocks multidrug transporters-mediated efflux activity in CD44high K562 cells, using a flow cytometric functional efflux assay based on the extrusion of rhodamine 123 (Rho 123), a fluorescent substrate of P-gp (Fig. 5A) and Hoechst 33342, a fluorescent substrate of BCRP (Fig. 5B). When CD44high K562 cells treated with 17-AAG, Rho123 efflux was rather increased, possibly due to induction of P-gp, but amurensin G significantly suppressed efflux of Rho 123. Moreover, combined treatment of amurensin G and 17-AAG suppressed remarkably the efflux of Rho 123 in CD44high K562 cells, which was consistent with the down-regulation of P-gp in CD44high K562 cells after combined treatment with 17-AAG and SIRT1 siRNA (Fig. 4). Since BCRP may also function to decrease cellular accumulation of 17-AAG in CD44high K562 cells, we examined the effect of amurensin G on BCRP-mediated efflux using Hoechst 33342 (Fig. 5B). When CD44high K562 cells treated with 17-AAG, Hoechst 33342 efflux was increased like Rho 123 efflux, possibly due to induction of BCRP, but amurensin G significantly suppressed efflux of Hoechst 33342. Moreover, combined treatment of amurensin G and 17-AAG suppressed remarkably the efflux of Hoechst 33342 in CD44high K562 cells, which was consistent with the down-regulation of BCRP in CD44high K562 cells after combined treatment with 17-AAG and amurensin G (Fig. 4). These results suggest that SIRT1 inhibitor is highly effective to increase an efficacy of Hsp90 inhibitors possibly through down-regulation of P-gp/BCRP.
Effect of SIRT1 inhibitors on P-gp (or BCRP)-mediated efflux activity of Hsp90 inhibitors in CD44high K562 cells. Cell suspension isolated from the cells treated with 17-AAG (5 μM) or in the presence or absence of AG (5 μg/ml) or for 12 h. And then these are incubated with rhodamine 123 (A) or Hoechst 33342 (B) and further incubated at 37 °C for 4 h to allow P-gp (or BCRP) transporter-mediated efflux. Cellular fluorescence was analyzed immediately by using flow cytometer.
Resistance to Hsp 90 inhibitors in CD44high K562 cells is overcome by SIRT1 inhibition
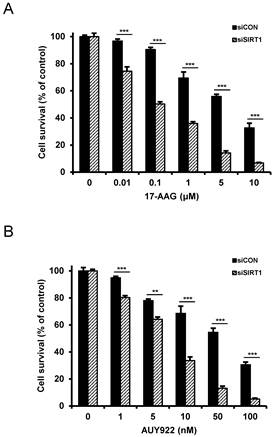
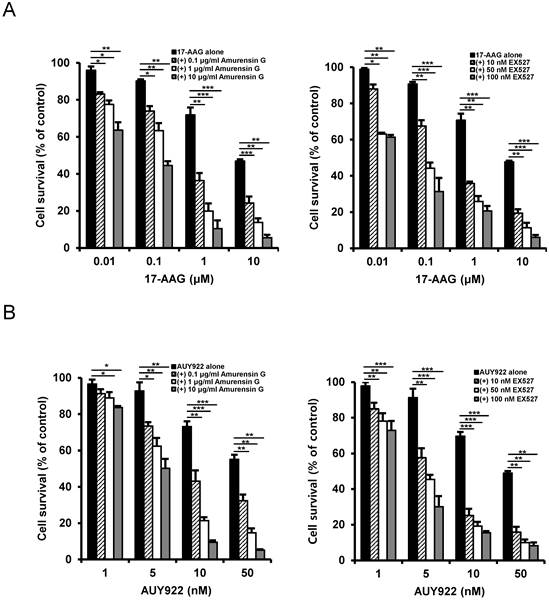
Since SIRT1 inhibition resulted in suppression of the up-regulated CSC-related molecules in CD44high K562 cells, it was determined if SIRT1 inhibition could reverse the resistance of CD44high K562 cells to Hsp90 inhibitors. When CD44high K562 cells were treated with Hsp90 inhibitor (17-AAG or AUY922) after SIRT1 knock-down, the sensitivity of the cells to 17-AAG or AUY922 was significantly enhanced in comparison with treatment with 17-AAG or AUY922 alone (Fig. 6A and B). To further examine the potential relationship between SIRT1 activity and sensitivity to Hsp90 inhibitor in CD44high K562 cells, we tested the combination effect of Hsp90 inhibitors with SIRT1 inhibitors in the cells. When CD44high K562 cells were co-treated with 17-AAG and amurensin G or EX527, a known chemical inhibitor of SIRT1 [37], there was a significant dose-dependent potentiation of 17-AAG-induced cytotoxicity against CD44high K562 cells by amurensin G or EX527 (Fig. 7A). Similarly, the combination effect of AUY922 and SIRT1 inhibitor (amurensin G or EX527) was observed in CD44high K562 cells (Fig. 7B). These results demonstrated that the susceptibility of CD44high K562 cells to Hsp90 inhibitors can be significantly enhanced by SIRT1 inhibitors.
