Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(10):1140-1149. doi:10.7150/ijbs.12657 This issue Cite
Research Paper
Activation of ER Stress and Autophagy Induced by TDP-43 A315T as Pathogenic Mechanism and the Corresponding Histological Changes in Skin as Potential Biomarker for ALS with the Mutation
Xuejing Wang1*, Shuang Zhou2*, Xuebing Ding1*, Mingming Ma3, Jiewen Zhang3, Yue Zhou4, Erxi Wu2 ![]() , Junfang Teng1
, Junfang Teng1 ![]()
1. Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, 450052, China
2. Department of Pharmaceutical Sciences, North Dakota State University, Fargo, ND, 58105, USA
3. Department of Neurology, People's Hospital of Zhengzhou University, Zhengzhou, Henan, 450003, China
4. Department of Statistics, North Dakota State University, Fargo, ND, 58105, USA
* Equal contribution
Received 2015-5-10; Accepted 2015-6-18; Published 2015-7-21
Abstract

TAR DNA binding protein 43 (TDP-43) A315T mutation (TDP-43A315T) has been found in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) as a disease causing mutation with enhanced protein aggregation, formation of protease-resistant fragments, and neurotoxicity. However, the molecular mechanisms for its pathogenic effects are largely unknown. In this study, we demonstrate that TDP-43A315T enhanced neuronal toxicity via activating endoplasmic reticulum (ER) stress-mediated apoptosis in SH-SY5Y cells. Moreover, autophagy was activated by overexpression of TDP-43A315T in a self-defensive manner to decrease neuronal toxicity. Inhibition of autophagy attenuates TDP-43A315T induced neuronal cell death. Furthermore, the expression levels of TDP-43, ER chaperone 78 kDa glucose-regulated protein (GRP-78), and autophagy marker microtubule-associated protein 1A/1B-light chain 3 (LC3) in the skin tissues from ALS patients with TDP-43A315T mutation were markedly higher than those from the healthy control. Thus, our findings provide new molecular evidence for TDP-43A315T neuropathology. In addition, the pathological change in the skin tissues of the patients with TDP-43A315T mutation can be used as a quick diagnostic biomarker.
Keywords: TDP-43A315T, Endoplasmic reticulum stress, Autophagy, Amyotrophic lateral sclerosis
Introduction
TDP-43 is a highly conserved and ubiquitously expressed DNA binding protein with multiple molecular functions including transcriptional regulation, alternative splicing, and RNA stabilization [1, 2]. So far, a few mutations have been identified in patients with amyotrophic lateral sclerosis (ALS) or frontotemporal lobar degeneration (FTLD) [3-6]. Among them, the Ala315Thr missense mutation of TARDBP (TDP-43A315T) gene which is located in a highly conserved region of exon 6, identified by Gitcho and colleagues in 2008 [7], is a very common one [3-6]. Overexpression of TDP-43A315T in cultured cells shows severe neuronal degeneration [8-15]. In addition, TDP-43A315T mice was found to develop features of ALS and FTLD [15] and exhibit motor dysfunctions and impaired learning and memory abilities during ageing [16]. However, despite the established link between TDP-43A315T and neurodegenerative diseases and the neurotoxicity of TDP-43A315T, the molecular underpinnings of the neuropathology of this mutation have yet to be explored.
Endoplasmic reticulum (ER) stress and autophagy are two critical players in the pathogenesis of ALS [17-21] and both have been recently correlated with TDP-43 proteinopathy [22-27]. ER is a vital cellular organelle required for protein folding and processing. Accumulation of unfolded or misfolded proteins within the ER results in ER stress triggers the unfolded protein response (UPR). The activation of UPR initially leads to adaptation and safeguarding cellular survival, while if homeostasis is disrupted, apoptosis and elimination of the abnormal cells will be initiated [28]. It has been demonstrated that overexpression of TDP-43 or its C-terminal fragments induced neuronal toxicity via activating ER stress [22, 23, 29]. Moreover, the ER chaperone protein disulphide isomerase (PDI) was found to be co-localized with TDP-43 in motor neurons in a greater proportion in TDP-43A315T transgenic mice compared with non-transgenic mice [22], suggesting the implication of ER stress in TDP-43A315T neuropathology. Similar to ER stress, autophagy being an intracellular lysosomal degradation process essentially associated with neuronal homeostasis, has both pro-death and pro-survival functions [30]. Previous studies suggested the existence of a feedback regulatory loop between TDP-43 and autophagy manifested by impairment of the autophagy resulted from the depletion of TDP-43 [24] while the induction of autophagy improved TDP-43 clearance and neuron survival [25-27]. However, the relationship between TDP-43A315T and autophagy as well as ER stress has not been established yet.
