Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(8):970-981. doi:10.7150/ijbs.11635 This issue Cite
Research Paper
The saponin DT-13 Attenuates Tumor Necrosis Factor-α-induced Vascular Inflammation Associated with Src/NF-кB/MAPK Pathway Modulation
Yuanyuan Zhang#, Minhui Sun#, Yuwei Han, Kefeng Zhai, Youmei Tang, Xiaoying Qin, Zhengyu Cao, Boyang Yu ![]() , Junping Kou
, Junping Kou ![]()
Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China Pharmaceutical University, 639 Longmian Road, Nanjing 211198, China
# Co-first authors (Y.Z.,& M.S.)
Received 2015-1-19; Accepted 2015-5-15; Published 2015-6-11
Abstract

This study aimed to explore the effect of DT-13 (25(R,S)-ruscogenin- 1-O- [β-d-glucopyranosyl- (1→2)][β-d-xylopyranosyl-(1→3)]-β -d- fucopyranoside) on tumor necrosis factor (TNF)-α-induced vascular inflammation and the potential molecular mechanisms. In vitro, DT-13 suppressed TNF-α-induced adhesion and migration of human umbilical vein endothelial cells (HUVECs) by inhibiting the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). DT-13 markedly suppressed NF-кB p65 phosphorylation, and when NF-кB p65 was over-expressed, the inhibitory effect of DT-13 on adhesion molecular decreased. DT-13 also suppressed TNF-α induced luciferase activities of ICAM-1 and VCAM-1 promoter containing NF-κB binding sites. Furthermore, DT-13 markedly suppressed p38 phosphorylation and Src degradation induced by TNF-α, whereas had no significant effect on ERK and JNK activation. In vivo, DT-13 at 4 mg/kg prevented vascular inflammation and the expression of adhesion molecules induced by TNF-α in mice. These findings suggest that DT-13 abrogates vascular inflammation by down-regulating adhesion molecules associated with modulating the NF-кB, p38MAPK, Src signaling pathways, and NF-κB binding site is at least one of the targets of DT-13. This study provides novel information regarding the mechanism by which DT-13 exerts its effects on vascular inflammation, which is important for the onset and progression of various diseases.
Keywords: DT-13, Endothelial inflammation, NF-кB, Src, MAPKs
1. Introduction
Endothelial cells (ECs) play a key role in the pathogenesis of a broad spectrum of many human diseases, including atherosclerosis, heart disease, stroke, diabetes, tumor growth and tumor metastasis [1]. The dysfunction of ECs is directly involved in vascular inflammation. The endothelium is a critical regulator of vascular tone; its function is impaired with atherosclerosis, with reduced vasodilation, and increased vascular tone associated with a proinflammatory and prothrombotic state. The reduction of inflammation localized in vascular ECs is therefore recognized as an important contributor to the pathophysiology of vascular inflammatory disease [2].
Numerous pathophysiological studies have demonstrated that the first step in the development of EC inflammation is monocytes adhesion to the vascular endothelium [3]. This pathological process depends on interactions between adhesion molecules expressed on the EC surface, including vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), E-selectin, and P-selectin [4-5]. As reported, these adhesion molecules are hallmarks of endothelial inflammation and/or endothelial dysfunction [6]. Generally, these adhesion molecules have been demonstrated to be critical for activation of endothelium and vascular inflammation [7].
The expression of these adhesion molecules is regulated by complex intracellular signaling pathways, which involve nuclear factor κB (NF-κB) and mitogen-activated protein kinases (MAPKs) [8-10]. It was reported that lactoferrin bound to a DNA region in the ICAM-1 promoter and inhibited the binding of NF-kappaB to a proximal NF-kappaB site in the ICAM-1 promoter [11]. Some chemical compounds have been shown to reduce TNF-α-induced VCAM-1 expression in endothelial cells via MAPK and/or NF-κB signaling pathways [12-13].
Additionally, Src activation is also involved in vascular inflammation. Previous reports suggest that Src tyrosine kinases promote inflammatory processes under various pathological conditions induced by TNF-α [14]. Src activity is regulated by tyrosine phosphorylation at two sites with opposing effects. When the C-terminal tyrosine (Y527) is phosphorylated, it is bound by the SH2 domain, thus holding Src in an inactive conformation [15]. In contrast, the phosphorylation of Tyr416, which is located in the activation loop of the kinase domain, increases enzyme activity [16]. Most importantly, Src activation is involved in cell adhesion. Several reports have demonstrated that TNF-α-induced vascular inflammation is mediated through the activation of alternative signaling molecules, including c-Src and ICAM-1 [17-18]. Overall, these signaling pathways play key roles in vascular inflammation.
The herb Liriope muscari (Decne.) Bailey, a traditional Chinese medicine, has been used to treat various diseases, such as pharyngitis, bronchitis, pneumonia, cough, and cardiovascular diseases, for many years in China. The steroidal saponin DT-13 (25(R,S)-ruscogenin- 1-O- [β-d-glucopyranosyl- (1→2)][β-d-xylopyranosyl-(1→3)]-β -d- fucopyranoside) (Fig. 1A), which is one of the major active compounds of L. muscari, exhibits significant anti-inflammatory, cardioprotective, and antitumor activities [19-21]. DT-13 also inhibits cancer cell adhesion and migration via the down-regulation of matrix metalloproteinases (MMPs) and p38 activation [22-23]. These activities of DT-13 suggest that it may have an effect on vascular inflammation. However, the mechanism by which DT-13 exerts its bioactivity remains uncharacterized.
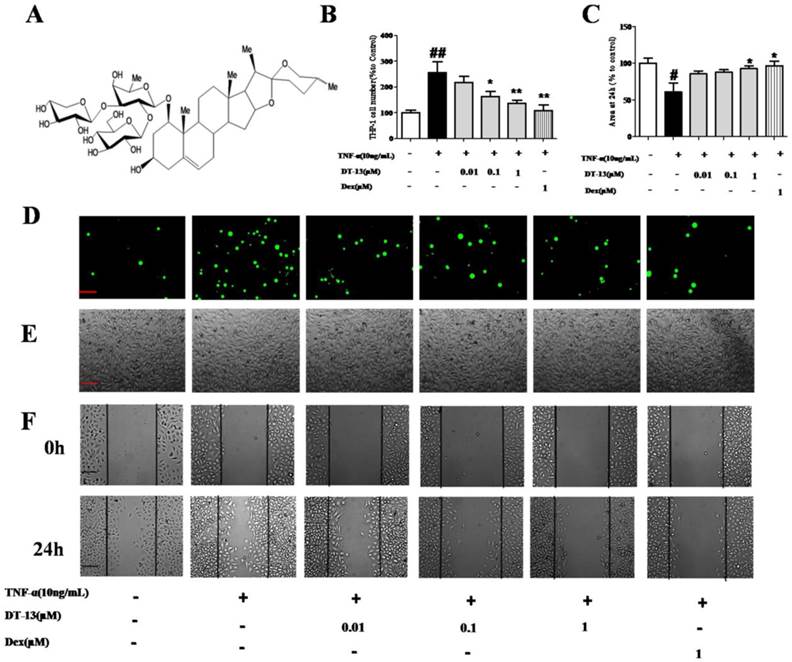
DT-13 inhibited TNF-α-activated endothelial cell adhesion and migration in vitro. (A) The chemical structure of DT-13. (B). THP-1 cells adhesion to HUVECs was measured using an ELISA reader at 488 nm. (C). Confluent monolayers were wounded and a wound assay was performed Materials and Methods section. The area of the wound was measured using Image J software. (D)&(E). HUVECs were pretreated with various concentrations of DT-13 (0.01, 0.1, and 1 μM) or dexamethasone (Dex, 1 μM) for 1 h followed by TNF-α (10 ng/mL) stimulation for 4 h. Subsequently, the cells were incubated with THP-1 cells (1×105/mL) for 30 min and washed three times with media. The images (×100) are the representative optical fields of various treatments. The cells were scanned under fluorescence at 488nm (D) and the light view (E). (F). Confluent monolayers were wounded and a wound assay was performed. HUVECs were pretreated with various concentrations of DT-13 (0.01, 0.1, and 1 μM) or dexamethasone (DEX, 1 μM) for 1 h followed by TNF-α (10 ng/mL) stimulation for 6 h. The wounded areas were imaged by fluorescence microscopy at 0- and 24-h time points. The data represent the mean ± SD from three experiments. ## P<0.01 vs. the control group; * P<0.05, ** P<0.01 νs. the TNF-α group.