Sensitization of CD44high K562 cells to Hsp90 inhibitors by SIRT1 depletion. Cells were transfected with of 20 nM SIRT1 siRNA (siSIRT1) or a nonspecific control siRNA (siCON). After 48 h of transfection, the cells were treated with serial concentrations of 17-AAG (A) or AUY922 (B). Percentage of cell survival was determined after 96 h of incubation using MTT assay. Each bar represents the mean ± S.D. of triplicate experiments. **p < 0.01 and ***p < 0.001.
Potentiation of cytotoxicity of Hsp90 inhibitor in CD44high K562 cells by SIRT1 inhibitors. Cells were treated with serial doses of 17-AAG (A) or AUY922 (B) in the presence or absence of amurensin G (0.1- and 10 μg/ml) or EX527 (10- and 50 nM). Percentage of cell survival was determined after 96 h of incubation using MTT assay. * p < 0.05, **p < 0.01 and ***p < 0.001.
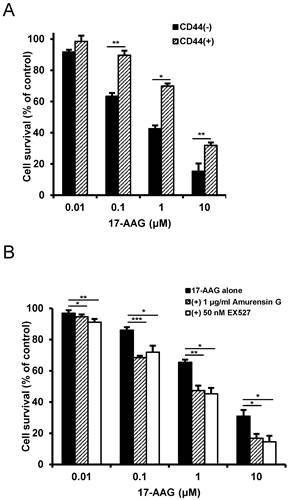
To confirm the reversal of resistance to Hsp90 inhibitor by SIRT1 inhibitor in other cancer stem-like cells, we used CD44+ HCT-15 cells with high colony forming activity and CD44- HCT-15 cells with poor colony forming activity separated from human colon cancer HCT-15 cells [29]. Like CD44high K562 cells, CD44+ HCT-15 cells were resistant to 17-AAG compared with CD44- HCT-15 cells (Fig. 8A), and amurensin G or EX527 potentiated sensitivity of CD44+ HCT-15 cells to 17-AAG (Fig. 8B). Therefore, our results strongly suggest a sensitization of cancer stem-like cells to Hsp90 inhibitors by SIRT1 inhibition.
Resistance of CD44+ HCT-15 cells to Hsp90 inhibitor and sensitization of CD44+ HCT-15 cells to Hsp90 inhibitor by SIRT1 inhibitor. A: CD44+ HCT-15 [CD44 (+)] and CD44- HCT-15 [CD44 (-)] cells were treated with serial doses of 17-AAG. B: CD44+ HCT-15 cells were treated with serial doses of 17-AAG in the presence or absence of amurensin G (1 μg/ml) or EX527 (50 nM). Percentage of cell survival was determined after 96 h of incubation using MTT assay. * p < 0.05, **p < 0.01 and ***p < 0.001.
Discussion
Despite the success of imatinib in controlling patients with CML, it has been shown that some patients become resistant to imatinib in the later stages of CML. The resistance to imatinib of CML cells is mainly mediated by mutations within the kinase domain or up-regulation of Bcr-Abl [38, 39]. However, in some patient disease progression may be mediated by BCR-ABL-independent mechanism caused by the high expression of tyrosine-phosphorylated Lyn, a member of the Src kinase family [31, 32]. It has been shown that Jak2 inhibition suppressed tumor formation caused by IM-resistant K562-R cells, which have reduced BCR-ABL expression and limited activation of BCR-ABL signaling cascades [31, 40, 41]. In the present study, we have demonstrated that combination of Hsp90 inhibitor and SIRT1 inhibitor could be an effective therapeutic approach to target CML-stem like cells exhibiting multidrug resistance to current therapies and high expression of several CSC-related molecules as well as reduced BCR-ABL expression and increased Lyn expression.