In this study, we demonstrate that TDP-43A315T induced neuronal toxicity via activating ER stress-mediated apoptosis in SH-SY5Y cells. Autophagy was enhanced by the expression of TDP-43A315T to attenuate neuronal toxicity in SH-SY5Y cells. More important, levels of ER stress protein marker 78 kDa glucose-regulated protein (GRP-78) and autophagy marker microtubule-associated protein 1A/1B-light chain 3 (LC3) were elevated in patients with TDP-43A315T mutation compared with the healthy control. Our findings elucidate the precise mechanism underlying the neuronal toxicity of TDP-43A315T, and may offer disease-specific biomarkers of skin tissue for early diagnosis for patients with TDP-43A315T mutation.
Materials and Methods
Cell culture, transfections, and chemicals
SH-SY5Y cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (GIBCO, USA) supplemented with 15% newborn calf serum (GIBCO, USA), 100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen, USA), at 37°C in a humidified incubator of 5% CO2. The mammalian expression plasmid pEGFP-TDP-43 and pEGFP-TDP-43A315T were gifts provided by Professor Guanghui Wang (Soochow University, China). The mammalian expression plasmids HA-TDP-43 and HA-TDP-43A315T were subclones of PCR products. Transfections were performed using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instructions. 3-Methyladenine (3-MA) was purchased from Sigma, USA. Plasmids were transiently transfected by Lipofectamine 2000 when the SH-SY5Y cells density was up to 40%. Briefly, the plasmids and Lipofectamine 2000 were premixed in OPTI-medium (GIBCO, USA) for 30 min and then applied to the cells. After transfection for 6 h, the medium was replaced with fresh medium containing 15% FBS, and cells were treated for another 24 h and harvested.
Immunoblotting
Immmunoblotting was performed as described before [31]. Briefly, whole cell lysates were prepared using TSPI buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mg/ml aprotinin, 10 mg/ml leupeptin, 0.5 mM Pefabloc SC, 10 mg/ml pepstatin, and 1% NP-40). The samples were subjected to SDS-PAGE. After transferred to nitrocellulose membranes, blots were blocked with 5% nonfat dry milk in TBST (0.25% Triton X-100 in PBS, pH 7.4) for 30 min, and then incubated with primary antibodies overnight at 4°C. After washing 3 times in TBST, the membrane was incubated with anti-rabbit IgG (Cell Signaling Technology, USA) or anti-mouse IgG (Cell Signaling Technology, USA) for 1 h. Membranes were washed three times and proteins were visualized after ECL (Pierce Chemical, USA) treatment. The primary antibodies used were rabbit polyclonal anti-Bcl-2 antibody (Santa Cruz Biotechnology, USA), rabbit polyclonal anti-Bcl-xL antibody (Cell Signaling Technology, USA), rabbit polyclonal anti-caspase-3 (Cell Signaling Technology, USA), rabbit polyclonal anti-caspase-9 (Abcam, UK), mouse monoclonal anti-CHOP antibody (Santa Cruz Biotechnology, USA), rabbit polyclonal anti-GRP-78 (Abcam, UK), rabbit polyclonal anti-phospho-eIF2α antibody (Cell Signaling Technology, USA), rabbit polyclonal anti-caspase-12 antibody (Abcam, UK), mouse monoclonal anti-Beclin-1 antibody (Cell Signaling Technology, USA), rabbit polyclonal anti-LC3 antibody (Cell Signaling Technology, USA), and rabbit monoclonal anti-GAPDH antibody (Cell Signaling Technology, USA).
MTT and LDH assays
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay was performed as previously described [32] to evaluate cell viability. After transfection with pEGFP, pEGFP-TDP-43 or pEGFP-TDP-43A315T for 72 h, 0.5 mg/ml MTT (Sigma-Aldrich, USA) was added to each well at 37 °C for 2 h. The formed formazan was dissolved in DMSO, and colorimetric determination was performed at 540 nm. Lactate dehydrogenase (LDH) activity was determined by using a commercial kit (Sigma-Aldrich, USA).
Flow cytometry assay
After transfection with HA, HA-TDP-43 or HA-TDP-43A315T for 72 h, cells washed twice with 1×PBS were double-stained with Annexin V conjugated to FITC and PI, using Annexin V-FITC apoptosis detection kit (Sigma-Aldrich, USA) according to the manufacturer's instruction. Samples were analyzed on a Cytomics FC 500 flow cytometer (Beckman Coulter, USA).
Loading of lysotracker red and MDC
Cultured cells were incubated with LysoTracker (Molecular Probes, USA) for 30 min. Each well was then washed three times with DMEM, and fixed with 2% paraformaldehyde for 10 min at 4°C. The red fluorescence of LysoTracker was visualized using a Nikon Labphoto-2 fluorescence microscope.
