Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(1):18-29. doi:10.7150/ijbs.13549 This issue Cite
Research Paper
Excess Nitric Oxide Activates TRPV1-Ca2+-Calpain Signaling and Promotes PEST-dependent Degradation of Liver X Receptor α
Jin-Feng Zhao1, Song-Kun Shyue2, Tzong-Shyuan Lee1,3 ![]()
1. Department of Physiology, National Yang-Ming University, Taipei, 11221, Taiwan.
2. Cardiovascular Division, Institute of Biomedical Sciences, Academia Sinica, Taipei, 11529, Taiwan.
3. Genome Research Center, National Yang-Ming University, Taipei, 11221, Taiwan.
Received 2015-8-13; Accepted 2015-11-5; Published 2016-1-1
Abstract
Excess nitric oxide (NO) deregulates cholesterol metabolism in macrophage foam cells, yet the underlying molecular mechanism is incompletely understood. To investigate the mechanism, we found that in macrophages, treatment with NO donors S-nitroso-N-acetyl-D,L-penicillamine (SNAP) or diethylenetriamine/nitric oxide induced LXRα degradation and reduced the expression of the downstream target of LXRα, ATP-binding cassette transporter A1 (ABCA1), and cholesterol efflux. In addition, SNAP induced calcium (Ca2+) influx into cells, increased calpain activity and promoted the formation of calpain-LXRα complex. Pharmacological inhibition of calpain activity reversed the SNAP-induced degradation of LXRα, down-regulation of ABCA1 and impairment of cholesterol efflux in macrophages. SNAP increased the formation of calpain-LXRα complex in a Pro-Glu-Ser-Thr (PEST) motif-dependent manner. Truncation of the PEST motif in LXRα abolished the calpain-dependent proteolysis. Removal of extracellular Ca2+ by EGTA or pharmacological inhibition of TRPV1 channel activity diminished SNAP-induced increase in intracellular Ca2+, calpain activation, LXRα degradation, ABCA1 down-regulation and impaired cholesterol efflux. In conclusion, excess NO may activate calpain via TRPV1-Ca2+ signaling and promote the recognition of calpain in the PEST motif of LXRα, thereby leading to degradation of LXRα and, ultimately, downregulated ABCA1 expression and impaired ABCA1-dependent cholesterol efflux in macrophages.
Keywords: nitric oxide, calcium, TRPV1, calpain, ABCA1, LXRα, macrophages
Introduction
Calpains, a family of non-lysosomal cysteine proteases play a crucial role in integrating cellular functions of mammals [1-3]. The activity of calpains is tightly regulated by calcium (Ca2+); increased intracellular Ca2+ ([Ca2+]i) level can increase calpain activity [1-3]. A protein with the polypeptide sequence enriched in proline (P), glutamate (E), serine (S) and threonine (T) (PEST motif) may be the target for degradation by calpain [4-6].
Growing evidence has revealed that calpains play a central role in regulating the pathophysiological function of vascular cells, including endothelial cells, smooth muscle cells and macrophage-foam cells [7-10]. For instance, inhibition of calpain activity ameliorates oxidative stress-induced endothelial dysfunction in diabetes [8]. In addition, calpain plays a pivotal role in regulating the cholesterol metabolism of macrophage-foam cells by modulating the protein stability of ATP-binding cassette transporter A1 (ABCA1) and ABCG1, two reverse cholesterol transporters with the PEST motif [11-13]. Thus, inhibiting calpain activity has been considered a therapeutic strategy in preventing or treating cardiovascular system diseases [7, 9]. However, much still remains to be learned about the molecular mechanism underlying calpain activation.
