Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(11):1248-1256. doi:10.7150/ijbs.13011 This issue Cite
Research Paper
Adapted Resistance to the Knockdown Effect of shRNA-Derived Srsf3 siRNAs in Mouse Littermates
Masahiko Ajiro1, Rong Jia1,3, Rui-Hong Wang2,4, Chu-Xia Deng2,4, Zhi-Ming Zheng1 ![]()
1. Tumor Virus RNA Biology Section, Gene Regulation and Chromosome Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, Maryland 21702, USA
2. Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, MD 20892, USA.
3. Wuhan University School of Stomatology, Wuhan, Hubei, China
4. Faculty of Health Sciences, University of Macau, Macau SAR, China
Received 2015-6-22; Accepted 2015-7-29; Published 2015-9-3
Abstract

Gene silencing techniques are widely used to control gene expression and have potential for RNAi-based therapeutics. In this report, transgenic mouse lines were created for conditional knockdown of Srsf3 (SRp20) expression in liver and mammary gland tissues by expressing Srsf3-specific shRNAs driven by a U6 promoter. Although a small portion of the transgenic mouse littermates were found to produce siRNAs in the targeted tissues, most of the transgenic littermates at two months of age failed to display a knockdown phenotype of Srsf3 expression in their liver and mammary gland tissues where an abundant level of Srsf3 siRNAs remained. We saw only one of four mice with liver/mammary gland expressing Srsf3 siRNA displayed a suppressed level of Srsf3 protein, but not the mRNA. Data indicate that the host resistance to a gene-specific siRNA targeting an essential gene transcript can be developed in animals, presumably as a physiological necessity to cope with the hostile perturbation.
Keywords: Srsf3, shRNAs, transgenic mouse
Introduction
Small interfering RNA (siRNA) has been widely used as a practical tool to induce a gene silencing through the RNA interference (RNAi) pathway. However, it is also reported that some siRNAs lose their gene silencing activity in certain conditions, through a functional suppression of RNAi [1-3]. The suppression of RNAi was initially described in plants, in which plant viruses escape from anti-viral siRNAs produced by host cells through the expression of viral suppressors of RNAi [4, 5]. Later on, a suppression of RNAi was also observed in human papillomavirus-infected cervical cancer cell lines, in which exposure of an shRNA transcribed from an H-1 promoter for a certain period of time renders the shRNA-derived siRNAs ineffective in knocking down the expression of human papillomavirus 16 (HPV16) E7 oncogene or cellular p53 tumor suppressor, in spite of the presence of proper processing and production of normal shRNA-derived siRNAs without any change in the shRNA sequence, suggesting the acquisition of an RNAi resistance in those cell populations [2, 3]. A similar observation was subsequently reported in a lentiviral vector-mediated siRNA delivery system, in which RNAi activity was lost even though a residual amount of the shRNA expression vector was still detectable [6]. Although a detailed mechanism underlying the development of RNAi resistance remains to be understood in animal cells, it has been proposed that a cellular protein at 50-kDa in size binds to siRNA and might be responsible for the resistance program [2]. Alternatively, ADAR1-mediated siRNA editing might contributes to siRNA dysfunction [7].
Although RNA silencing suppression by plant pathogens via viral suppressors of RNA silencing has been known for years [4, 5] and development of RNAi resistance was also observed in animal cells in cultures, there is no report of such siRNA resistance being developed in animals. In this article, we report our observation of RNAi resistance in mouse liver and mammary gland tissues with the conditional expression of shRNA-derived siRNAs specifically targeting serine/arginine-rich splicing factor 3 (Srsf3 [SRp20]), a proto-oncogenic splicing factor[8], in transgenic mouse lines. Liver and mammary gland-specific expression of the Srsf3 siRNA was achieved by albumin promoter (Alb)- and murine mammary tumor virus promoter (MMTV)-dependent Cre-recombinase, respectively. In both cases, the activities of Srsf3 siRNAs, which are highly potent for RNAi induction in cell cultures, were lost in liver and mammary gland tissues at two months of age. Our observation raises a potential caveat in application of siRNA-expression systems in transgenic mice and clinical application of RNAi.
Materials and Methods
Cell culture
NK and 69 cell lines are derived from mouse breast tumor cells with MMTV-c-neu and BRCA1-/-, respectively [9]. NIH3T3 mouse fibroblast cells and HCT116 human colorectal cancer cells are obtained from American Type Culture Collection (Manassas, VA). Cell culture is maintained in 5% CO2 at 37oC. Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT) was used for 69 and NK cells, DMEM with 10% calf serum (HyClone) for NIH3T3, and McCoy's 5a medium (Life Technologies) supplemented with 10% FBS for HCT116. All medium are also supplemented with 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Antibody
Anti-SRSF3 mouse monoclonal antibody (clone 7B4) was purchased from EMD Millipore (Billerica, MA). Anti-SRSF3 rabbit polyclonal antibody (ab125124) was purchased from abcam (Cambridge, MA). Anti-GAPDH rabbit monoclonal antibody (14C10) was purchased from Cell Signaling Technology (Danvers, MA). Anti-β-tubulin mouse monoclonal antibody was purchased from SIGMA Aldrich (St. Louis, MO).