0.05 mM monodansylcadaverine (MDC) (Sigma-Aldrich, USA) were added to cultured cells at 37°C for 1 h, and the changes of fluorescence were detected by Nikon Labphoto-2 fluorescence microscope at excitation wave length 380 nm with emission filter 525 nm.
Immunohistochemistry
Skin tissue sections were washed in 1×PBS for three times, and pre-treated with H2O2 for 5 min at room temperature. After blocking the non-specific binding sites using 1×PBS containing 2% BSA, the skin tissue sections were incubated with the primary antibodies (anti-TDP-43 rabbit antibody or anti-GRP-78 rabbit antibody or anti-LC3 rabbit antibody) overnight at 4°C. Following three washes with 1×PBS containing 0.3% triton-100, a biotinylated anti-rabbit IgG secondary antibody (Vector Laboratories, USA) was applied to the skin tissue sections for 2 h at room temperature. The signal was visualized using ABC reagent from the Vector Laboratories by following the company's instructions. Images were captured by using Nikon Labphoto-2 fluorescence microscope. The human subject studies were approved by the ethical standards committee of Zhengzhou University. Written informed consent for skin biopsy was obtained from all patients and healthy individuals participating in the study.
Statistical analysis
All statistical analyses were performed using SPSS statistical software package (SPSS version 8.0). Data were shown as mean±SD. A p value less than 0.05 was regarded for statistical significance.
Results
TDP-43A315T induces severer neuronal toxicity than wild-type TDP-43
To determine whether TDP-43A315T induces neuronal toxicity, we selected SH-SY5Y cells as an in vitro model of neuronal cells. SH-SY5Y cells were transiently transfected with pEGFP-TDP-43 or pEGFP-TDP-43A315T or pEGFP vectors. We examined the expression levels of both anti-apoptotic proteins Bcl-2 and Bcl-xL and pro-apoptotic proteins cleaved caspase-3 and cleaved caspase-9 by using Western blotting analysis (Figure 1A, B). The results show that the levels of anti-apoptotic proteins Bcl-2 and Bcl-xL were markedly suppressed, while the levels of the pro-apoptotic cleaved caspase-3 and cleaved caspase-9 were noticeably up-regulated in cells overexpressing TDP-43 or TDP-43A315T compared with control.
Expression of TDP-43A315T induces neuronal cell death. (A) Western blotting was performed using lysates of SH-SY5Y cells following transfection of pEGFP, pEGFP-TDP-43, or pEGFP-TDP-43A315T for 72 h. Each sample was probed with the indicated antibodies. (B) Quantification showing significant increase in cleaved caspase-3 and cleaved caspase-9 levels in pEGFP-TDP43 and pEGFP-TDP-43A315T transfected cells in comparison to pEGFP transfected cells, whereas expression levels of Bcl-2 and Bcl-xL were significantly decreased. Values shown are the mean ± SD from three experiments. (C) SH-SY5Y cells viability was measured by MTT assay after cells were transfected with the indicated constructs. Values shown are the mean ± SD from three experiments. (D) The levels of LDH released into the media were measured 72 h after cells were transfected with the indicated constructs. Values shown are the mean ± SD from three experiments. (E) Neuronal apoptosis was detected by Annexin V-FITC/PI after cells were transfected with the indicated constructs. Values shown are the mean ± SD from three experiments. Level of statistical significance: *p < 0.05, ** p < 0.01.
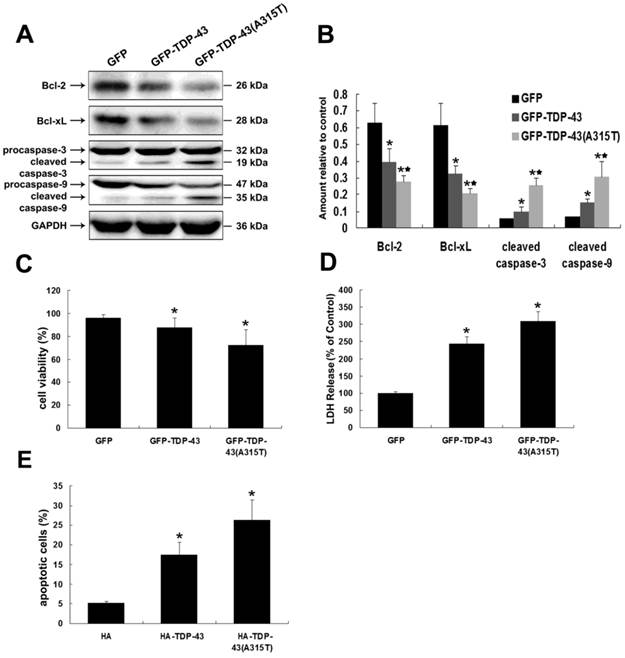
The neuronal toxicity of TDP-43 and TDP-43A315T was further confirmed by MTT and LDH assays. As shown in Figure 1C, overexpression of GFP-TDP-43 or GFP-TDP-43A315T evidently suppressed the cell viability of SH-SY5Y cells compared with pEGFP transfected control cells. Similarly, LDH levels were increased in cells expressing GFP-TDP-43 or GFP-TDP-43A315T compared with control samples (Figure 1D). Apoptosis was further analyzed using annexin V/PI staining. Based on quantitative analysis, percentage of apoptotic cells in these transfected with HA, HA-TDP-43, and HA-TDP-43A315T were 5.1%, 17.41%, and 26.35%, respectively (Figure 1E). Furthermore, TDP-43A315T induced severer neuronal damage than TDP-43 in both cell viability and percentage of apoptotic cells. These findings suggest that TDP-43A315T induces neuronal toxicity via activating apoptosis and inhibiting cell viability.