In the present study, we evaluated the effect of DT-13 on the adhesion of monocytes to TNF-α-induced ECs, which is a common model to investigate the pathogenesis of vascular inflammation [24-25]. We also demonstrate the effect of DT-13 on NF-κB p65 activation as well as p38MAPK and Src (Tyr527 and Tyr416) phosphorylation and confirmed its inhibitory activity on vascular inflammation in vivo. Our results provide novel information regarding the potential mechanism by which DT-13, the major active compound from L. muscari (Decne.), exerts its activity on EC adhesion and migration, which are critical steps of vascular inflammatory diseases.
2. Materials and methods
2.1 Extraction and isolation of DT-13
DT-13 was prepared according to a previously described method and was determined to be (25(R,S)-ruscogenin-1-O-[β-d-glucopyranosyl-(1→2)][β-d-xylopyranosyl-(1→3)]-β-d-fucopyranoside) by comparing its physical data (1H NMR, 13C NMR and MS) with published values. The purity of DT-13 was determined to be 98.5% by a high-performance liquid chromatography-evaporative light scattering detector (HPLC-ELSD) assay according to a previously reported method [26].
2.2 Materials and chemicals
Dexamethasone (Dex) was purchased from Zhejiang Xianju Pharmaceutical Co. Ltd (Hangzhou, Zhejiang, China). Human and mouse TNF-α was obtained from Bioworld Technology Inc. (Minneapolis, MN, USA). In addition, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was obtained from Sigma-Aldrich (St. Louis, MO, USA). The bicinchoninic acid (BCA) protein assay and enhanced chemiluminescence (ECL) kits were obtained from Beyotime Institute of Biotechnology (Shanghai, China). Antibodies against human p65, phospho-p65 (Ser536), Src, phospho-Src (Tyr527 and Tyr416), IкB-α, phospho-IкB-α, ERK, phospho-ERK, JNK, phospho-JNK, p38, phospho-p38, ICAM-1, and VCAM-1 were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Mouse glyceroldehyde-3-phosphate dehydrogenase (GAPDH) and horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from Bioworld Technology Inc. (Minneapolis, MN, USA).
2.3 Cell culture
Human umbilical vein endothelial cells (HUVECs) and human monoblastic leukemia cells (THP-1) were purchased from Shanghai FuMeng Gene Bio-technology (Shanghai, China). HUVECs were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2.0 g/L sodium bicarbonate. THP-1 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 50 μM β-mercaptoethanol, 2.0 mM glutamine, 100 U/mL penicillin and streptomycin. The cells were maintained at 37°C with 5% CO2 and 95% humidity.
2.4 Monocyte-HUVEC adhesion assay
An adhesion assay was performed as previously described [27]. HUVECs plated in a 96-well culture plate at a concentration of 104 cells/well. At 70-80% confluence, the cells were treated with DT-13 (0.01, 0.1 and 1 μM) or dexamethasone (1 μM) for 1 h followed by 4 h incubation with TNF-α (10 ng/mL) at 37 °C. The media aspirated, and 100 µL of RPMI 1640 medium or THP-1 cells (1×105) were added to different wells. The THP-1 cells were fluorescently labeled with BCECF AM (Molecular Probes, Beyotime, Jiangsu, China) for 1 h according to the manufacturer's instructions. After incubation for 60 min in a CO2 incubator, the plates were washed with fresh RPMI 1640 medium to remove the non-adherent THP-1 cells. Images were obtained, and the absorbance was measured at 488 nm. The experiments were repeated three times, each with six replicates.
2.5 HUVECs migration assay
HUVECs plated in a 12-well plate at a density of 105cells/well. When the HUVECs were 50 to 60% confluent, a scratch wound was produced using a 200-µL plastic tip. After three washes with RPMI 1640 medium, the cells were incubated with serum-free RPMI 1640 medium containing various concentrations of DT-13 (0.01, 0.1, and 1 µM) followed by 6h TNF-α (10 ng/mL) exposure. The cells were then cultured in the desired concentrations of DT-13 or vehicle (DMSO, 0.1%) in RPMI 1640 medium with 0.5% serum. The wounded areas were imaged by fluorescence microscopy at 0h and 24h time points. The area of the wound was measured using ImageJ software (National Institute of Health, Bethesda, MA).
2.6 Flow cytometry
After washing twice with PBS, HUVECs were incubated in 50 µL of staining buffer (PBS, 1% BSA and 0.1% NaN3) containing phycoerythrin (PE)-anti-VCAM-1 and PE-anti-ICAM-1 (eBioscience, San Diego, CA) for 30 min at 4°C in the dark. After three washes, the cells were resuspended in 0.5 mL of staining buffer, and the fluorescence intensity was measured using a FACScan instrument (Becton Dickinson, San Jose, CA, USA).
2.7 Transient transfection of NF-κB in HUVECs
HUVECs were seeded into 6cm culture dishes (Corning, Lowell, MA) for overnight. Subsequently, 1μg of pNF-κB-M98 (pReceiver-M98, Funeng, Guangdong, China) or pcDNA was transfected into HUVECs using Endofectin-Plus transfection Reagent (Genecopoeia, Guangdong, China), according to the manufacturer's instruction. After transfection for 43h, cells were pretreated with or without DT-13 at 1μM for 1h prior to TNF-α (10 ng/mL) induction for 4h. The cells were collected for Western blot analysis.
2.8 ICAM-1and VCAM-1-promoter luciferase reporter gene assay
The luciferase assay was designed as previously described with minor modification [28]. Briefly, the human ICAM-1 promoter (-230/+45, -85/+45) and VCAM-1 promoter (-213/+119, -44/+119) were ligated into the pGL3-Basic luciferase reporter vector (Promega,WI,USA). Cells at 70-80% confluence were transfected with various human ICAM-1/VCAM-1 promoter-luciferase vectors (1 μg) or scramble siRNA as designed (Shanghai, China). β-Gal plasmid was co-transfected with ICAM-1/VCAM-1 promoter-luciferase plasmids to serve as the control for transfection efficiency. After transfection for 36h, cells were pretreated with or without DT-13 at 1μM for 1h prior to TNF-α (10 ng/mL) induction for 4h. Luciferase activities were determined using a luciferase assay kit (Promega, Madison, WI) in according to the manufacturer's instructions.