Preparation and transfection of the conditional shRNA expression plasmids
Conditional Srsf3 shRNA expression plasmids, pJR22, pMA13 and pMA14, are prepared from pBS/U6-ploxPneo with ApaI and EcoRI as described previously [10]. shRNA-coding inserts were prepared by annealing oligo DNAs: oJR 102 (5'-GGAACTAGATGGAAGAACAAAGCTTTGTTCTTCCATCTAGTTCCCTTTTTG-3') and oJR 103 (5'-AATTCAAAAAGGGAACTAGATGGAAGAACAAAGCTTTGTTCTTCCATCTAGTTCC-3') for pJR22 by two steps (stick-end and then blunt-end) of ligation; oMA 7 (5'-CTAGATGGAAGAACACTGTTTCAAGAGAACAGTGTTCTTCCATCTAGTTTTTT-3') and oMA 8 (5'-AATTAAAAAACTAGATGGAAGAACACTGTTCTCTTGAAACAGTGTTCTTCCATCTAGGGCC-3') for pMA13 by stick-end ligation; oMA 13 (5'-GCTATTATGGACCACTCAGAATTCAAGAGATTCTGAGTGGTCCATAATAGCTTTTTT-3') and oMA 14 (5'-GCTATTATGGACCACTCAGAATTCAAGAGATTCTGAGTGGTCCATAATAGCTTTTTT-3') for pMA14 by stick-end ligation. For the expression of shRNAs in mouse cell lines, we prepared re-pJR22, re-pMA13 and re-pMA14, which are Cre/loxP recombination products of pJR22, pMA13 and pMA14 produced in Cre-expressing E. coli BNN 132 strain. Plasmid transfection to cell lines was conducted by Lipofectamine 2000 (Life Technologies).
Northern blotting
Northern blotting for small size RNAs were performed as described previously [11]. Briefly, total RNAs were separated in 15% polyacrylamide gel with 7.5 M Urea and transferred to a nylon membrane for small size RNAs. Probe hybridization was carried out for 16 h at 42oC in Super-Hybridization buffer (Sigma Aldrich). oMA23 (5'-GGAACTAGATGGAAGAACA-3', for pJR22 guide-strand siRNA), oMA 25 (5'-CTAGATGGAAGAACACTGT-3', for pMA13 guide-strand siRNA) and oMA 27 (5'-GCTATTATGGACCACTCAGAA-3', for pMA14 guide-strand siRNA) are end-labeled by γ-32P ATP and used as individual probes.
Real-time RT-PCR
Total RNA was extracted by TriPure (Roche diagnostics) from mouse cell cultures or tissues and treated with TURBO DNase (Life Technologies). Then, cDNA was synthesized by MuLV reverse transcriptase (Life Technologies), and applied to real-time RT-PCR with TaqMan probes and primers for Srsf3 and Gapdh (Life Technologies). Relative expression levels of Srsf3 were calculated by ΔΔCt from real-time RT-PCR.
Generation and maintenance of transgenic mice
Transgenic mice for the conditional expression of Srsf3 shRNA was generated by pro-nuclear injection of 1 ng/μl of the linearized ~2.7 kb fragment of pMA14, resulting from Asp718 and AflIII digestion, into oocytes isolated from FVB/N mice. Founder mice are screened by genotyping PCR for tail snips. Resulting shRNA/40+/- strain mice were subsequently crossed with Alb-Cre+/+ or MMTV-Cre+/+ strain mice to obtain shRNA/40+/-:Alb-Cre+/-, shRNA/40-/-:Alb-Cre+/-, shRNA/40+/-:MMTV-Cre+/- and shRNA/40-/-:MMTV-Cre+/- mice. Male and female mice were kept for continuous mating, and the resulting offsprings were genotyped by PCR for DNA from tail snips. For those mice genotyped, penetration rate of shRNA genotype was determined as following: 47% (9/19) for male, 52% (15/29) for female and 50% (24/48) in total for shRNA/40:Alb-Cre mouse strain; 44% (7/16) for male, 41% (9/22) for female and 42% (16/38) in total for shRNA/40:MMTV-Cre mouse strain. Thus, the inheritance of Srsf3 shRNA/40 followed Mendelian manner. A subset of littermates were then analyzed by Western blot, Northern blot, and real-time RT-PCR. All of transgenic mice are maintained in a mixed background of 129, FVB/N, C57Bl/6, and Black Swiss. All animals are maintained and treated according to the protocol approved by NIH. PCR was performed for 100 ng of purified tissue genomic DNA with a primer pair of F1 (oJR120, 5'-CCATCAGAAGCTGACTCTAGC-3') or F2 (oMA94, 5'-GCACAGACTTGTGGGAGAAG-3') and R1 (oJR119, 5'-GCTATGACCATGATTACGCCA-3').