Liver X receptors (LXRs), including LXRα and LXRβ, are lipid-sensing nuclear receptors that regulate cholesterol metabolism and inflammation [14-16]. LXRα is mainly expressed in the liver, intestine and macrophages, whereas LXRβ expression is ubiquitous [14, 16]. As a sensor for oxysterols, LXRα physiologically modulates cholesterol homeostasis by promoting transcriptional regulation of several ABC transporters [14, 16]. Hepatic and intestinal ABCG5 and ABCG8, two key players in excess cholesterol export from liver and intestine, are tightly regulated by LXRα[14, 15, 17]. Moreover, LXRα-drived ABCA1 and ABCG1 expression coordinates reverse cholesterol efflux from macrophages [18, 19]. A wealth of evidence implicates deregulated LXRα activation in the pathogenesis of hyperlipidemia and atherosclerosis [14-19]. Several lines of evidence suggest that LXRα protein level can be controlled by transcriptional and posttranslational modification including sumoylation, phosphorylation and ubiquitination [20, 21]. We recently demonstrated that excess nitric oxide (NO), generated from inducible NO synthase (iNOS), has pro-atherogenic effects by downregulating LXRα expression, thereby decreasing ABCA1-dependent cholesterol efflux from macrophages [23]; however, whether posttranslational modification is involved in NO-mediated downregulation of LXRα protein and the underlying mechanism remain elusive.
Transient receptor potential vanilloid receptor 1 (TRPV1), a non-selective cation channel, plays a central role in the integration of pain and inflammatory responses in multiple chemical and physical stimuli in neurons [24-26]. The activation of TRPV1 channels results in increased [Ca2+]i level and activation of downstream signaling cascades and, eventually, excitation of sensory neurons [24, 26]. In addition to its vital role in regulating neuron function, the TRPV1 channel was recently found to be a key player in regulating the physiological functions of cardiovascular cells and the pathogenesis of cardiovascular diseases [27-30]. For instance, activation of TRPV1 channels in endothelial cells protects against atherosclerosis, hypertension and stroke [27, 28] and ameliorates oxidized low-density lipoprotein (oxLDL)-induced lipid accumulation in macrophages [31]. However, the contribution of TRPV1 channels and the underlying molecular mechanism in excess NO-downregulated LXRα and the related cholesterol metabolism in macrophages is less well defined.
Here, we aimed to examine the requirement for calpain in excess NO-downregulated LXRα in macrophages. We first investigated the role of calpain in excess NO-downregulated LXRα and the involvement of the PEST motif in macrophages, then delineated the role of TRPV1-Ca2+ signaling in SNAP-activated calpain, downregulation of LXRα and ABCA1, and impaired cholesterol efflux.
Materials and methods
Reagents
S-nitroso-N-acetyl-D,L-penicillamine (SNAP), diethylenetriamine/nitric oxide (DETA/NO), 3-hexanoly-7- nitro-2,1,3-benzoxadial-4-yl (NBD) cholesterol, capsazepine (CPZ) and SB366791 were from Cayman Chemical (Ann Arbor, MI, USA). Rabbit anti-LXRα antibody and TurboFect transfection reagent were from Thermo Scientific (Rockford, IL, USA). Mouse anti-ABCA1 antibody was from Abcam (Cambridge, MA, USA). Mouse anti-α-tubulin, anti-Flag and anti-GAPDH antibodies; Tri reagent; cycloheximide (CHX); ethylene glycol tetraacetic acid (EGTA); calpeptin; and apolipoprotein AI (apoAI) were from Sigma (St Louis, MO, USA). Dil-oxLDL was from Biomedical Technologies (Stoughton, MA, USA). The Fluo-8 calcium assay kit was from AAT Bioquest (Sunnyvale, CA, USA). RPMI-1640 medium and Dulbecco's modified Eagle's medium (DMEM) were from Hyclone (Logan, Utah, USA). The calpain activity assay kit was from BioVision (Milpitas, CA, USA). μ-calpain was from Merck (Merck KGaA, Darmstadt, Germany).
Cell culture
Murine macrophage J774.A1 cells (American Type Culture Collection, TIB-67) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml) and streptomycin (100 μg/ml) (HyClone, Logan, UT, USA). Human embryonic kidney 293 (HEK293) cells (American Type Culture Collection, CRL-3216) were maintained in DMEM supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml).