Overexpression of TDP-43A315T induces ER stress-associated apoptosis in neuronal cells
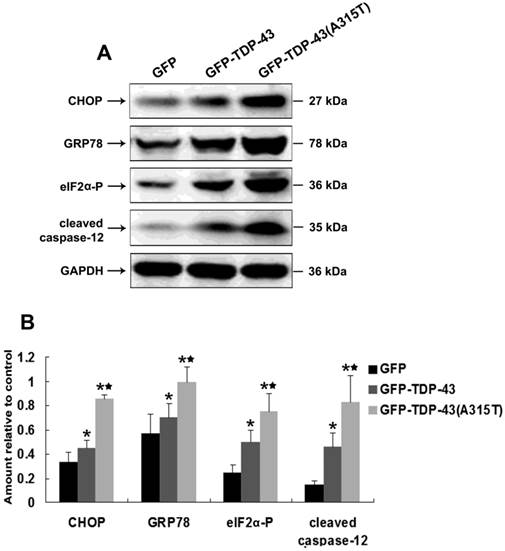
ER stress was found to be a key player in the pathogenesis of ALS [17, 18, 20] and implicated with TDP-43 proteinopathy [22, 23]. To examine whether TDP-43A315T can induce ER stress, SH-SY5Y cells were transiently transfected with pEGFP-TDP-43, pEGFP-TDP-43A315T or pEGFP vectors. Then, the expression levels of several key mediators of ER stress, including ER stress-mediated cell death regulator C/EBP homologous protein (CHOP) [33] and ER chaperone GRP-78 [34], and the levels of ER stress responsive phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (eIF2α) [35] and activation of pro-apoptotic caspase-12 [36] were examined by Western blotting. Our results show that the expression of CHOP and GRP-78 was markedly up-regulated and the levels of phosphorylation of eIF2α and cleaved caspase-12 were significantly increased by overexpression of TDP43 or TDP-43A315T, especially in the cells expressing TDP-43A315T (Figure 2A, B). These data demonstrate that TDP-43A315T induced neuronal cell death is associated with the activation of ER stress-associated apoptosis.
Overexpression of TDP-43A315T induces ER stress. (A) Western blotting was performed using lysates of SH-SY5Y cells following transfection of pEGFP, pEGFP-TDP43, or pEGFP-TDP-43A315T for 72 h. Each sample was probed with the indicated antibodies. (B) Quantification showing significant increase in CHOP, GRP-78, phosphorylated eIF2α, and cleaved caspase-12 levels in pEGFP-TDP43 and pEGFP-TDP-43A315T transfected cells in comparison to pEGFP-transfected cells. Values shown are the mean ± SD from three experiments. Level of statistical significance: *p < 0.05, ** p < 0.01.
Autophagy attenuates TDP-43A315T induced neuronal cell death
To determine the roles of TDP-43A315T in autophagic activity, we examined the expression levels of macroautophagic markers, LC3 and Beclin-1, using Western blotting analysis in cells transfected with pEGFP-TDP-43, pEGFP-TDP-43A315T or pEGFP vectors (Figure 3A, B). Our results show that the expression of Beclin-1 and the conversion from LC3 I to LC3 II were increased by overexpression of TDP-43 and TDP-43A315T (Figure 3A, B), especially in cells expressing GFP-TDP-43A315T.