2.9 Western blot analysis
The western blotting was designed as described with minor modification [29]. HUVECs were treated with various concentrations (0.01, 0.1, and 1 µM) of DT-13 for 1 h following TNF-α (10 ng/mL) stimulation for 4 h or 15 min. After washing with PBS, the cells were lysed in lysis buffer (20 mM Tris (pH 7.5), 150 mM NaCl, 1% TritonX-100, sodium pyrophosphate, β-glycerophosphate, EDTA, Na3VO4, and leupeptin) for 30 min on ice. The protein concentration was determined using a BCA protein assay kit. Equal amounts of protein (40 μg) were loaded on an SDS-PAGE gel for electrophoresis, and then transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was then blocked with 3% bovine serum albumin (BSA) for 1.5 h at room temperature followed by an overnight incubation at 4˚C in the primary antibody. After washing, an HRP-conjugated secondary antibody was added and incubated for 1.5 h. The bands were detected using an ECL kit and quantified by Quantity One software. The antibodies against the respective total proteinswere diluted 1:1000, and the antibodies against phosphorylated proteins were diluted 1:500.
2.10 Monocytes adhesion ex vivo
The monocytes adhesion ex vivo for mouse was assayed according to the described previously with minor modifications [30]. Ten-weeks-old male C57BL/6 mice were purchased from the Model Animal Research Centre of Yangzhou University (Yangzhou, certificate no. SCXK 2012-0004). The mice were randomly divided into 4 groups with 6 mice per group (control, TNF-α, TNF-α+DT-13 and TNF-α+dexamethasone) based on weight. The mice were pretreated with Saline (0.9% NaCl, 10ml/kg, intragastric (i.g.)), DT-13 (4.0 mg/kg, i.g.) or dexamethasone (2 mg/kg, i.g.) daily for 7 days. One week later, the mice were given an intraperitoneal (i.p.) injection of 25 μg/kg TNF-α daily for 7 consecutive days, or PBS for control mice. Mice were continually treated with the Saline, DT-13 or dexamethasone during this period. All of the mice were euthanized with Nembutal (Zhejiang Xianju Pharmaceutical Co. Ltd, Hangzhou, Zhejiang, China) 2 h after the last TNF-α injection at the last day. Aortas from the mice were rapidly excised, and fat was carefully trimmed to remove. The aortas were washed twice with ice-cold PBS and placed into DMEM for 10 min at 37°C. Then, the endothelium was exposed after aortas were opened longitudinally. Then the aortas were pinned onto 4% agar in 35-mm plates containing 1 mL of heat-inactivated DMEM and 1% FBS. WEHI3 mouse monocytes were fluorescently labeled with BCECF AM (Molecular Probes, Beyotime, Jiangsu, China) according to the manufacturer's instructions for 2h. The aortas were then incubated with 1×106 fluorescently labeled WEHI3 mousemonocytes for 30 min at 37°C. After 30min later, the aortas were washed twice with pre-heated PBS to remove unbound monocytes. The picture of aortas and the number of monocytes bound to the aortas were determined by confocal microscopy. The data are presented as the means± standard deviation (S.D.) of five areas of the aorta. Meanwhile, blood samples were collected and the serum was ready for ELISA analysis. All procedures and assessments were approved by the Animal Ethics Committee of the School of Chinese Materia Medica, China Pharmaceutical University.
2.11 In vivo leukocyte infiltration assay
The leukocyte infiltration was assayed according to the described previously with a few changes [31]. Ten-weeks-old male C57BL/6 mice were pretreated with Saline (0.9% NaCl, 10ml/kg, intragastric (i.g.)), DT-13 (1.0, 2.0, or 4.0 mg/kg, i.g.) or dexamethasone (2 mg/kg, i.g.) daily for 7days. On the 7th day, TNF-α (25 μg/kg, i.p.) was administered in normal saline. The mice were sacrificed after 6 h, and the peritoneal cavities were washed with 5 mL of normal saline. The samples (20 μL) of the obtained peritoneal fluids were mixed with 0.38 mL of Turk's solution (0.01% crystal violet in 3% acetic acid), and the number of leukocytes was counted under a light microscope.
2.12 Measurements of adhesion molecules
Soluble forms of ICAM-1 (sICAM-1) and VCAM-1 (sVCAM-1) in the serum were measured using the Quantikine ELISA Kit (Senbeijia Tech., China) according to the manufacturer's instructions. The samples were plotted against standard curves to determine serum concentrations of sICAM-1 and sVCAM-1.
2.13 Immunofluorescence staining
For immunofluorescence (IF) staining, tissues were harvested after perfusion, dehydrated in 30% sucrose, embedded into optimal cutting temperature (OCT) compound, and cut into 10-μm thick slices. The sections were blocked in 10% goat serum with 0.1% BSA solution (in TBS) for 1 h at room temperature and then incubated with a primary antibody overnight at 4°C. Rabbit anti-ICAM-1 (Santa Cruz) was diluted at 1:200, whereas goat anti-CD31 (BD Corporation) was diluted at 1:100. Fluorescent secondary antibodies were diluted at 1:200 and incubated for 1 h at room temperature. All of the sections were mounted and imaged using a fluorescence microscope (Leica, Germany).
2.14 Statistical analysis
GraphPad Prism software (Version 4.0, GraphPad Software Inc., San Diego, CA) was used to perform the data analysis. The data are expressed as the means ± SD. Significance between different groups was determined using a one-way analysis of variance (ANOVA) where appropriateor Student's two-tailed t-test or Dunnett's test when the data involved three or more groups.P-values less than 0.05 were considered significant.
3. Results
3.1 DT-13 inhibits cell adherence and migration in TNF-α- activated HUVECs
To investigate the effect of DT-13 on monocytes adhesion to TNF-α-activated HUVECs, THP-1 cells were incubated with HUVECs pretreated with various concentrations of DT-13 followed by TNF-α stimulated. DT-13 has no effect on the HUVECs without the TNF-α activated (Supplementary Figure S1.B). Fig. 1D and E indicate that a minimal number of THP-1 cells adhered to the HUVECs in the absence of TNF-α stimulation. TNF-α (10 ng/mL, 4 h) exposure induced a robust increase in THP-1 cells adherence to activated HUVECs. Pretreatment of TNF-α-exposed HUVECs with DT-13 at nontoxic concentrations of 0.01 to 1 μM markedly attenuated the THP-1 cell adherence in a concentration-dependent manner (Fig. 1B). As a positive control, 1 μM Dex decreased THP-1 cells adhesion to TNF-α-activated HUVECs.
On the other hand, the ability of DT-13 to reduce TNF-α-activated EC migration was confirmed using a scratch wound-healing mobility assay. As shown in Fig. 1C and F, incubation with TNF-α (10 ng/mL) for 24 h significantly enhanced endothelial migration. Against this, pretreatment with 1 µM DT-13 significantly reduced TNF-α-enhanced HUVEC migration.