CD44high K562 cells exhibited higher expression of CSC-related molecules such as CD44, Oct4, Nanog, CD34, c-Myc, mut p53 as well as ABC transporters BCRP and P-gp, and were more resistant to various chemotherapeutic drugs and Hsp90 inhibitors compared with K562wt cells. There are several evidences showing that the stemness genes are involved indirectly in drug resistance. Hyaluronan-CD44 interaction activates Nanog-Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells, and human mesenchymal stem cells [17, 42], and suppression of CD44 inhibits the tumorigenicity and multidrug resistance in colon and ovarian cancer-initiating cells [43, 44]. Oct4 also mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway [20]. Myc protein positively regulates MDR1 gene promoter by interaction with E-box-related motifs and alters ABC transporter expression [45], and β-catenin binds to the MDR1 promoter and increases P-gp expression [18]. In addition, mut p53 may be involved transcriptional activation of the MDR1 gene and drug resistance [46].
Recently, Hsp90 inhibitors are being considered as promising anticancer drugs, since Hsp90 can stabilize a wide range of signaling molecules implicated in cancers [3]. However, they have limitations as effective anticancer drugs due to drug efflux and rebound activation of HSF1 by Hsp 90 inhibitors. SIRT1 is known to be necessary to maintain the stability of HSF1 after activation and to prevent its rapid degradation, and down-regulation of SIRT1 results in a release of the acetylated HSF1 from its cognate promoter elements [47, 48]. In our experiments, depletion of SIRT1 by siRNA in CD44high K562 cells resulted in a significant reduction of the MDR-related CSC molecules such as CD44, Oct-4, β-catenin, c-Myc and mut p53, BCRP and P-gp, which were increased in CD44high K562 cells compared with K562wt cells, and additionally decreased the levels of HSF1 and Hsp70/Hsp27. Therefore, our results suggest that inhibition of SIRT1 may repress directly or indirectly the expression of the MDR-related CSC molecules and inhibit the expression of Hsps, possibly through down-regulation of HSF1, and consequently may enhance the anti-cancer effect of Hsp90 inhibitors. This hypothesis was supported by findings showing the significant suppression of the 17-AAG-mediated induction of Hsp70/Hsp27 as well as ABC transporters-mediated efflux activity after treatment with SIRT1 siRNA or amurensin G in CD44high K562 cells. Currently, we do not understand all aspects of the reasons why depletion of SIRT1 resulted in a significant reduction of the MDR-related stem cell markers, which were increased in CD44high K562 cells. Recently, it has been shown that the expression of CD44 and BCRP was decreased by treatment with HDAC inhibitors including suberoylanilide hydroxamic acid and trichostatin A in squamous cell carcinoma of the head and neck [49], indicating that the expression of CD44 and BCRP can be regulated by various HDACs including SIRT1. The level of β-catenin can be increased by SIRT1 [50], and indirectly by HSF1 via induction of the RNA-binding protein HuR, which stabilize mRNA and/or enhance translation [51]. It has been reported that depletion of HSF1 induces destabilization of mut p53, which may contribute to the evolvement of CSCs [7, 52]. Therefore, these findings might support a significant reduction of various MDR-related CSC molecules in CD44high K562 cells by depletion of SIRT1.