Inhibition of autophagy enhances TDP-43A315T induced neuronal cell death. (A) Western blotting was performed using cell lysates of SH-SY5Y cells following transfection of pEGFP, pEGFP-TDP-43, or pEGFP-TDP-43A315T for 72 h. Each sample was probed with the indicated antibodies. (B) Quantification showing significant increase in LC3 II/LC3 I ratio and Beclin 1 levels in pEGFP-TDP-43 and pEGFP-TDP-43A315T transfected cells in comparison to pEGFP-transfected cells. Values shown are the mean ± SD from three experiments. (C) SH-SY5Y cells viability was measured by MTT assay in cells treated with 3-MA. 3-MA treatment enhanced TDP43 and TDP-43A315T induced neuronal death. Values shown are the mean ± SD from three experiments. (D) The release of LDH was used as an indicator of neuronal toxicity following 3-MA treatment. 3-MA treatment caused increase of LDH release in cells overexpressing TDP-43 and TDP-43A315T. Values shown are the mean ± SD from three experiments. (E) Neuronal apoptosis was detected by Annexin V-FITC/PI after cells treated with 3-MA. Values shown are the mean ± SD from three experiments. Level of statistical significance: *p < 0.05, ** p < 0.01. (F) Autophagy was evaluated by fluorescence microscopy with Lysotracker and MDC staining in HA-TDP43 and HA-TDP-43A315T transfected cells.
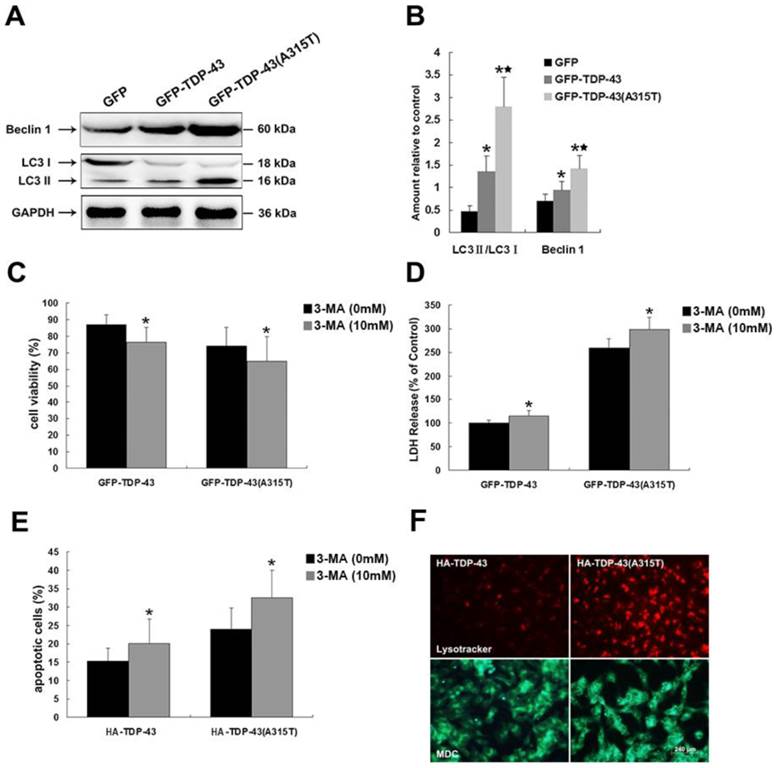
To elucidate the functions of autophagy in TDP-43A315T induced neuronal cell death, we treated SH-SY5Y cells expressing pEGFP-TDP-43, pEGFP-TDP-43A315T or HA-TDP-43, HA-TDP-43A315T with autophagy inhibitor 3-MA. As shown in Figure 3C, 3-MA treatment suppressed the viability of TDP-43 and TDP-43A315T expressing SH-SY5Y cells shown by MTT assay. Similarly, 3-MA treatment increased LDH release (Figure 3D). The percentage of apoptotic cells was increased by 3-MA in cells expressing HA-TDP-43 or HA-TDP-43A315T (Figure 3E). Furthermore, the staining of the autolysosome markers LysoTracker Red and MDC confirmed the activation of autophagy in SH-SY5Y cells transfected with HA-TDP-43 or HA-TDP-43A315T, while the stronger activation of autophagy was induced by HA-TDP-43A315T in comparison to that by HA-TDP-43 (Figure 3F). Taken together, these data indicate that the activation of autophagy is involved in a self-defensive mechanism for neuron survival in cells overexpressing TDP-43 or TDP-43A315T, especially in neuronal cells overexpressing TDP-43 A315T.