3.2 DT-13 inhibits the expression of ICAM-1 and VCAM-1 in TNF-α- activated HUVECs
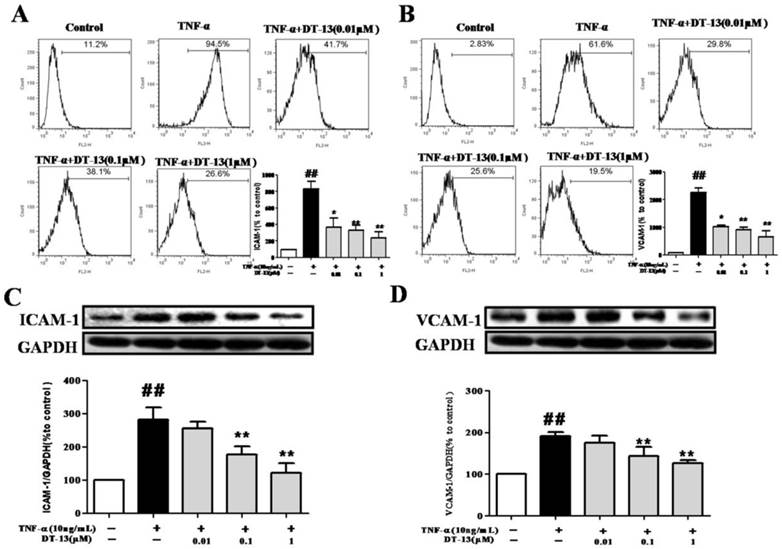
Cell adhesion is controlled by several molecules on the cell surface of HUVECs, including ICAM-1 and VCAM-1 [32]. To determine the possible molecular mechanism by which DT-13 influenced THP-1 cells adherence to TNF-α-activated HUVECs, ICAM-1 and VCAM-1 expression was evaluated by flow cytometry. Incubation of HUVECs with TNF-α dramatically increased ICAM-1 (Fig. 2A) and VCAM-1 expression on the cell surface (Fig. 2B). Pretreatment with DT-13 markedly suppressed the expression of TNF-α-induced ICAM-1 (Fig. 2A) and VCAM-1 (Fig. 2B) on the surface of HUVECs in a concentration-dependent manner. The maximum suppression of TNF-α-induced ICAM-1 and VCAM-1 expression by DT-13 was 72% and 67%, respectively. The expression of ICAM-1 and VCAM-1 in HUVECs following DT-13 treatment was also evaluated by Western blot analysis. Non-activated HUVECs exhibited low levels of ICAM-1 (Fig.2C) and VCAM-1 (Fig.2D). However, the expression was dramatically increased by TNF-α (10 ng/mL) treatment. Pretreatment with DT-13 (0.1, and 1 μM) markedly reduced ICAM-1 and VCAM-1 expression. The maximum inhibitory rates of DT-13 on ICAM-1 and VCAM-1 expression were 42.5% and 36.8%, respectively.
DT-13 suppressed the expression of ICAM-1 and VCAM-1 in TNF-α-activated HUVECs. (A-B). HUVECs were pretreated with DT-13 (0.01, 0.1, and 1 μM) for 1 h followed by TNF-α (10 ng/mL) exposure for 4 h. ICAM-1(A) and VCAM-1(B) surface expression were detected by flow cytometry. (C-D). The expression of ICAM-1(C) and VCAM-1(D) was detected by Western blotting. Representative blots normalized to GAPDH expression are presented. The data represent the mean ± SD from three experiments. ##P<0.01 vs. the control group; * P<0.05, ** P<0.01 vs. the TNF-α group.
3.3 DT-13 suppressed ICAM-1 and VCAM-1 expression dependent with NF-κB pathway
TNF-α activates NF-кB, which subsequently translocates into the nucleus, and promotes the gene transcription of adhesion molecules [33]. We therefore evaluated the influence of DT-13 on TNF-⍺ activated NF-кB using Western blot analysis. HUVECs exposed to TNF-α (10 ng/mL) exhibited dramatic increases in NF-кB p65 and IкB-α phosphorylation, whereas the expression of total p65 and IкB-α remained unchanged. DT-13 inhibited p65 (Fig. 3A) phosphorylation, with maximum inhibitory rates of 43.3%. Meanwhile, DT-13 inhibited the IкB-α (Supplementary Figure S2.A&B) phosphorylation with maximum inhibitory rates of 41.2%.
DT-13 inhibited TNF-α-induced ICAM-1 & VCAM-1 activity dependent with NF-κB pathway. (A). DT-13 inhibited TNF-α-induced p-65 phosphorylation in HUVECs. HUVECs were pretreated with DT-13 (0.01, 0.1 or 1 μM) for 1 h followed by TNF-α (10 ng/mL) exposure. Expression and activation of p-65 were detected by western blotting. ## P<0.01 νs. the control group; * P<0.05,** P<0.01 νs. the TNF-α group. (B) & (C). The over-expression of p65 overcomes the inhibitory effect of DT-13 on ICAM-1 (B) and VCAM-1 (C) expression. (D) Human ICAM-1 promoter or (E) VCAM-1 promoter were ligated into pGL3 basic luciferase vectors. The binding sites for transcription factors are also shown. Right: Cells were transfected with indicated forms of (D) ICAM-1 or (E) VCAM-1 promoter for 24 h, and then incubated with DT-13 for 1 h followed by TNF-α (10 ng/mL) stimulation for 4 h. The promoter activity is represented by the level of luciferase activity indicated by relative light units (RLU). pRL-TK plasmid was also transfected into cells and used as an internal control. The data represent the mean ± SD of three experiments. # P<0.05, ## P<0.01 νs. the control group; * P<0.05,** P<0.01 νs. the TNF-α group.
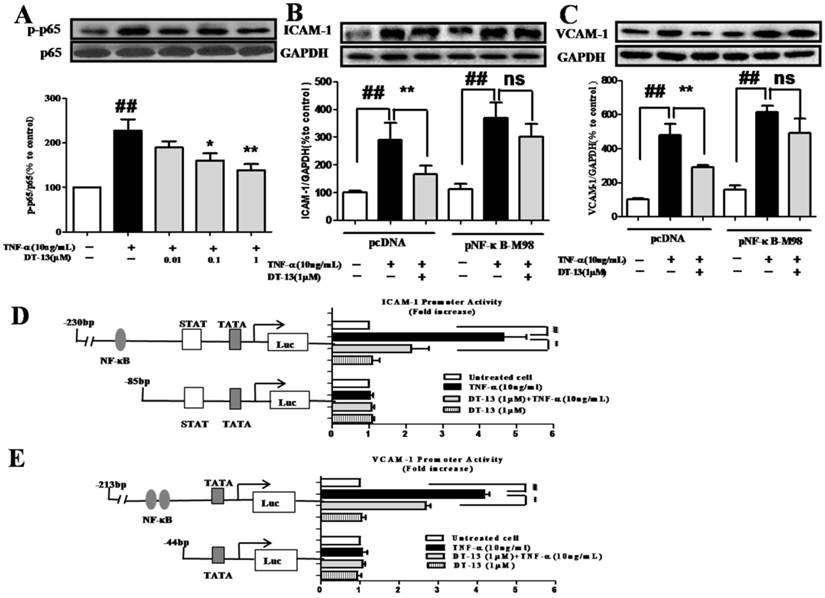
To further investigate the role of NF-κB in the inhibitory activity of DT-13, the pNF-κB-M98 or pcDNA were transfected into HUVECs. Following the over-expression of p65 (Supplementary Figure S3), the suppression of ICAM-1 (Fig. 3B) and VCAM-1 (Fig.3C) by DT-13 decreased. As shown in Fig.3D&E, TNF-α induces the activity of the ICAM-1 promoter containing NF-κB, TATA, STAT, and the VCAM-1 promoter containing NF-κB, GTATA without obvious effect on the promoters of ICAM-1 and VCAM-1 that lack NF-κB binding sites. In contrast, pretreatment with DT-13 (1 μM) before TNF-α stimulation reversed this increased expression. Moreover, 1 μM DT-13 alone did not affect the basic activity of both ICAM-1 and VCAM-1 promoters and had no effects on the promoters of ICAM-1 and VCAM-1 that lack NF-κB binding sites. These findings suggest DT-13 suppressed ICAM-1 and VCAM-1 expression dependent with NF-κB pathway.