Western blot analysis
Cells were washed with PBS, then lysed with immunoprecipitation (IP) lysis buffer (50 mM Tris pH 7.5, 5 mM EDTA, 300 mM NaCl, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin and 10 μg/mL aprotinin). After centrifugation at 13,000 rpm for 5 min at 4°C, the supernatant was harvested as cell lysates. A 50-μg sample was fractionated by SDS-PAGE, then transblotted onto Immobilon-P membrane (Millipore, Bedford, MA, USA), which was incubated with primary antibodies, then secondary antibodies. The samples were detected by use of an enhanced chemiluminescence kit (PerkinElmer, Boston, MA, USA) and quantified by use of ImageQuant 5.2 (Healthcare Bio-Sciences, Philadelphia, PA, USA).
Quantitative real-time RT-PCR
Total cellular RNA was extracted by use of Tri reagent. A 5-μg amount of total RNA was converted to cDNA with reverse transcriptase (Fermentas, MD, USA) for use as templates in quantitative real-time RT-PCR (qPCR) with the TaqMan probe-based real-time quantification system (Applied Biosystems, Foster, CA, USA). mRNA levels were normalized to that of GAPDH and expressed as fold change of control levels.
Measurement of calpain activity
Calpain activity was measured by use of a kit. Briefly, cellular lysates (100 μg) were mixed with reaction buffer and Ac-LLY-AFC (fluorogenic substrate) for 1 h at 37°C. The level of free AFC was measured by fluorometry at 400 nm excitation and 505 nm emission.
Immunoprecipitation
Macrophages were washed with PBS and lysed with lysis buffer (25 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA and 0.1% NP-40, 1% Triton X-100, 0.1 mM Na3PO4 and protease inhibitors). After centrifugation at 13,000 rpm for 5 min at 4°C, 1000 μg supernatant was incubated with 1 mg specific primary antibody overnight at 4°C, then protein A/G-Sepharose beads for 2 h at 4°C. The protein A/G-antibody complexes were precipitated by centrifugation at 4000 rpm for 5 min. The immune complexes were washed three times with cold PBS for 20 min, then eluted in SDS lysis buffer (1% Triton, 0.1% SDS, 0.2% sodium azide, 0.5% sodium deoxycholate, 1 mM PMSF, 10 μg/mL aprotinin and 10 μg/mL leupeptin). Samples were heated at 100°C for 10 min and examined by western blot analysis.
Plasmid constructs
The coding region for human LXRα (hLXRα) was amplified from cDNA by PCR (2 min at 94°C, then 15 sec at 94°C, 30 sec at 61°C and 90 sec at 68°C for 35 cycles) with the primer sequences 5'-TTA CGC GTA TGT CCT TGT GGC TGG GGG C-3' and 5'-TCT GGG ATG TGC ACG AAT GAT CTA GAG C-3'. The amplified DNA fragments were digested with MluІ and XbaІ, cloned into pCMV5N-Flag vector, and confirmed by sequencing. hLXRα with truncated PEST motif (hLXRα-dPEST) was constructed by deleting hLXRα cDNA from base pairs 208 to 234. The DNA fragments were amplified by PCR (2 min at 94°C, then 15 sec at 94°C, 30 sec at 61°C and 90 sec at 68°C for 35 cycles) from cDNA with the primer sequences 5'-TTA CGC GTA TGT CCT TGT GGC TGG GGG C-3' and 5'-CCA CAG CCC TGC TCA CCA GGA AGC TTA A-3' for N-terminal DNA fragment, and 5'-GCA AGC TTA TCC GTC CAC AAA AGC GGA A-3' and 5'-TCT GGG ATG TGC ACG AAT GAT CTA GAG C-3' for C-terminal DNA fragment. The N- or C-terminal PCR products were digested with MluІ and HindIII or HindIII and XbaІ, respectively, then cloned into the pCMV5N-Flag vector. The plasmid was confirmed by sequencing. The coding regions for TRPV1 wild type (TRPV1-WT), TRPV1-Y671D mutant (TRPV1-D) and TRPV1-Y671K mutant (TRPV1-K) with the pcDNA3.1(+) plasmid were kindly provided by Dr. Carla Nau (Department of Anesthesiology, Friedrich-Alexander-University, Erlangen-Nuremberg, Erlangen, Germany) [32]. Each TRPV1 DNA fragment was digested with EcoRI and XbaI, then cloned into the pCMV5N-Flag vector. DNA sequences were verified by sequencing analysis.