Patients with TDP-43A315T mutation have elevated levels of TDP-43 and protein markers for ER stress and autophagy in the skin tissue
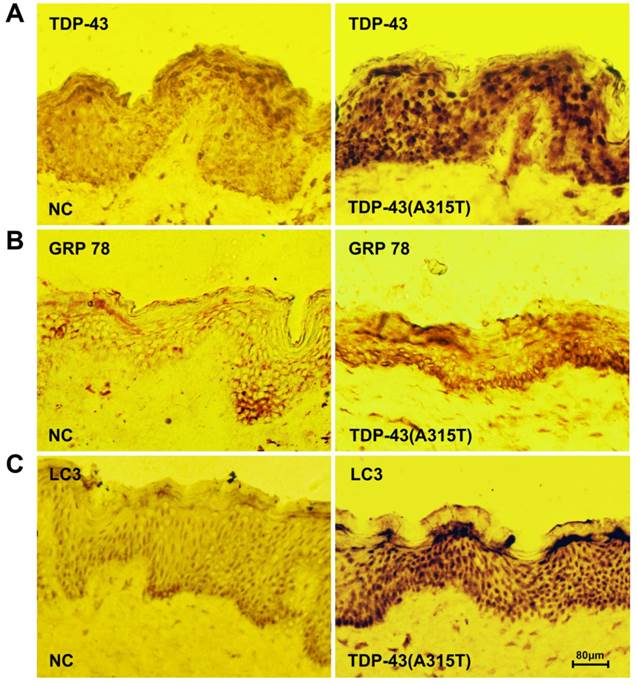
To further determine the roles of TDP-43A315T in ALS pathology, we analyzed the skin biopsy from ALS patients harboring TDP-43A315T and healthy controls. We found that an elevated level of TDP-43 expression was detected in patients with TDP-43A315T mutation compared with control samples (Figure 4A). To analyze the impact of ER stress and autophagy in TDP-43A315T causing disease progression and pathogenesis, we next examined the levels of ER chaperone GRP-78 and macroautophagic marker LC3, in ALS patients using immunohistochemistry. Interestingly, the levels of both GRP-78 (Figure 4B) and LC3 (Figure 4C) were dramatically increased in skin tissue of patients with TDP-43A315T compared with healthy control. These results confirmed the close relationship of TDP-43A315T with ER stress and autophagy in ALS pathology, and suggest that expression levels of TDP-43, GRP-78 and LC3 in skin tissue as potential disease-specific biomarkers for early diagnosis for patient with TDP-43A315T mutation.
Elevated levels of TDP-43 and protein markers for ER stress and autophagy in the skin tissue of patients with TDP-43A315T mutation. (A) Immunochemistry staining for the expression of TDP-43 in skin tissue from healthy individuals (left) and patients harboring TDP-43A315T mutation (right). (B) Immunochemistry staining for the expression of GRP-78 in skin tissue from healthy individuals (left) and patients harboring TDP-43A315T mutation (right). (C) Immunochemistry staining for the expression of LC3 in skin tissue from healthy individuals (left) and patients harboring TDP-43A315T mutation (right).
Discussion
Missense mutations in TDP-43 have been associated with TDP-43 proteinopathies in ALS and FTLD, and the A315T mutant has been identified as an ALS causing mutation with enhanced protein aggregation and neurotoxicity to neurons [3-6, 37-40]. However, the molecular mechanism of the TDP-43A315T neuropathology is not clear. In this study, we demonstrate that TDP-43A315T induces enhanced neuronal toxicity via activating ER stress-mediated apoptosis in SH-SY5Y cells. Autophagy was activated by the expression of TDP-43A315T to attenuate neuronal toxicity. In addition, ER stress protein marker GRP-78 and autophagy marker LC3 were observed in elevated levels in the skin tissue from patients with TDP-43A315T mutation, suggesting a diagnostic value of these molecules. Our findings, for the first time, reveal the roles of both ER stress and autophagy in TDP-43A315T neuropathology.
One of the common features shared by several neurodegenerative disorders is the accumulation of protein aggregates and the inclusions containing misfolded proteins, which is also a common trigger for ER stress [28, 41, 42]. In fact, the frequent observation of ER stress activation and its crosstalk with other important processes implicated in neurodegenerative diseases, including oxidative stress, mitochondrial-mediated apoptosis, and autophagy, imply an essential role of ER stress in neurodegeneration [19, 43-45]. Our data showing the significant up-regulated levels of phosphorylated eIF2α in TDP-43A315T expressing neuronal cells indicates that the protein kinase RNA-like endoplasmic reticulum kinase (PERK) in ER stress pathway is activated by TDP-43A315T. When the UPR fails to rescue the ER stress and to restore cell homeostasis, cell death is initiated by the ER stress specific pro-apoptotic proteins and caspases, and the interaction between the ER stress pathways and the mitochondria apoptotic pathway [43, 45]. One major pro-apoptotic protein for ER stress activated cell death is CHOP, whose expression level is substantially stimulated by activation of the PERK and ATF6 branches of the UPR [33, 46]. In this study, we observe significant increase of CHOP expression in neuronal cells expressing TDP-43 or TDP-43A315T compared with control cells, with the highest level of CHOP in TDP-43A315T cells (Figure 2A, B), which reveals the involvement of CHOP in TDP-43A315T induced neuronal cell death. Whether CHOP is activated via the PERK pathway only or its combination with ATF6 pathway requires further investigation.