WST-8 cell proliferation assay
WST (Water soluble Tetrazolium salts)-8 cell proliferation assay was performed by Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD) as described previously [12, 13]. Briefly, cells were incubated with 10% WST-8 in culture medium for 40 min at 37oC and the cell culture medium was subjected to 450 nm absorbance measurement to determine cell growth and viability.
Results
Functional validation of Srsf3 shRNA in mouse cells
In order to avoid an embryonic lethality owing to an ubiquitous knockout of Srsf3 [14], we attempted to knockdown Srsf3 in a tissue-specific manner by Cre/loxP system. In this strategy, we apply a conditional shRNA expression system with a U6 promoter, divided into two segments by floxed neor (neomycin-resistant gene) sequence (Fig. 1A) [10, 15, 16]. In this system, ~2 kb of neor insertion disrupts U6 promoter activity in the absence of Cre recombinase. Then, Cre/loxP recombination removes neor to activate U6 promoter and initiate Srsf3 shRNA expression (Fig. 1A). To select an optimal shRNA for Srsf3 knockdown, we constructed three shRNA expression plasmids, pJR22, pMA13 and pMA14, by inserting Srsf3 shRNAs into pBS/U6-ploxPneo plasmid (Fig. 1B)[10] to target a splice junction (pJR22 and pMA13) or exon region (pMA14) of Srsf3 (Fig. 1C) and compared them for their processing efficiencies into siRNAs in mouse cells. In order to achieve shRNA expression in mouse cell lines that do not express Cre recombinase, pJR22, pMA13 and pMA14 were processed for Cre/loxP recombination by transformation of Cre-expressing E. coli BNN 132 strain. The resulting post-recombination forms of individual plasmids purified from the BNN 132 strain were renamed as re-pJR22, re-pMA13 and re-pMA14. Then, we transfected re-pJR22, re-pMA13 and re-pMA14 into NIH3T3 mouse fibroblast and 69 mouse breast cancer cells, with pBS/U6-loxP, a Cre/loxP recombination form of pBS/U6-ploxPneo plasmid [10], as a negative transfection control. Forty-eight h after the transfection, total RNAs were analyzed by Northern blotting for precursor shRNA expression and production of guide-strand siRNA (Fig. 2A). Notably, we found an efficient production of the guide-strand siRNA from re-pMA14 (Fig. 2A, lanes 6 and 11), but not from re-pJR22 (Fig. 2A, lanes 4 and 9) or re-pMA13 (Fig. 2A, lanes 5 and 10). Given an efficient processing of pMA14-derived shRNA, we further examined knockdown effect of Srsf3 following transfection of re-pMA14 in NIH3T3, NK and 69 cells. As a result, we found re-pMA14 transfection consistently suppresses Srsf3 expression at protein (Fig. 2B) and mRNA (Fig. 2C) levels over the pBS/U6-loxP transfected cells. In addition, we also found a significant retardation of cell growth after the re-pMA14 transfection of NK or NIH3T3 cells when compared with the pBS/U6-loxP transfected cells (Fig. 2D), consisting with the previous observations that Srsf3 plays an essential role in cell proliferation [8, 17].
Conditional shRNA expression system targeting Srsf3. (A) A diagram of the conditional shRNA expression system. In this system, U6 promoter is inactivated by the insertion of ~2 kb of floxed neor sequence. Cre/loxP recombination remove the neor sequence and reactivates U6 promoter to initiate transcription of Srsf3 shRNA. SPH, SphI post-octamer homology; OCT, octamer motif; PSE, proximal sequence element; TATA, TATA box. (B) Construction of the conditional shRNA expression plasmids, pJR22, pMA13 and pMA14. Srsf3 shRNA was inserted downstream of the U6-neor in pBS/U6-ploxPneo[10]. Guide- and passenger-strand sequences are indicated by open boxes. Nucleotide positions of Srsf3 target sites of each shRNA are indicated according to Mus musculus Srsf3 mRNA (GenBank: NM_013663.5). The maturation process of transcribed shRNA is illustrated below. (C) Diagram of shRNA target sites on mouse Srsf3 mRNA and comparison with human SRSF3 mRNA sequence (GenBank: NM_003017.4). Plasmid pJR22 and pMA13 are designed to express a shRNA targeting the exon 2 and 3 junction, and pMA14 for targeting the exon 2 of Srsf3. Nucleotides not conserved in human SRSF3 mRNA are underlined.