Transient transfection
In the calpain-catalyzed proteolysis assay, HEK293 cells were transfected with hLXRα or hLXRα-dPEST by use of TurboFect reagent for 48 h. In the Ca2+ influx assay, HEK293 cells were transfected with TRPV1-WT, TRPV1-D or TRPV1-K for 48 h, then treated with SNAP for the indicated times.
Calpain-catalyzed proteolysis assay
The calpain-catalyzed proteolysis assay was performed as described [33]. Transfected HEK293 cells were lysed with IP lysis buffer, then coincubated with 1.5 μg μ-calpain plus 5 mM CaCl2 at 31°C for 1 h. SDS lysis buffer was added to stop the reaction, then samples were examined by western blot analysis.
Cholesterol efflux assay
Cholesterol efflux was determined as described [31]. Macrophages were cultured with SNAP (200 μM) in the presence of various inhibitors for 12 h, then equilibrated with NBD-cholesterol (1 μg/ml) for 6 h. After a washing with PBS, the NBD-cholesterol-labeled cells were cultured in medium for another 6 h with SNAP, various inhibitors and apoAI (10 μg/ml). The fluorescence-labeled cholesterol released from cells into the medium was analyzed by use of a multilabel counter (PerkinElmer, Waltham, MA, USA) with 485 nm excitation and 535 nm emission.
Dil-oxLDL fluorescent assay
Macrophages were treated with SNAP in the presence of calpeptin for 12 h, then equilibrated with fluorescent oxLDL (Dil-oxLDL) for an additional 18 h in the presence of drugs. Cells were washed and lysates were analyzed by fluorometry (Molecular Devices) at 514 nm excitation and 550 nm emission.
[Ca2+]i detection
With the calcium assay kit, cells were preincubated with fluorescent dye Fluo-8 at 37°C for 1 h, then test compounds were added for the indicated times. Cell lysates were harvested and analyzed by fluorometry at 490 nm excitation and 525 nm emission.
Statistical analysis
Data are presented as mean ± SEM from 4 independent experiments. Mann-Whitney test was used to compare two independent groups and Kruskal-Wallis followed by Bonferroni posthoc analysis to compare multiple groups. SPSS v20.0 (SPSS Inc, Chicago, IL) was used for analysis. Differences were considered statistically significant at P < 0.05.