In the current work, we have also analyzed whether and how caspases, well-known pro-apoptotic components, were involved in TDP-43A315T associated ER stress induced cell death. ER stress has been reported to increase cytosolic calcium levels and trigger the activation of calpains, which subsequently cleave Bcl-XL and activates caspase-12 [47]. The cleaved caspase-12 further stimulates the activation of caspase-9, which in turn activates caspase-3 and lead to apoptosis [48]. The association of TNF receptor-associated factor 2 (TRAF2) with phosphorylated IRE1 also results in the cleavage of caspase-12 [49]. These are in line with our findings that TDP-43A315T induced increased levels of cleaved caspase-12, cleaved caspase-3, and cleaved caspase-9, and down-regulated pro-apoptotic Bcl-2 family proteins. These data demonstrate that TDP-43A315T induces neuronal cell death via ER stress.
Autophagy has been shown as an essential process for neuronal homeostasis, and may be neuro-protective in some setting [50-52], or if excessively induced or deregulated, can lead to autophagic cell death [53]. In this study, to further evaluate the role of autophagy in TDP-43A315T neuropathology, SH-SY5Y cells were treated with an autophagy inhibitor. Our results showed that overexpression of TDP-43 or TDP-43A315T induces macroautophagy and the inhibition of autophagy enhances TDP-43 or TDP-43A315T induced neuronal cell death. Our data together with previous observations indicate that activation of autophagy is a self-defensive mechanism for neuronal survival and support the recent assumption that autophagy activation might have therapeutic potential for ALS treatment. Future research on the therapeutic implication of autophagy activation and its cross-talk with ER stress in ALS is warranted.
Currently, there is no a simple specific diagnostic test for most motor neuron diseases (MNDs). The various symptoms among different individuals and different stages of the disease, and the similarity of MNDs with other diseases significantly complicate the diagnosis [54]. Identification of specific and simple diagnostic markers for ALS and other MNDs is urgently needed. Previous studies revealing TDP-43A315T as a disease causing mutation for ALS and our finding that ER stress and autophagy were significantly activated by TDP-43A315T in neuronal cells prompted us to evaluate the histological changes in patients harboring TDP-43A315T mutation and possible diagnostic potential of our findings from the current study. By analyzing the skin biopsy from ALS patients harboring TDP-43A315T, we observed elevated levels of TDP-43 expression in patients with TDP-43A315T compared with healthy individuals. The ER chaperone GRP-78 stands out from the many ER-resident proteins because of its protein processing and calcium binding activities and its critical role as a master regulator of early ER stress and UPR signaling [55]. Previous studies have also revealed that the depletion of GRP-78 inhibited both ER stress and nutrient starvation-induced autophagosome formation, and the suppression of GRP-78 mildly increased the association of Class III phosphatidylinositol 3-kinase (PI3KC3) with the macroautophagic marker Beclin1, which suggested GRP-78 as a functional mediator in both ER stress and the intriguingly regulated cross-talk between ER stress and autophagy [55, 56]. LC3, on the other hand, is currently the major protein marker specifically associated with autophagosome in eukaryotes [57]. In our study, we observe that levels of GRP-78 and LC3 in neuronal cells expressing TDP-43A315T were elevated compared with control. More intriguingly, the levels of both GRP-78 and LC3 were dramatically increased in skin tissues from patients with TDP-43A315T mutation compared with healthy individuals (Figure 4). These results confirm our in vitro finding for the activation of both ER stress and autophagy by TDP-43A315T in ALS pathology and further suggest TDP-43, GRP-78, and LC3 as potential diagnostic markers for ALS patients. Future clinical studies with larger amount of patients to further evaluate the diagnostic potential of these markers involved in the ER stress and autophagy pathways are warranted and would be beneficial for the disease diagnosis and therapeutics.
Conclusions
In summary, our results demonstrate that TDP-43A315T induced neuronal death via ER stress-mediated apoptosis. In addition, autophagy was activated by the expression of TDP-43A315T in a self-defensive manner to attenuate neuronal toxicity. Furthermore, the expression levels of ER stress protein marker GRP-78 and autophagy marker LC3 were elevated in the skin tissue from patients with TDP-43A315T mutation compared with control. Our findings provide a therapeutic rationale of suppressing ER stress and enhancing autophagy for patients with ALS and suggest the potential of TDP-43, GRP-78, and LC3 as biomarkers in skin tissue for ALS diagnosis.