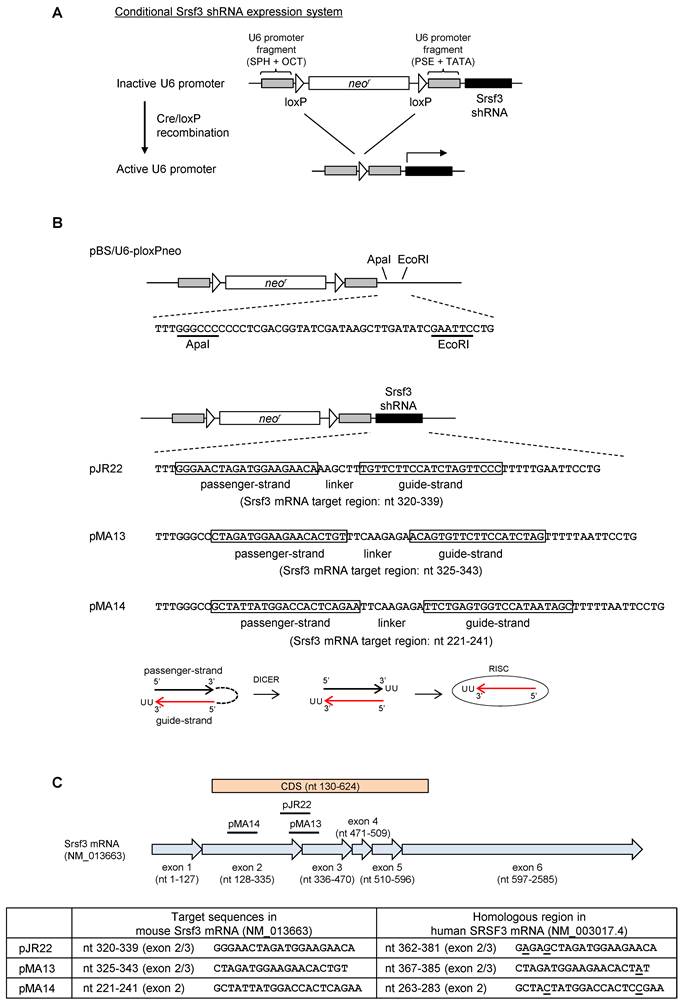
Confirmation of the processing and knockdown effect of Srsf3 from pMA14 in mouse cell lines. (A) NIH3T3 mouse fibroblast and 69 mouse breast cancer cells were transfected with pBS/U6-loxP (Cre/loxP recombined form of the pBS/U6-ploxPneo plasmid [10]), re-pJR22, re-pMA13 and re-pMA14, which are post-recombination forms of pBS/U6-ploxPneo, pJR22, pMA13 and pMA14. 48 h after the transfection, 40 μg of total RNAs were analyzed by Northern blot with a denaturing 15% polyacrylamide gel. Pre-shRNAs and guide-strand siRNAs were detected by pooled 32P-labeled probes complementary to guide-strand siRNA sequences of pJR22, pMA13 and pMA14. Identities of each band are indicated on the right. (B-D) Confirmation of the knockdown effect of Srsf3 shRNA from re-pMA14 in mouse cell lines. NK, NIH3T3 and 69 mouse cells are transfected twice with pBS/U6-loxP or re-pMA14 plasmid for 96 h with an interval of 48 h between transfections, and analyzed by Western blot for protein expression (B), real-time RT-PCR for mRNA expression (C), and WST-8 cell proliferation assay (D). Relative intensity of Srsf3 signal (%) is indicated in (B), with the expression level from pBS/U6-loxP vector-transfected cells set to 100%. Mean ± SD is shown in (C) and (D). *, p<0.05; **, p<0.01 by Student's t-test (n = 3).
Generation of the transgenic mice conditionally expressing Srsf3 shRNAs
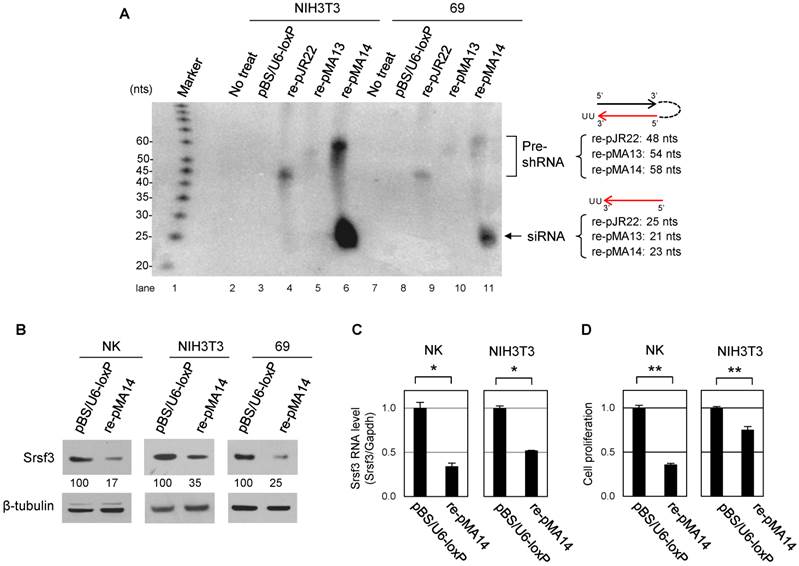
After confirming the function of shRNA derived from pMA14 in mouse cells, we next generated the transgenic mice for its conditional expression. We conducted a pronuclear injection of linearized pMA14 plasmid into FVB/N mouse oocytes, and the founder mice were screened by genotyping PCR with a primer set of F1 and R1 for genomic DNA extracted from tail snips (Fig. 3A). As a result, from 47 mice derived from the injected oocytes, we identified three founder female mice, which we named shRNA/34, shRNA/37 and shRNA/40 (Fig. 3B). In addition, we found the offspring of shRNA/37 founder mouse do not express shRNA even in the presence of Cre recombinase, as discussed later, and only shRNA/40 strain showed both reproductive function and conditional shRNA expression.