Results
Involvement of calpain in SNAP-increased proteolytic degradation of LXRα
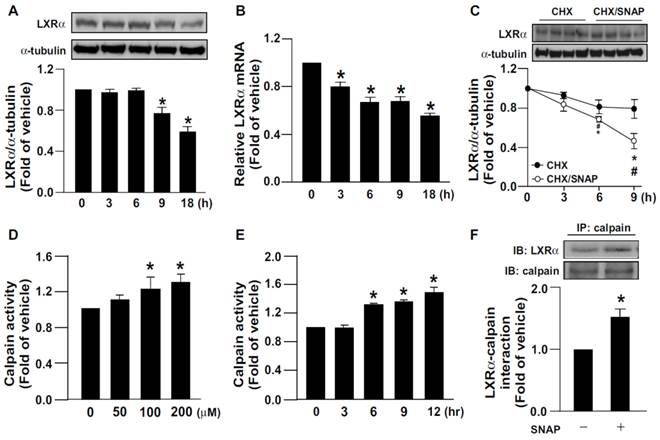
We first investigated the effect of SNAP-released NO on LXRα expression. Treating macrophages with SNAP decreased the protein and mRNA expression of LXRα (Fig. 1A, B). Additionally, the proteolytic degradation of LXRα was time-dependently enhanced with SNAP treatment as compared with vehicle treatment (Fig. 1C). Since calpain is involved in regulating proteolysis [1, 2], we elucidated the role of calpain in the SNAP-induced downregulation of LXRα. Treatment with SNAP dose- and time-dependently increased calpain activity (Fig. 1D, E). As well, SNAP promoted the formation of an LXRα-calpain complex on immunoprecipitation assay (Fig. 1F). Similar results were also observed in NO donor DETA/NO-treated macrophages (Supplementary Fig. 1A-D). Therefore, calpain may participate in excess NO-induced downregulation of LXRα.
SNAP increases calpain activity and promotes degradation of LXRα protein in macrophages. J774.A1 macrophages were treated with SNAP (200 μM) for the indicated times. (A) Western blot analysis of protein levels of LXRα and α-tubulin. (B) qRT-PCR of mRNA levels of LXRα relative to that of GAPDH. (C) Cells were treated with or without SNAP (200 μM) in the presence of cycloheximide (CHX, 2 μg/mL) for the indicated times. (D) Macrophages were treated with various concentrations of SNAP for 12 h or (E) SNAP (200 μM) for the indicated times. Calpain activity was calculated by the assay kit. (F) Macrophages were incubated with SNAP (200 μM) for 6 h. Cellular lysates was immunoprecipitated (IP) with anti-calpain antibody and then immunoblotted (IB) with LXRα Ab. Data are mean ± SEM from 4 independent experiments *, P <0.05 vs. vehicle-treated, #, P <0.05 vs. SNAP-treated CHX.
PEST motif of LXRα is critical for calpain-mediated proteolysis
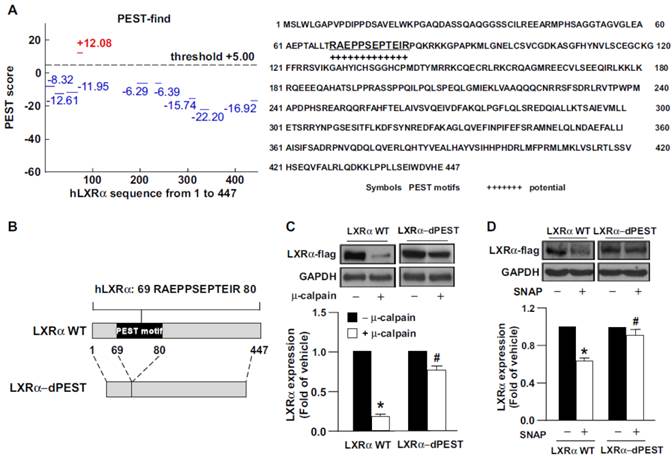
Growing evidence indicates that protein sequences enriched in PEST motif are easily decomposed by calpain protease [4-6]. Use of the PEST-FIND system (http://at.embnet.org/embnet/tools/bio/PESTfind/; protein with PEST score > +5 considered to have a potential PEST motif) [5, 34] suggested that LXRα may be recognized by calpain. The PEST score for LXRα was +12.08, and a conserved PEST sequence was found in LXRα at amino acids 69-80 (Fig. 2A). To provide further evidence that the PEST motif within LXRα is critical for calpain proteolysis, we created a construct expressing PEST motif-deleted LXRα (LXRα-dPEST) (Fig. 2B). We found that μ-calpain effectively degraded LXRα-WT but not LXRα-dPEST protein (Fig. 2C). Additionally, treatment with SNAP failed to induce degradation of PEST-deleted LXRα in macrophages (Fig. 2D). These results suggest the importance of the PEST motif of LXRα for calpain-dependent proteolysis.