Acknowledgements
This work was supported by project grants from the National Natural Science Foundation of China (No. 81471307, 81301086, 81100881, and 81100949) and the Youth Innovation Fund of The First Affiliated Hospital of Zhengzhou University (Xuejing Wang and Xuebing Ding) as well as 5451 Project of Health Department of Henan Province (201201007 and Xuebing Ding).
Authors' contributions
XW, XD, EW and JT conceived the research design and carried out the experiments, as well as analyzed the data. SZ, XW, MM, EW, and XD wrote, reviewed and/or revised this manuscript. MM, JY, JZ, YZ, and JT provided administrative, technical, and material support. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337-43
2. Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584-96
3. Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P. et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470-3
4. Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ. et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843-7
5. Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S. et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat. 2009;30:E974-83
6. Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C. et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 2010;13:509-17
7. Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D. et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535-8
8. Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E. et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010;19:3206-18
9. Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P. et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19:671-83
10. Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413-24
11. Laird AS, Van Hoecke A, De Muynck L, Timmers M, Van den Bosch L, Van Damme P. et al. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One. 2010;5:e13368
12. Kabashi E, Daoud H, Riviere JB, Valdmanis PN, Bourgouin P, Provencher P. et al. No TARDBP mutations in a French Canadian population of patients with Parkinson disease. Arch Neurol. 2009;66:281-2
13. Vaccaro A, Patten SA, Ciura S, Maios C, Therrien M, Drapeau P. et al. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS One. 2012;7:e42117
14. Vaccaro A, Tauffenberger A, Aggad D, Rouleau G, Drapeau P, Parker JA. Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. PLoS One. 2012;7:e31321
15. Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809-14
16. Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J. et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134:2610-26
17. Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J. et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152-65
18. Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400-7
19. Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R. et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294-306
20. Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627-36
21. Cheung ZH, Ip NY. Autophagy deregulation in neurodegenerative diseases - recent advances and future perspectives. J Neurochem. 2011;118:317-25
22. Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA. et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One. 2013;8:e81170
23. Vaccaro A, Patten SA, Aggad D, Julien C, Maios C, Kabashi E. et al. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol Dis. 2013;55:64-75
24. Bose JK, Huang CC, Shen CK. Regulation of autophagy by neuropathological protein TDP-43. The Journal of biological chemistry. 2011;286:44441-8
25. Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM. et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677-85
26. Wang X, Fan H, Ying Z, Li B, Wang H, Wang G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neuroscience letters. 2010;469:112-6
27. Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H. et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. 2014;127:1263-78
28. Yoshida H. ER stress and diseases. FEBS J. 2007;274:630-58
29. Vaccaro A, Tauffenberger A, Ash PE, Carlomagno Y, Petrucelli L, Parker JA. TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 2012;8:e1002806
30. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861-73
31. Wang X, Ma M, Teng J, Zhang J, Zhou S, Zhang Y. et al. Chronic exposure to cerebrospinal fluid of multiple system atrophy in neuroblastoma and glioblastoma cells induces cytotoxicity via ER stress and autophagy activation. Oncotarget. 2015May30;6(15):13278-94
32. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55-63
33. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381-9
34. Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373-81
35. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H. et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230-9
36. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98-103
37. Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF. et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193
38. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668-72
39. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C. et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572-4
40. Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X. et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol. 2011;18:822-30
41. Imaizumi K, Katayama T, Tohyama M. Presenilin and the UPR. Nat Cell Biol. 2001;3:E104
42. Zhao L, Longo-Guess C, Harris BS, Lee JW, Ackerman SL. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat Genet. 2005;37:974-9
43. Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608-18
44. Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M. et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111-23
45. Walker AK, Atkin JD. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB life. 2011;63:754-63
46. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R. et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066-77
47. Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887-94
48. Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287-94
49. Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T. et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935-40
50. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585-95
51. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I. et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880-4
52. Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008;1782:691-9
53. Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B. et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11-22
54. McDermott CJ, Shaw PJ. Diagnosis and management of motor neurone disease. BMJ. 2008;336:658-62
55. Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15:1460-71
56. Wirth M, Joachim J, Tooze SA. Autophagosome formation--the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol. 2013;23:301-9
57. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-8
Author contact
![]() Corresponding authors: Junfang Teng: E-mail: dxb515481com, Tel: +0018613838210077 or Erxi Wu: E-mail: Erxi.wuedu, Tel: +0017012317250
Corresponding authors: Junfang Teng: E-mail: dxb515481com, Tel: +0018613838210077 or Erxi Wu: E-mail: Erxi.wuedu, Tel: +0017012317250