Identification of the founder mice for conditional Srsf3 shRNA expression and confirmation of the tissue-specific recombination. (A) Primers (F1, F2 and R1) used for the founder screening and Cre/loxP recombination. (B) PCR screening for 47 mice derived from oocytes with a pronuclear injection of linearized pMA-14. Mouse genomic DNAs from tail snips were analyzed by PCR with a primer set of F1 and R1. Three (#34, #37 and #40) out of 47 mice were identified as the founder mice, which we named as shRNA/34, shRNA/37 and shRNA/40. P, plasmid pMA14 positive control. (C) shRNA/37 and shRNA/40 were bred with Alb-Cre or MMTV-Cre mice, and the resulting Alb-Cre:shRNA/40 (male), MMTV-Cre:shRNA/40 (female), and MMTV-Cre:shRNA/37 (female) were analyzed for the Cre/loxP recombination in individual tissues at 2 months of age. PCR was performed for genomic DNAs extracted from individual tissues with primer sets of F2+R1 to detect a post-recombination form and F1+R1 to detect a pre-recombination form of pBS/U6-ploxPneo-derived pMA14 plasmid, respectively. (D) Summary of reproductive function and Cre-mediated recombination of the founder mice of shRNA/34, shRNA/37 and shRNA/40.
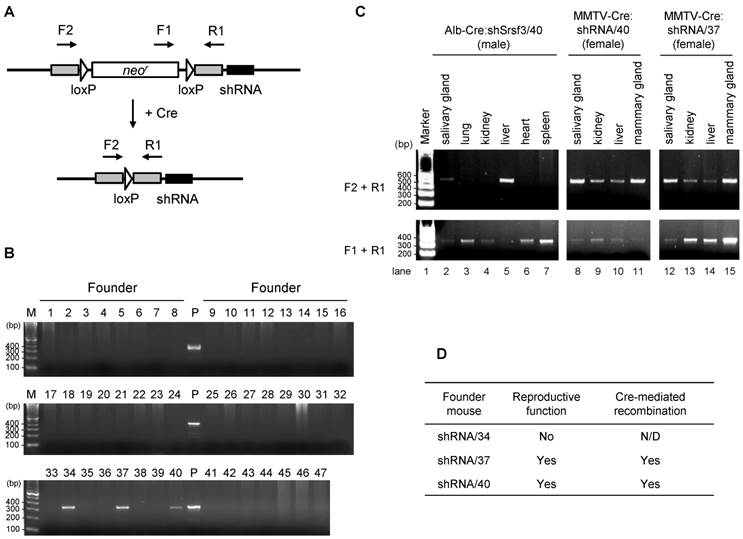
In order to achieve a conditional Srsf3 shRNA expression, subsequent breeding of these founder mice revealed that shRNA/34 failed to be pregnant even after a year of mating period with multiple male mice, suggesting shRNA/34 founder mouse has a defect in reproductive function. However, we had successfully bred shRNA/37 and shRNA/40 strains with albumin promoter- or murine mammary tumor virus promoter-dependent Cre expression strains (Alb-Cre and MMTV-Cre, respectively). We confirmed tissue-specific induction of Cre/loxP recombination in Alb-Cre:shRNA/40, MMTV-Cre:shRNA/40 and MMTV-Cre:shRNA/37 mice by PCR for genomic DNA extracted from individual tissues at 2 months of age (Fig. 3C) by showing that F1 and R1 primers led to detect pre-recombination form of shRNA cassette and F2 and R1 primer pair to detect post-recombination form (Fig. 3A and 3C). Alb-Cre:shRNA/40 mouse showed Cre/loxP recombination predominantly in liver (Fig. 3C, lane 5) and weakly in salivary gland (Fig. 3C, lane 2), but not in lung, kidney, heart or spleen (Fig. 3C, lanes 3, 4, 6 and 7). MMTV-Cre:shRNA/40 and MMTV-Cre:shRNA/37 showed Cre/loxP recombination primarily in mammary gland and salivary gland (Fig. 3C, lanes 8 and 11 and Fig. 3C, lanes 12 and 15), but also in kidney and liver in a weaker extent (Fig. 3C, lanes 9-10 and Fig. 3C, lane 13-14).
In summary, we obtained three founders from 47 screened mice and two of them can be successfully bred with Alb-Cre or MMTV-Cre strain mouse (Fig. 3D) for mouse reproduction.