Here, we showed that the sensitivity of CD44high K562 cells to Hsp90 inhibitors was profoundly enhanced by SIRT1 knock-down or treatment with SIRT1 inhibitors. Since SIRT1 inhibition suppressed 17-AAG-mediated Hsp70/Hsp27 induction and P-gp (or BCRP)-mediated 17-AAG efflux activity and also accelerated down-regulation of the MDR-related CSC molecules, we suggest that these complicated mechanism of SIRT1 inhibition may be involved in overcoming resistance of CD44high K562 cells against Hsp90 inhibitor, and therefore combination treatment with Hsp90 inhibitor and SIRT1 inhibitor is more effective than each drug alone in eliminating CD44high K562 cells. Taken together, our data showed for the first time that combined treatment with Hsp90 inhibitor and SIRT1 inhibitor would be a novel therapeutic approach to target CSCs that are resistant to current therapies.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2014R1A1A2053321) and the National R&D Program for Cancer Control, Ministry for Health, Welfare, and Family affairs (No. 0920050), Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther. 2011;89:491-502
2. Sampieri K, Fodde R. Cancer stem cells and metastasis. Semin Cancer Biol. 2012;22:187-93
3. Whitesell L, Lin NU. HSP90 as a platform for the assembly of more effective cancer chemotherapy. Biochim Biophys Acta. 2012;1823:756-66
4. Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat Rev. 2013;39:375-87
5. Regan PL, Jacobs J, Wang G. et al. Hsp90 inhibition increases p53 expression and destabilizes MYCN and MYC in neuroblastoma. Int J Oncol. 2011;38:105-12
6. Bradley E, Bieberich E, Mivechi NF, Tangpisuthipongsa D, Wang G. Regulation of embryonic stem cell pluripotency by heat shock protein 90. Stem Cells. 2012;30:1624-33
7. Li D, Marchenko ND, Schulz R. et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res. 2011;9:577-88
8. Kamal A, Thao L, Sensintaffar J. et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407-10
9. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761-72
10. Piper PW, Millson SH. Mechanisms of Resistance to Hsp90 Inhibitor Drugs: A Complex Mosaic Emerges. Pharmaceuticals. 2011;4:1400-22
11. Vilaboa NE, Galan A, Troyano A, de Blas E, Aller P. Regulation of multidrug resistance 1 (MDR1)/P-glycoprotein gene expression and activity by heat-shock transcription factor 1 (HSF1). J Biol Chem. 2000;275:24970-6
12. Negi LM, Talegaonkar S, Jaggi M, Ahmad FJ, Iqbal Z, Khar RK. Role of CD44 in tumour progression and strategies for targeting. J Drug Target. 2012;20:561-73
13. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167-74
14. Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175-80
15. Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia. 2009;14:3-9
16. Caldera V, Mellai M, Annovazzi L, Monzeglio O, Piazzi A, Schiffer D. MGMT hypermethylation and MDR system in glioblastoma cancer stem cells. Cancer Genomics Proteomics. 2012;9:171-8
17. Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem. 2008;283:17635-51
18. Correa S, Binato R, Du Rocher B, Castelo-Branco MT, Pizzatti L, Abdelhay E. Wnt/beta-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer. 2012;12:303
19. Marques DS, Sandrini JZ, Boyle RT, Marins LF, Trindade GS. Relationships between multidrug resistance (MDR) and stem cell markers in human chronic myeloid leukemia cell lines. Leuk Res. 2010;34:757-62
20. Wang XQ, Ongkeko WM, Chen L. et al. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. 2010;52:528-39
21. Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci U S A. 2010;107:22745-50
22. Sampath J, Sun D, Kidd VJ. et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem. 2001;276:39359-67
23. Godar S, Ince TA, Bell GW. et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62-73
24. Abdelalim EM, Tooyama I. Knockdown of p53 suppresses Nanog expression in embryonic stem cells. Biochem Biophys Res Commun. 2014;443:652-7
25. Menssen A, Hydbring P, Kapelle K. et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A. 2012;109:E187-96
26. You JS, Kang JK, Seo DW. et al. Depletion of embryonic stem cell signature by histone deacetylase inhibitor in NCCIT cells: involvement of Nanog suppression. Cancer Res. 2009;69:5716-25