The PEST motif is essential for calpain-mediated proteolysis of LXRα. (A) The PEST score of human LXRα (hLXRα) is +12.8 determined by use of the computer program PEST-FIND. +, PEST potential motif. (B) Schematic model of creating LXRα WT or LXRα PEST deletion mutant (LXRα-dPEST) constructs. (C and D) Flag-tag LXRα WT or LXRα-dPEST plasmid was transiently transfected into HEK293 cells or macrophages for 48 h. Western blot analysis of protein levels of Flag and GAPDH in cellular lysates (50 μg) incubated with 5 mM CaCl2 and μ-calpain at 31°C for 1 h. Data are mean ± SEM from 4 independent experiments. *, P < 0.05 vs. LXRα-WT vehicle-treated. #, P < 0.05 vs. LXRα-WT calpain-treated.
Calpain mediates SNAP-impaired LXRα-ABCA1-dependent cholesterol efflux in macrophages
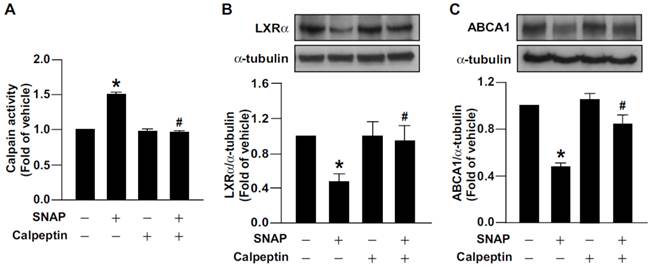
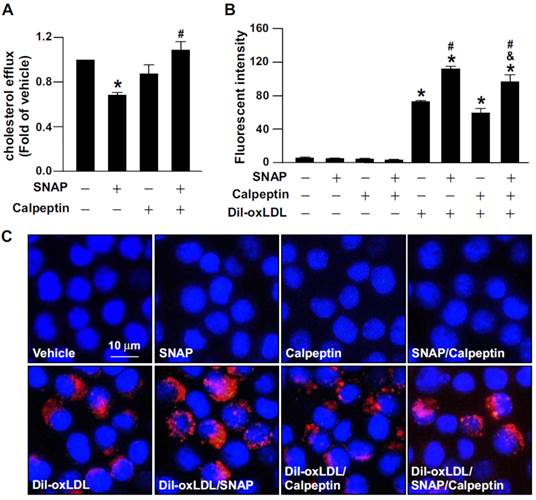
We previously reported that excess NO downregulates the level of ABCA1, the main reverse cholesterol transporter in macrophages, by decreasing LXRα protein and mRNA expression [31]. We then investigated the functional significance of calpain in excess NO-promoted lipid accumulation in macrophage-foam cells. Inhibition of calpain by the pharmacological inhibitor calpeptin abolished the SNAP-increased calpain activity and prevented its inhibitory effects on the protein expression of LXRα and ABCA1 (Fig. 3A-C). More importantly, suppression of calpain activity rescued SNAP-impaired cholesterol efflux (Fig. 4A) and thus ameliorated SNAP-augmented lipid accumulation induced by oxLDL (Fig. 4B, C). These results suggest that calpain is a key player in the SNAP-induced unfavorable effects on cholesterol metabolism of macrophage-foam cells.
Calpain is crucial in SNAP-induced downregulation of LXRα and ABCA1. (A) Calpain activity in macrophages treated with SNAP (200 μM) in the absence or presence of calpain inhibitor calpeptin (10 μM) for 12 h. (B and C) Cells were coincubated with calpeptin and SNAP for 18 h. Western blot analysis of LXRα and ABCA1 protein levels. Data are mean ± SEM from 4 independent experiments *, P < 0.05 vs. vehicle-treated, #, P < 0.05 vs. SNAP-treated.