Development of resistance to Srsf3 knockdown by Srsf3-specific shRNA-derived siRNA in mouse liver and mammary gland tissues
To confirm tissue-specific Srsf3 shRNA expression in Alb-Cre:shRNA/40, Alb-Cre:shRNA/37, MMTV-Cre:shRNA/37, and MMTV-Cre:shRNA/40 mice, total RNA was extracted from individual tissues from 2-month-old mice and Srsf3 shRNA expression was evaluated by Northern blotting with a probe complementary to the guide-strand siRNA from pMA14. In consistent with the presence of efficient Cre/loxP recombination (Fig. 3C), Alb-Cre:shRNA/40 showed Srsf3 siRNA production primarily in liver and weakly in salivary gland, but not in kidney, lung, heart and spleen tissues (Fig. 4A, lanes 2-4, Fig. 4C, lanes 2-7). MMTV-Cre:shRNA/40 showed Srsf3 siRNA expression in salivary gland, kidney, liver and mammary gland (Fig. 4A, lanes 9-12). In contrast, MMTV-Cre:shRNA/37 and Alb-Cre:shRNA/37 did not show Srsf3 siRNA expression in all tissues examined (Fig. 4A, lanes 5-8, and Fig. 4B, lanes 2-9) in spite of successful Cre/loxP recombination in these tissues in MMTV-Cre:shRNA/37 (Fig. 3C) and Alb-Cre:shRNA/37 was positive in genotyping for Alb-Cre. These observations suggest that the shRNA expression construct had integrated into a chromosomal locus which was accessible by Cre recombinase, but was transcriptionally inactive in shRNA/37 strain. Thus, the offsprings of shRNA/37 founder mouse did not express shRNAs even in the presence of Cre recombinase. In the subsequent studies, only the shRNA/40 strain was used for further studies. We investigated the liver-specific Srsf3 shRNA expression in Alb-Cre:shRNA/40 mice (Fig. 4C). In Alb-Cre:shRNA/40 and Alb-Cre male littermates, there was no significant difference in body weight (Fig. 4D), liver weight (Fig. 4E), and liver morphology (Fig. 4F) at 2 months of age. By comparing individual Alb-Cre:shRNA/40 littermates for their expression levels of Srsf3 shRNA, Srsf3 mRNA, and Srsf3 protein in liver tissues (Fig. 4G-I), we confirmed the production of Srsf3-specific siRNAs in Alb-Cre:shRNA/40 littermates (Fig. 4G, lanes 2, 5 and 7), but not in Alb-Cre littermates (Fig. 4G, lanes 3, 4, 6 and 8). However, Srsf3 shRNA expression was found not always being accompanied with a reduced level of Srsf3 protein in these liver tissues.
Conditional expression of Srsf3 shRNAs in transgenic mice and their knockdown efficiency in liver tissues. (A-C) Tissue-specific expression of Srsf3 shRNAs in transgenic mice. Northern blot was conducted for 40 μg of total RNAs extracted from individual tissues from Alb-Cre:shRNA/40 (male) (A, lanes 2-4 and C, lanes 2-7), Alb-Cre:shRNA/37 (male) (B, lanes 2-9), MMTV-Cre:shRNA/37 (female) (A, lanes 5-8) and MMTV-Cre:shRNA/40 (female) (A, lanes 9-12), at 2 months of age. The shRNA and its precursor were detected by a 32P-labeled probe complementary to the guide-strand shRNA from pMA14. Eight μg of total RNA from re-pMA14-transfected NIH3T3 cells served as a positive control (A, lane 13, and B, lane 10). Total RNA was separated by a denaturing 15% polyacrylamide gel. (D-F) Alb-Cre:shRNA/40 (n=3) and Alb-Cre (n=2) male littermates were analyzed for their body weight (D) and liver weight (E) at 2 months of age. Representative pictures of livers from 2-month-old Alb-Cre:shRNA/40 and Alb-Cre male littermates are shown in (F). Scale bar, 1 cm. (G-I) Liver tissues of two-month-old littermates of Alb-Cre:shRNA/40 and Alb-Cre (a, b, c, d, e, f, and g indicate individual mice) were analyzed by Northern blotting for Srsf3 shRNA expression (G), by real-time RT-PCR for Srsf3 mRNA expression (H), and by Western blotting for protein expression (I). Relative intensity (%) of Srsf3 protein signal is indicated in (I), with Srsf3 protein level in Alb-Cre control liver set to 100%, after normalizing with Gapdh. (J) Mammary gland tissues of two-month-old MMTV-Cre:shRNA/40 and adult MMTV-Cre female mice are analyzed by Western blotting for Srsf3 protein expression after normalizing with Gapdh which served as a control for sample loading, with Srsf3 level in MMTV-Cre mammary gland tissues set to 100%. HCT116 cell lysate served as a positive control in (I). Mean±SD is shown in (D), (E) and (H). NS, p>0.05 by Student's t-test.