3.4 DT-13 modulated MAP kinase phosphorylation in TNF-α-stimulated HUVECs
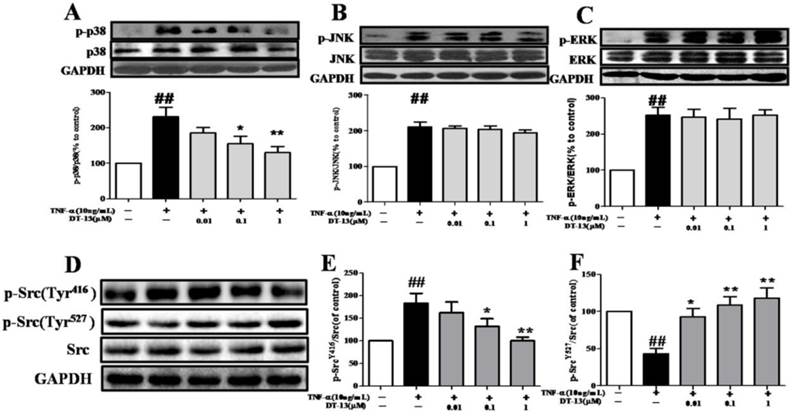
HUVECs exposed to TNF-α exhibit increased phosphorylation of mitogen-activated protein kinases (MAPKs), such as p38, ERK1/2, and JNK. We therefore examined the effects of DT-13 on p38, p-ERK1/2, and p-JNK in TNF-α-activated HUVECs. As shown in Fig. 4A, TNF-α-induced phosphorylation of p38 MAPK was significantly suppressed by pretreatment with DT-13 (0.1 or 1 μM) for 5 h in HUVECs. The maximum inhibitory rate was 50.2%. However, DT-13 did not significant affect ERK and JNK phosphorylation (Fig. 4B and 4C).
DT-13 inhibited the TNF-α-induced activation of MAPK and Src in HUVECs. HUVECs were pretreated with DT-13 (0.01, 0.1 or 1 μM) for 1 h followed by TNF-α (10 ng/mL) exposure. Activation of p38 and phospho-p38 (A), JNK and phospho-JNK (B), and ERK and phospho-ERK (C) , phospho-Src (Tyr416) (G&H)and phospho-Src (Tyr527) (G&I) were detected by western blotting. The data represent the mean ± SD of three experiments. # P<0.05, ## P<0.01 vs. the control group; * P<0.05,** P<0.01 vs. the TNF-α group.
3.5 DT-13 modulates Src phosphorylation in TNF-α-stimulated HUVECs
Given the important role of Src activation in EC inflammation [34], we therefore investigated the effects of DT-13 on p-Src (Tyr416 and Tyr527) in TNF-α-activated HUVECs. As shown in Fig. 4D, E and F, TNF-α activated Src via a decrease in Src (Tyr527) phosphorylation and an increase in Src (Tyr416) phosphorylation, whereas DT-13 modulated Src activation by increasing Src (Tyr527) phosphorylation and decreasing Src (Tyr416) phosphorylation.
3.6 DT-13 decreases cell adherence and leukocyte infiltration in TNF-α-induced mice
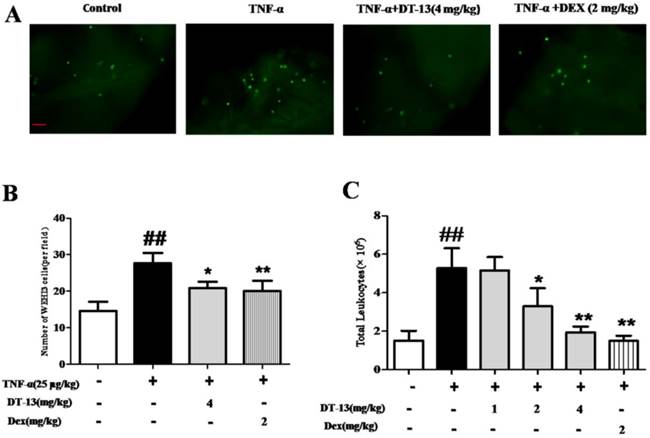
Inflammation-induced mononuclear cell adhesion to ECs is an important step in the development of vascular disease. This study determined whether DT-13 exerts a regulatory effect on the adhesion of monocytes to ECs by identifying whether DT-13 prevents TNF-α-induced vascular inflammation in vivo. We examined monocytes binding to intact endothelium in mouse aortic vessels to evaluate the significance of the DT-13 effect. An ex vivo monocytes adhesion assay was performed using cultured mouse WEHI3 monocytes binding to isolated mouse aortic endothelium. As shown in Fig. 5A and B, the number of mouse monocytes WEHI3 cells bound to the endothelium of mouse aortas isolated from TNF-α-treated mice was significantly increased compared with that of control mice, indicating that the vessels in the TNF-α-treated mice were activated and exhibited inflammation. Dex (2 mg/kg) was used as a positive control and decreased WEHI3 cell adhesion. DT-13 (i.g.) reversed this adverse effect (Fig. 5B). Moreover, DT-13 did not affect ECs adhesion in the absence of TNF-α stimulation (Supplemental Data). TNF-α stimulated the migration of mouse leukocytes, and 2 and 4 mg/kg DT-13 treatment via intragastric administration resulted in a significant reduction in peritoneal leukocyte counts (Fig. 5C). Dex (2 mg/kg) was used as a positive control and decreased leukocyte migration. DT-13 (i.g.) at 4mg/kg reversed this adverse effect (Fig. 5C).
DT-13 prevented ECs adhesion and leukocytic infiltration in TNF-α-treated mice. (A) A significantly increased number of mouse monocyte WEHI3 cells bound to the endothelium of mouse aortas isolated from TNF-α-treated mice compared with control mice, and DT-13 administered by i.g. injection reversed this adverse effect. (B) The number of adhered fluorescently labeled WEHI3 mouse cells was counted in each field. (C) DT-13 inhibited leukocyte migration in vivo at the concentrations of 2 and 4 mg/kg with TNF-α treatment. The data representthe means ± SD of five areas of one aorta. Bar=100 μm. ##P<0.01 vs. the control group; * P<0.05, ** P<0.01 vs. the TNF-α group.
3.7 DT-13 reduced the expression of adhesion molecules in TNF-α-induced mice
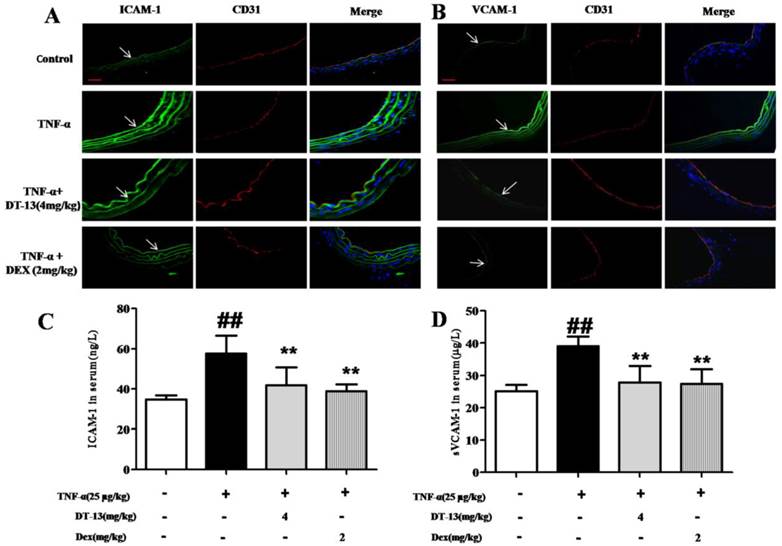
As shown in Fig. 6A and B, the expression of ICAM-1 and VCAM-1 in aorta ECs was activated by TNF-α compared with the control. DT-13 (4 mg/kg) decreases ICAM-1 and VCAM-1 expression. The serum concentrations of ICAM-1 and sVCAM-1 were significantly increased in TNF-α-treated mice compared with control mice. Intragastric administration of DT-13 significantly suppressed the TNF-α-induced increase in ICAM-1 and sVCAM-1 (Fig. 6C and D). As these adhesion molecules are critically involved in the firm adhesion of monocytes to activated ECs and subsequent monocytes recruitment into the subendothelial space [35], these results suggest that DT-13 exhibits an anti-inflammatory effect in vivo via the inhibition of adhesion molecules.