27. Herbert KJ, Cook AL, Snow ET. SIRT1 inhibition restores apoptotic sensitivity in p53-mutated human keratinocytes. Toxicol Appl Pharmacol. 2014;277:288-97
28. Perez RE, Knights CD, Sahu G. et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J Cell Physiol. 2010;225:394-405
29. Lee SH, Kim MJ, Kim DW, Kang CD, Kim SH. Amurensin G enhances the susceptibility to tumor necrosis factor-related apoptosis-inducing ligand-mediated cytotoxicity of cancer stem-like cells of HCT-15 cells. Cancer Sci. 2013;104:1632-9
30. Oh WK, Cho KB, Hien TT. et al. Amurensin G, a potent natural SIRT1 inhibitor, rescues doxorubicin responsiveness via down-regulation of multidrug resistance 1. Mol Pharmacol. 2010;78:855-64
31. Donato NJ, Wu JY, Stapley J. et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690-8
32. Donato NJ, Wu JY, Stapley J. et al. Imatinib mesylate resistance through BCR-ABL independence in chronic myelogenous leukemia. Cancer Res. 2004;64:672-7
33. Yuan H, Wang Z, Li L. et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012;119:1904-14
34. Wang Z, Chen W. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer. 2013;4:82-90
35. Pham PV, Phan NL, Nguyen NT. et al. Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med. 2011;9:209
36. Slomiany MG, Dai L, Bomar PA. et al. Abrogating drug resistance in malignant peripheral nerve sheath tumors by disrupting hyaluronan-CD44 interactions with small hyaluronan oligosaccharides. Cancer Res. 2009;69:4992-8
37. Napper AD, Hixon J, McDonagh T. et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48:8045-54
38. Lamontanara AJ, Gencer EB, Kuzyk O, Hantschel O. Mechanisms of resistance to BCR-ABL and other kinase inhibitors. Biochim Biophys Acta. 2013;1834:1449-59
39. Jabbour EJ, Cortes JE, Kantarjian HM. Resistance to tyrosine kinase inhibition therapy for chronic myelogenous leukemia: a clinical perspective and emerging treatment options. Clin Lymphoma Myeloma Leuk. 2013;13:515-29
40. Samanta AK, Chakraborty SN, Wang Y. et al. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene. 2009;28:1669-81
41. Samanta A, Perazzona B, Chakraborty S. et al. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia. 2011;25:463-72
42. Liu CM, Chang CH, Yu CH, Hsu CC, Huang LL. Hyaluronan substratum induces multidrug resistance in human mesenchymal stem cells via CD44 signaling. Cell Tissue Res. 2009;336:465-75
43. Subramaniam V, Vincent IR, Gilakjan M, Jothy S. Suppression of human colon cancer tumors in nude mice by siRNA CD44 gene therapy. Exp Mol Pathol. 2007;83:332-40
44. Cheng W, Liu T, Wan X, Gao Y, Wang H. MicroRNA-199a targets CD44 to suppress the tumorigenicity and multidrug resistance of ovarian cancer-initiating cells. FEBS J. 2012;279:2047-59
45. Blanc E, Goldschneider D, Ferrandis E. et al. MYCN enhances P-gp/MDR1 gene expression in the human metastatic neuroblastoma IGR-N-91 model. Am J Pathol. 2003;163:321-31
46. Bush JA, Li G. Cancer chemoresistance: the relationship between p53 and multidrug transporters. Int J Cancer. 2002;98:323-30
47. Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063-6
48. Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089-115
49. Chikamatsu K, Ishii H, Murata T. et al. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci. 2013;104:1468-75
50. Simic P, Zainabadi K, Bell E. et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol Med. 2013;5:430-40
51. Chou SD, Murshid A, Eguchi T, Gong J, Calderwood SK. HSF1 regulation of beta-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene. 2014
52. Shetzer Y, Solomon H, Koifman G, Molchadsky A, Horesh S, Rotter V. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis. 2014;35:1196-208
Author contact
![]() Corresponding authors: Sun-Hee Kim and Chi-Dug Kang, Department of Biochemistry, Pusan National University School of Medicine, Yangsan 626-870, Republic of Korea. Tel.: +82-51-510-8081. Fax: +82-51-510-8086, E-mail address: ksh7738ac.kr (Sun-Hee Kim), kcdshbwac.kr (Chi-Dug Kang).
Corresponding authors: Sun-Hee Kim and Chi-Dug Kang, Department of Biochemistry, Pusan National University School of Medicine, Yangsan 626-870, Republic of Korea. Tel.: +82-51-510-8081. Fax: +82-51-510-8086, E-mail address: ksh7738ac.kr (Sun-Hee Kim), kcdshbwac.kr (Chi-Dug Kang).