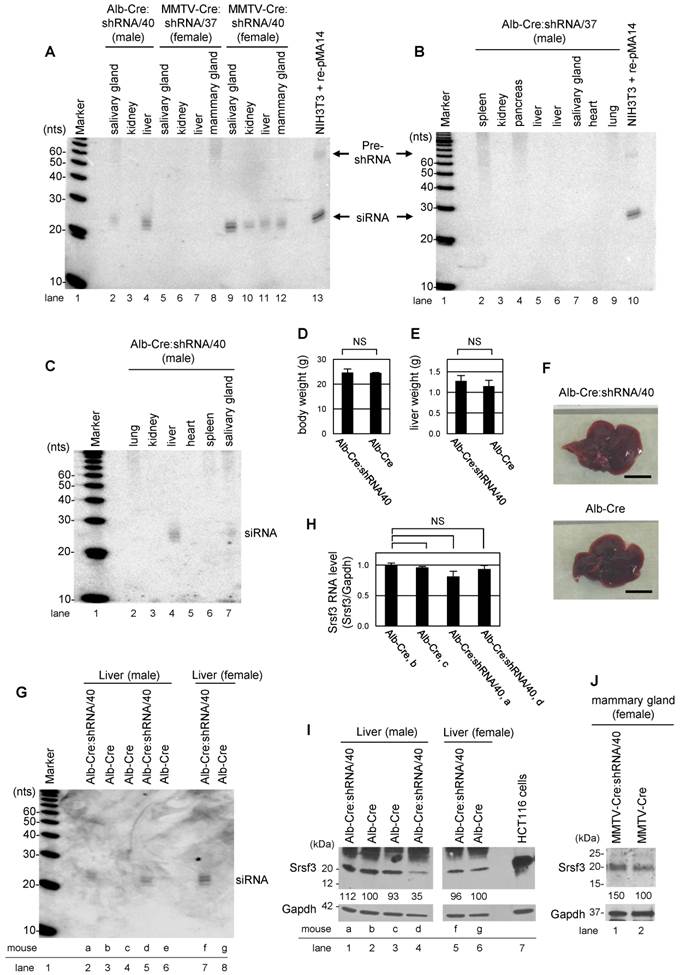
We found that only one of three littermates showed a suppressed level of Srsf3 protein in their liver tissues (Fig. 4I, compare lanes 1 and 5 with lane 4), but this mouse, mouse d, did not have a significant decrease in Srsf3 mRNA level (Fig. 4H). Data suggest that the reduction of Srsf3 protein level in the liver of the mouse d could be due to Srsf3 siRNA-mediated inhibition of Srsf3 protein translation, consisting with the report that siRNAs function as miRNAs [18]. In examination of MMTV-Cre:shRNA/40 littermates, we also failed to detect a reduced level of Srsf3 protein in the Srsf3 siRNA-expressing mammary gland tissues (Fig. 4J).
Discussion
Endogenous production of a gene-specific siRNA via shRNAs expressed from a U6 or H1 promoter has been widely used in various studies [2, 19-21]. It has been thought that the expressed shRNA-derived siRNAs would mediate long-term, stable knockdown of their target transcripts as long as transcription of the shRNAs takes place, whereas exogenous siRNAs introduced into cells lead to only transiently silence of gene expression and their intracellular concentrations are diluted over the course of successive cell divisions [22]. However, many RNA viruses escape RNAi-mediated suppression by counteracting the RNAi machinery through mutation of the targeted region, by encoding viral suppressors or both [3, 23]. In this report, we provided the evidence that the endogenous Srsf3-specific siRNAs derived from shRNAs does not provide long-term, stable knockdown of their target transcripts in liver and mammary gland tissues of transgenic mouse lines at two months of age while transcription of the shRNAs and normal production of the processed siRNAs take place in these tissues. This data is consistent with the findings that endogenous shRNA-derived siRNAs stably expressed in mammalian cells could also lead to only a short-term effect depending on its target transcripts [2, 6]. It has been our notion that shRNA-derived siRNAs targeting an essential gene transcript are more likely ended up with a short-term effect as a result of the host cells battling to survive [2] and adapting to this hostile perturbation by this gene-specific siRNA. As of today, we have no clue on how cells respond initially, but over time, return to non-responding status even when the shRNA-derived siRNAs that caused the initial response, persist in the cells.
Although many RNA viruses escape RNAi-mediated suppression by counteracting the RNAi machinery through mutation of the targeted region, by encoding viral suppressors, or both [3, 5, 23], the adapted resistance to a gene-specific siRNA in mammalian cells is distinguishable from the target region mutation-mediated resistance observed in the RNA viruses. Our previous study showed that neither the targeted viral E7 region, nor shRNA-derived HPV16 E7 siRNAs, displayed any mutation in the cervical cancer cell line where the development of siRNA resistance was observed. Conceivably, the cellular machinery must be involved from the early responsive to the later irresponsive status in the continuing presence of a harmful siRNA to host cells. The process of shRNA into functional siRNA involves the cellular RNAi machinery that naturally processes microRNA (miRNA) production to regulate gene expression by modulating mRNA stability and translation. Therefore, all checkpoints regulating miRNA production and function [3, 24] would also apply to regulate shRNA and siRNA production and function. A miRNA precursor surveillance system in quality control of miRNA synthesis was recently introduced by the finding of that cellular TUT7 and TUT4, two terminal uridylyl transferases, uridylate pre-miRNA 3' ends to facilitate their degradation by the exosome [25]. However, this surveillance mechanism would not interpret our finding that the resistance happened when the shRNA-derived siRNAs remained at a high level.