DT-13 supressed ICAM-1 and VCAM-1 expression in vivo. (A-B). DT-13 inhibited ICAM-1 (A) or VCAM-1 (B) expression in aortic endothelial cells activated by TNF-α. ICAM-1 or VCAM-1 was labeled using anti-ICAM-1 or anti-VCAM-1 antibodies (FITC-labeled secondary antibodies, 488 nm) and anti-CD31 (TRITC- FITC-labeled secondary antibody, 594 nm). The arrow denotes ICAM-1 and VCAM-1 on ECs. Bar=100 μm. The data are represented as the mean ± SD of five areas of one aorta. (C-D). DT-13 decreased serum concentrations of sICAM-1 (C) or sVCAM-1 (D) in TNF-α- treated mice by an ELISA assay. ##P<0.01 νs. the control group; * P<0.05, ** P<0.01 νs. the TNF-α group.
4. Discussion
The adherence of monocytes to ECs is a major early event in vascular inflammation. Monocytes bind to the activated endothelium through adhesion molecules. Upon relocating to the intima, monocytes become intimal macrophages and internalize modified lipoproteins to become foam cells. Eventually, these macrophages congregate to the central core of activated vessels [36]. In the present study, we used a monocytes cell model of adhesion to TNF-α-activated ECs, which is a common model to mimic the interaction between monocytes and ECs. We found that DT-13 significantly inhibited adhesion as detected by fluorescence and ELISA assays in vitro and in vivo. These results indicated the potential application of DT-13 in the prevention of early inflammation in vascular diseases.
The inhibition of monocytes adherence to TNF-α-activated ECs was likely mediated through the regulation of the cell surface expression of adhesion molecules, such as ICAM-1 and VCAM-1, given that we observed that DT-13 significantly inhibited TNF-α-activated cell surface expression of ICAM-1 and VCAM-1. These adhesion molecules have been implicated in the onset and progression of vascular diseases [37]. Numerous studies have demonstrated that ICAM-1 and VCAM-1 are key molecules that firmly adhere between inflammatory cells and the vascular surface [38]. For example, ICAM-1 mediates the adhesion of leukocytes to an activated endothelium by establishing strong interactions with integrins and inducing the firm arrest of inflammatory cells at the vascular surface, participating in leukocyte extravasation [39]. VCAM-1 is also the dominant adhesion molecule on the endothelial surface of the vascular wall in the initiation of atherosclerosis [40]. Previous studies demonstrated that VCAM-1 expression increased in the early stages of atherosclerotic lesion formation and correlated well with monocytes adhesion to ECs. A reduction in the expression of these adhesion molecules is hypothesized to be one of the logical mechanisms to ameliorate the early stages of vascular disease [41-42]. Our results demonstrated that DT-13 inhibited monocytes adhesion to TNF-α-activated ECs at least in part by reducing ICAM-1 and VCAM-1 expression in vivo and in vitro.
NF-κB, a major transcription factor in the development of atherosclerotic injury and inflammation, plays critical roles in endothelial cell activation as well as the transcription of numerous cytokines and adhesion molecule-related proteins [43-44]. As previously reported, NF-κB subunits are retained in the cytoplasm by binding to the inhibitory IκB protein in unstimulated cells. Upon stimulation, phosphorylation and the subsequent degradation of IκB-α liberates the NF-κB subunit p65 to enter the nucleus and bind to DNA target sites [45]. Our results demonstrating that DT-13 inhibits TNF-α-induced p65 and IκB phosphorylation suggested that the NF-κB signaling pathway was responsible for the effect of DT-13on EC adhesion. NF-κB is one of the transcription factors that have been reported to be responsible for ICAM-1 and VCAM-1 expression [46-47]. Following the over-expression of p65, the effect of DT-13 on ICAM-1/VCAM-1 decreased. Moreover, in the promoter activity assay, deletion of the NF-κB binding sites in VCAM-1 and ICMA-1 affected TNF-α-induced promoter activity and DT-13 activity. The over-expression of NF-kB/p65 overcomes the inhibitory effect of DT-13 on ICAM-1 and VCAM-1 expression, and DT-13 inhibited the p65 DNA binding activities. These results indicated that NF-κB played a key role in DT-13-inhibited VCAM-1 and ICAM-1 transcription and NF-κB binding site was at least one of the potential targets of DT-13.
In addition to the NF-κB pathway, MAP kinases also play an important role in regulating the expression of adhesion molecules on ECs [48-49]. Studies suggest that various cells preferentially utilize MAPK signaling pathways in response to TNF-α stimulation or inflammation [50-51]. We demonstrated that DT-13 inhibited TNF-α-induced phosphorylation of p38 but not ERK1/2 or JNK.
Src plays essential roles in the regulation of angiogenesis, cell survival, proliferation, adherence, and movement [52]. The activation of Src is involved in TNF-α-induced inflammatory processes, such as cell adhesion, resulting in the activation of downstream components, such as ICAM-1, and MAPK activation [17, 53]. Thus, the effect of DT-13 on Src activation was investigated in TNF-α-induced HUVECs. As previously reported, phosphorylation of the C-terminal tyrosine(Y527) leads to an intra molecular interaction with the SH2domain, resulting in an inactive Src conformation [54]. According to the report, the destabilization of the phosphorylation Tyr416 was linked with the activation of Src and the phosphorylation of Tyr527. The assembly/disassembly of the SH3-SH2 regulatory apparatus plays an important role in the stable conformation of the activated loop. The insertion of Tyr527 into the SH2 domain results in the stabilization of assembled regulatory domains. At the same time, the kinase domain and the transient exposure of Tyr416 would help the phosphorylation of Tyr527 [55]. This study demonstrated that DT-13 suppresses the activation of Src via the downstream phosphorylation of Src (Tyr527). Anti-inflammatory herbal medicines, such as Archidendron clypearia, Polygonum hydropiper, Cinnamomum cassia, and Sorbus commixta, directly suppress the kinase activities of Src [56-58]. Radicicol and nitidine chloride, which have been identified as potential anti-tumor natural products, also inhibit Src tyrosine kinases [59-60]. Diosgenin is the primary steroidal saponin found in several plants, including Dioscorea species (yams), fenugreek, and Costus speciosus. Diosgenin suppresses phospho-Src (Tyr416) levels in a time-dependent manner [61]. Hence, our results strongly suggest that the inhibition of the critical regulatory enzyme Src by DT-13 potentially contributes to inhibition of ECs adhesion by inhibiting the NF-κB and MAPK pathways and the subsequent expression of adhesion molecules.
Notably, ruscogenin, the sapogenin of DT-13, exhibits analogous anti-inflammatory effects in vivo or in vitro on certain targets, such as ICAM-1 and NF-κB [62-63]. In addition, DT-13 exhibits significant anti-inflammatory and cardioprotective activities in vascular disease. These activities suggest that DT-13 potentially affects vascular disease. Moreover, we have demonstrated that DT-13 exhibits low acute toxicity and is genotoxically safe. Ninety days of repeated oral administration of DT-13 at levels of up to 360 mg/kg/day did not cause toxic effects in male or female rats [26]. These results indicated that DT-13 may be safely used as a potential drug for vascular diseases.
In summary, we demonstrate that DT-13, the major active component isolated from Liriope muscari, suppresses the adhesion of TNF-α-stimulated ECs in vitro and in vivo by decreasing ICAM-1 and VCAM-1 expression, which possibly correlates with the inhibition of the NF-κB and p38 MAPK signaling pathways by modulating Src activation. These findings also indicate that NF-κB plays a key role in DT-13-inhibited VCAM-1 and ICAM-1 transcription, and NF-κB binding site is at least one of the potential targets of DT-13. In addition, we demonstrate that DT-13 inhibits TNF-α-induced phosphorylation of p38 but not ERK1/2 or JNK. Our data provide some new insights into the possible mechanism of the effect of DT-13 on cell adhesion and migration, which is important for the onset and progression of cardiovascular diseases, such as atherogenesis. Therefore, DT-13 may serve as a safe candidate drug for vascular disease intervention.
Supplementary Material
Supplementary Figures S1-S3.
Acknowledgements
This study was supported by the Major National Science and Technology Project of China for Significant New Drugs Development (No. 2012ZX09102201-015), the National Natural Science Foundation of China (No.81274131), the General Financial Grant from the China Postdoctoral Science Foundation (No.2012M521150), 2011' Program for Excellent Scientific and Technological Innovation Team of Jiangsu Higher Education, and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
1. Rajendran P, Rengarajan T, Thangavel J. et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9:1057-1069
2. Savoia C, Sada L, Zezza L. et al. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int J Hypertens. 2011;2011:281240
3. Liu YC, Hsieh CW, Weng YC. et al. Sulforaphane inhibition of monocyte adhesion via the suppression of ICAM-1 and NF-kappaB is dependent upon glutathione depletion in endothelial cells. Vascul Pharmacol. 2008;48:54-61
4. Sun B, Zou X, Chen Y. et al. Preconditioning of carbon monoxide releasing molecule-derived CO attenuates LPS-induced activation of HUVEC. Int J Biol Sci. 2008;4:270-278
5. Golias C, Tsoutsi E, Matziridis A. et al. Review. Leukocyte and endothelial cell adhesion molecules in inflammation focusing on inflammatory heart disease. In Vivo. 2007;21:757-769
6. Sen T, Aksu T. Endothelial progenitor cell and adhesion molecules determine the quality of the coronary collateral circulation/Endothelial progenitor cells (CD34+KDR+) and monocytes may provide the development of good coronary collaterals despite the vascular risk factors and extensive atherosclerosis. Anadolu Kardiyol Derg. 2012;12:447
7. Rathouska J, Nemeckova I, Zemankova L. et al. Cell adhesion molecules and eNOS expression in aorta of normocholesterolemic mice with different predispositions to atherosclerosis. Heart Vessels. 2015;30:241-248
8. Tsai HY, Huang PH, Lin FY. et al. Ginkgo biloba extract reduces high-glucose-induced endothelial reactive oxygen species generation and cell adhesion molecule expression by enhancing HO-1 expression via Akt/eNOS and p38 MAP kinase pathways. Eur J Pharm Sci. 2013;48:803-811
9. Kesanakurti D, Chetty C, Bhoopathi P. et al. Suppression of MMP-2 attenuates TNF-alpha induced NF-kappaB activation and leads to JNK mediated cell death in glioma. PLoS One. 2011;6:e19341
10. Kim DS, Kim MS, Kang SW. et al. Pine bark extract enzogenol attenuated tumor necrosis factor-alpha-induced endothelial cell adhesion and monocyte transmigration. J Agric Food Chem. 2010;58:7088-7095
11. Kim CW, Lee TH, Park KH. et al. Human lactoferrin suppresses TNF-alpha-induced intercellular adhesion molecule-1 expression via competition with NF-kappaB in endothelial cells. FEBS Lett. 2012;586:229-234
12. Liang CJ, Lee CW, Sung HC. et al. Magnolol reduced TNF-alpha-induced vascular cell adhesion molecule-1 expression in endothelial cells via JNK/p38 and NF-kappaB signaling pathways. Am J Chin Med. 2014;42:619-637
13. Wan M, Liu J, Ouyang X. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-kappaB pathway. J Periodontal Res. 2015;50:189-196
14. van Vliet C, Bukczynska PE, Puryer MA. et al. Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat Immunol. 2005;6:253-260
15. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012;8:1385-1397
16. Zhu M, Chen D, Li D. et al. Luteolin inhibits angiotensin II-induced human umbilical vein endothelial cell proliferation and migration through downregulation of Src and Akt phosphorylation. Circ J. 2013;77:772-779
17. Liu G, Vogel SM, Gao X. et al. Src phosphorylation of endothelial cell surface intercellular adhesion molecule-1 mediates neutrophil adhesion and contributes to the mechanism of lung inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1342-1350
18. Tamizhselvi R, Koh YH, Sun J. et al. Hydrogen sulfide induces ICAM-1 expression and neutrophil adhesion to caerulein-treated pancreatic acinar cells through NF-kappaB and Src-family kinases pathway. Exp Cell Res. 2010;316:1625-1636
19. Zhao RP, Sun L, Lin SS. et al. The saponin monomer of dwarf lilyturf tuber, DT-13, inhibits angiogenesis under hypoxia and normoxia via multi-targeting activity. Oncol Rep. 2013;29:1379-1386
20. Tao J, Wang H, Zhou H. et al. The saponin monomer of dwarf lilyturf tuber, DT-13, reduces L-type calcium currents during hypoxia in adult rat ventricular myocytes. Life Sci. 2005;77:3021-3030
21. Liu J, Chen T, Yu BY. et al. Ruscogenin glycoside (Lm-3) isolated from Liriope muscari inhibits lymphocyte adhesion to extracellular matrix. J Pharm Pharmacol. 2002;54:959-965
22. Zhang YY, Liu JH, Kou JP. et al. DT-13 suppresses MDA-MB-435 cell adhesion and invasion by inhibiting MMP-2/9 via the p38 MAPK pathway. Mol Med Rep. 2012;6:1121-1125
23. Sun L, Lin SS, Zhao RP. et al. The saponin monomer of dwarf lilyturf tuber, DT-13, reduces human breast cancer cell adhesion and migration during hypoxia via regulation of tissue factor. Biol Pharm Bull. 2010;33:1192-1198
24. Yamawaki H, Kuramoto J, Kameshima S. et al. Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem Biophys Res Commun. 2011;408:339-343
25. Lee YJ, Hwang SM, Yoon JJ. et al. Inhibitory effect of Thuja orientalis on TNF-alpha-induced vascular inflammation. Phytother Res. 2010;24:1489-1495
26. Ma ST, Kou JP, Yu BY. Safety evaluation of steroidal saponin DT-13 isolated from the tuber of Liriope muscari (Decne.) Baily. Food Chem Toxicol. 2011;49:2243-2251
27. Kou JP, Sun Y, Lin Y. et al. Anti-inflammatory activities of aqueous extract from Radix Ophiopogon japonicus and its two constituents. Biol Pharm Bull. 2005;28:1234-1238
28. Yan S, Zhang X, Zheng H. et al. Clematichinenoside inhibits VCAM-1 and ICAM-1 expression in TNF-alpha-treated endothelial cells via NADPH oxidase-dependent IkappaB kinase/NF-kappaB pathway. Free Radic Biol Med. 2015;78:190-201