Our observation raises the caution on today's application of siRNA-expression system in transgenic mice and in clinical application of RNAi for therapeutic purpose. Due to required long-term exposure to a gene-specific siRNA, these approaches might trigger the unwanted RNAi resistance response for the cells to cope with siRNA-induced changes. Many biological systems have such homeostatic mechanisms that deal with fluctuating environments to maintain core functions at a steady level [26].
Acknowledgements
We thank Haiyan Lu in NIDDK transgenic Core for her performance of oocyte pro-nuclear injection. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and National Institute of Diabetes, Digestive and Kidney Diseases, NIH.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lage H. Therapeutic potential of RNA interference in drug-resistant cancers. Future Oncol. 2009;5:169-185
2. Tang S, Tao M, McCoy JP. et al. Short-term induction and long-term suppression of HPV16 oncogene silencing by RNA interference in cervical cancer cells. Oncogene. 2006;25:2094-2104
3. Zheng ZM, Tang S, Tao M. Development of Resistance to RNAi in Mammalian Cells. Ann N Y Acad Sci. 2005;1058:105-118
4. Voinnet O, Pinto YM, Baulcombe DC. Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc Natl Acad Sci U S A. 1999;96:14147-14152
5. Pumplin N, Voinnet O. RNA silencing suppression by plant pathogens: defence, counter-defence and counter-counter-defence. Nat Rev Microbiol. 2013;11:745-760
6. Gu W, Payne E, Sun S. et al. Inhibition of cervical cancer cell growth in vitro and in vivo with dual shRNAs. Cancer Gene Ther. 2011;18:219-227
7. Yang W, Wang Q, Howell KL. et al. ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J Biol Chem. 2005;280:3946-3953
8. Jia R, Li C, McCoy JP. et al. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci. 2010;6:806-826
9. Fishler T, Li YY, Wang RH. et al. Genetic instability and mammary tumor formation in mice carrying mammary-specific disruption of Chk1 and p53. Oncogene. 2010;29:4007-4017
10. Shukla V, Coumoul X, Deng CX. RNAi-based conditional gene knockdown in mice using a U6 promoter driven vector. Int J Biol Sci. 2007;3:91-99
11. Ajiro M, Jia R, Zhang L. et al. Intron definition and a branch site adenosine at nt 385 control RNA splicing of HPV16 E6*I and E7 expression. PLoS ONE. 2012;7:e46412
12. Ajiro M, Katagiri T, Ueda K. et al. Involvement of RQCD1 overexpression, a novel cancer-testis antigen, in the Akt pathway in breast cancer cells. Int J Oncol. 2009;35:673-681
13. Ajiro M, Zheng ZM. E6^E7, a novel splice isoform protein of human papillomavirus 16, stabilizes viral E6 and E7 oncoproteins via HSP90 and GRP78. MBio. 2015;6:e02068-14
14. Jumaa H, Wei G, Nielsen PJ. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr Biol. 1999;9:899-902
15. Shukla V, Coumoul X, Wang RH. et al. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007;39:1145-1150
16. Coumoul X, Li W, Wang RH. et al. Inducible suppression of Fgfr2 and Survivin in ES cells using a combination of the RNA interference (RNAi) and the Cre-LoxP system. Nucleic Acids Res. 2004;32:e85
17. He X, Arslan AD, Pool MD. et al. Knockdown of splicing factor SRp20 causes apoptosis in ovarian cancer cells and its expression is associated with malignancy of epithelial ovarian cancer. Oncogene. 2011;30:356-365
18. Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438-442
19. Paddison PJ, Caudy AA, Bernstein E. et al. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948-958
20. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550-553
21. Pluta K, Diehl W, Zhang XY. et al. Lentiviral vectors encoding tetracycline-dependent repressors and transactivators for reversible knockdown of gene expression: a comparative study. BMC Biotechnol. 2007;7:41
22. Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet. 2007;8:173-184
23. Konishi M, Wu CH, Kaito M. et al. siRNA-resistance in treated HCV replicon cells is correlated with the development of specific HCV mutations. J Viral Hepat. 2006;13:756-761
24. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509-524
25. Liu X, Zheng Q, Vrettos N. et al. A MicroRNA precursor surveillance system in quality control of microRNA synthesis. Mol Cell. 2014;55:868-879
26. Hoeller O, Gong D, Weiner OD. How to understand and outwit adaptation. Dev Cell. 2014;28:607-616
Author contact
![]() Corresponding author: E-mail: zhengtnih.gov; Phone: 301-846-7634 (office); Fax: 301-846-6846
Corresponding author: E-mail: zhengtnih.gov; Phone: 301-846-7634 (office); Fax: 301-846-6846