29. Zhang YY, Han YW, Zhai KF. et al. OphiopogoninD suppresses MDAMB435 cell adhesion and invasion by inhibiting matrix metalloproteinase9. Mol Med Rep. 2015;12:1493-1498
30. Nallasamy P, Si H, Babu PV. et al. Sulforaphane reduces vascular inflammation in mice and prevents TNF-alpha-induced monocyte adhesion to primary endothelial cells through interfering with the NF-kappaB pathway. J Nutr Biochem. 2014;25:824-833
31. Lee W, Ku SK, Min BW. et al. Vascular barrier protective effects of pellitorine in LPS-induced inflammation in vitro and in vivo. Fitoterapia. 2014;92:177-187
32. Wu K, Tian S, Zhou H. et al. Statins protect human endothelial cells from TNF-induced inflammation via ERK5 activation. Biochem Pharmacol. 2013;85:1753-1760
33. Mankan AK, Lawless MW, Gray SG. et al. NF-kappaB regulation: the nuclear response. J Cell Mol Med. 2009;13:631-643
34. Liu G, Place AT, Chen Z. et al. ICAM-1-activated Src and eNOS signaling increase endothelial cell surface PECAM-1 adhesivity and neutrophil transmigration. Blood. 2012;120:1942-1952
35. Fu R, Yan T, Wang Q. et al. Suppression of endothelial cell adhesion by XJP-1, a new phenolic compound derived from banana peel. Vascul Pharmacol. 2012;57:105-112
36. Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292-2301
37. Jayakumar T, Chang CC, Lin SL. et al. Brazilin ameliorates high glucose-induced vascular inflammation via inhibiting ROS and CAMs production in human umbilical vein endothelial cells. Biomed Res Int. 2014;2014:403703
38. Hu X, Sun A, Xie X. et al. Rosuvastatin changes cytokine expressions in ischemic territory and preserves heart function after acute myocardial infarction in rats. J Cardiovasc Pharmacol Ther. 2013;18:162-176
39. Reglero-Real N, Marcos-Ramiro B, Millan J. Endothelial membrane reorganization during leukocyte extravasation. Cell Mol Life Sci. 2012;69:3079-3099
40. Lv YY, Jin Y, Han GZ. et al. Ursolic acid suppresses IL-6 induced C-reactive protein expression in HepG2 and protects HUVECs from injury induced by CRP. Eur J Pharm Sci. 2012;45:190-194
41. Zhao Y, Yuan Z, Liu Y. et al. Activation of cannabinoid CB2 receptor ameliorates atherosclerosis associated with suppression of adhesion molecules. J Cardiovasc Pharmacol. 2010;55:292-298
42. Iiyama K, Hajra L, Iiyama M. et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res. 1999;85:199-207
43. Rebai O, Le Petit-Thevenin J, Bruneau N. et al. In vitro angiogenic effects of pancreatic bile salt-dependent lipase. Arterioscler Thromb Vasc Biol. 2005;25:359-364
44. Dai J, Peng L, Fan K. et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene. 2009;28:3412-3422
45. Wang T, Wu F, Jin Z. et al. Plumbagin inhibits LPS-induced inflammation through the inactivation of the nuclear factor-kappa B and mitogen activated protein kinase signaling pathways in RAW 264.7 cells. Food Chem Toxicol. 2014;64:177-183
46. Hadad N, Tuval L, Elgazar-Carmom V. et al. Endothelial ICAM-1 protein induction is regulated by cytosolic phospholipase A2alpha via both NF-kappaB and CREB transcription factors. J Immunol. 2011;186:1816-1827
47. Uzzo RG, Crispen PL, Golovine K. et al. Diverse effects of zinc on NF-kappaB and AP-1 transcription factors: implications for prostate cancer progression. Carcinogenesis. 2006;27:1980-1990
48. Deregibus MC, Buttiglieri S, Russo S. et al. CD40-dependent activation of phosphatidylinositol 3-kinase/Akt pathway mediates endothelial cell survival and in vitro angiogenesis. J Biol Chem. 2003;278:18008-18014
49. Ho AW, Wong CK, Lam CW. Tumor necrosis factor-alpha up-regulates the expression of CCL2 and adhesion molecules of human proximal tubular epithelial cells through MAPK signaling pathways. Immunobiology. 2008;213:533-544
50. Kang JS, Yoon YD, Han MH. et al. Glabridin suppresses intercellular adhesion molecule-1 expression in tumor necrosis factor-alpha-stimulated human umbilical vein endothelial cells by blocking sphingosine kinase pathway: implications of Akt, extracellular signal-regulated kinase, and nuclear factor-kappaB/Rel signaling pathways. Mol Pharmacol. 2006;69:941-949
51. Wu Z, Xiong Y, Gajanayake T. et al. p38 Mitogen-activated protein kinase is required for glucosamine-induced endothelial nitric oxide synthase uncoupling and plasminogen-activator inhibitor expression. Circ J. 2012;76:2015-2022
52. Byeon SE, Yi YS, Oh J. et al. The role of Src kinase in macrophage-mediated inflammatory responses. Mediators Inflamm. 2012;2012:512926
53. Yang CM, Lee IT, Lin CC. et al. c-Src-dependent MAPKs/AP-1 activation is involved in TNF-alpha-induced matrix metalloproteinase-9 expression in rat heart-derived H9c2 cells. Biochem Pharmacol. 2013;85:1115-1123
54. Abram CL, Courtneidge SA. Src family tyrosine kinases and growth factor signaling. Exp Cell Res. 2000;254:1-13
55. Xu W, Doshi A, Lei M. et al. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629-638
56. Jeong HY, Moh SH, Yang Y. et al. Src and Syk are targeted to an anti-inflammatory ethanol extract of Aralia continentalis. J Ethnopharmacol. 2012;143:746-753
57. Yang Y, Hyun Moh S, Yu T. et al. Methanol extract of Osbeckia stellata suppresses lipopolysaccharide- and HCl/ethanol-induced inflammatory responses by inhibiting Src/Syk and IRAK1. J Ethnopharmacol. 2012;143:876-883
58. Yu T, Lee S, Yang WS. et al. The ability of an ethanol extract of Cinnamomum cassia to inhibit Src and spleen tyrosine kinase activity contributes to its anti-inflammatory action. J Ethnopharmacol. 2012;139:566-573
59. Xu Y, Oliverson BG, Simmons DL. Trifunctional inhibition of COX-2 by extracts of Lonicera japonica: direct inhibition, transcriptional and post-transcriptional down regulation. J Ethnopharmacol. 2007;111:667-670
60. Wang Z, Jiang W, Zhang Z. et al. Nitidine chloride inhibits LPS-induced inflammatory cytokines production via MAPK and NF-kappaB pathway in RAW 264.7 cells. J Ethnopharmacol. 2012;144:145-150
61. Li F, Fernandez PP, Rajendran P. et al. Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett. 2010;292:197-207
62. Huang YL, Kou JP, Ma L. et al. Possible mechanism of the anti-inflammatory activity of ruscogenin: role of intercellular adhesion molecule-1 and nuclear factor-kappaB. J Pharmacol Sci. 2008;108:198-205
63. Guan T, Liu Q, Qian Y. et al. Ruscogenin reduces cerebral ischemic injury via NF-kappaB-mediated inflammatory pathway in the mouse model of experimental stroke. Eur J Pharmacol. 2013;714:303-311
Author contact
![]() Corresponding authors: Dr. Boyang Yu & Dr. Junping Kou, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China, Pharmaceutical University, 639 Longmian Road, Nanjing 211198, China. Tel&Fax: +86- 25- 86185158; E-mail: boyangyu59com& junpingkouedu.cn
Corresponding authors: Dr. Boyang Yu & Dr. Junping Kou, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China, Pharmaceutical University, 639 Longmian Road, Nanjing 211198, China. Tel&Fax: +86- 25- 86185158; E-mail: boyangyu59com& junpingkouedu.cn