Inhibition of calpain abrogates the detrimental effect of SNAP on cholesterol efflux and oxLDL-induced lipid accumulation in macrophages. (A) J774.A1 macrophages were treated with SNAP (200 μM) in the presence of calpeptin (10 μM) for 12 h, then underwent equilibration of NBD-cholesterol (1 μg/ml) with apoAI (10 μg/ml) for an additional 6 h. The medium and cell lysates were collected for the measurement of fluorescence. Fold induction was defined as the level of cholesterol efflux relative to that in vehicle-treated cells set to 1. (B and C) Macrophages were incubated with SNAP in the presence of fluorescence-labeled oxLDL (Dil-oxLDL) or calpeptin (10 μM) for 18 h. Fluorescence was measured by fluorometry and photographed by fluorescence microscopy. Cellular nuclei were stained with DAPI (blue). Data are mean ± SEM from 4 independent experiments. *, P < 0.05 vs. vehicle-treated, #, P < 0.05 vs. oxLDL-treated cells, &, P < 0.05 vs. SNAP-treated group with oxLDL treatment.
TRPV1 channels are required for SNAP-increased [Ca2+]i level in macrophages
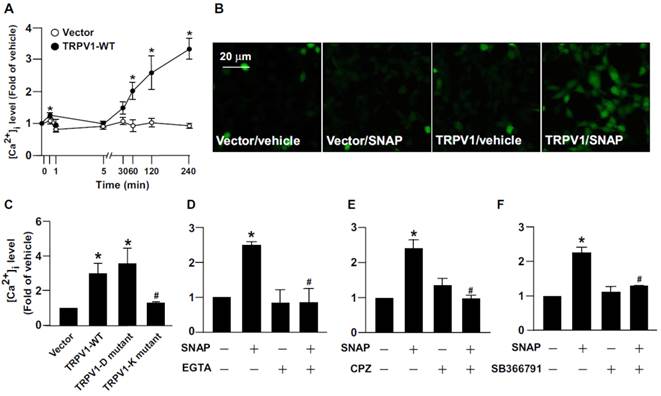
The contribution of [Ca2+]i in the regulation of calpain activity is well defined [1-3]. We delineated the effect of Ca2+ influx on NO-activated calpain. As compared with vehicle treatment, SNAP or DETA/NO treatment rapidly increased Ca2+ influx within 240 min (Fig. 5A and Supplementary Fig. 1E), which occurred as early as 1 min after treatment, slightly decreased at 5-60 min, then gradually increased to peak again at 4 h (Fig. 5A). Removal of extracellular Ca2+ by EGTA prevented SNAP-induced Ca2+ influx (Fig. 5B, E). To address the implication of TRPV1 in SNAP-induced Ca2+ influx, macrophages were pretreated with the TRPV1 pharmacological inhibitor CPZ or SB366791 (both 10 μM) to inhibit the Ca2+ channel activity of TRPV1. Pre-treatment with CPZ or SB366791 abrogated the SNAP-increased [Ca2+]i level (Fig. 5C-E). To provide further evidence that TRPV1 activation is critical for SNAP-evoked Ca2+ influx in macrophages, we used gain- and loss-of-function strategies to block the Ca2+ permeability of TRPV1 channels by re-expressing full-length TRPV1 WT, TRPV1-K (TRPV1 mutant with defective Ca2+ permeability) or TRPV1-D (TRPV1 mutant with normal Ca2+ permeability) in HEK293 cells, a cell line lacking TRPV1 channels. Compared to vector transfection, SNAP treatment increased [Ca2+]i level in TRPV1 WT- and TRPV1-D- but not TRPV1-K-reerexpressed HEK293 cells (Fig. 6A-C). Removal of extracellular Ca2+ or inhibiting TRPV1 with specific antagonists diminished the SNAP-induced Ca2+ influx in TRPV1 WT-reerexpressed HEK293 cells (Fig. 6D-E). TRPV1-mediated Ca2+ influx might be an important event in SNAP-activated calpain.
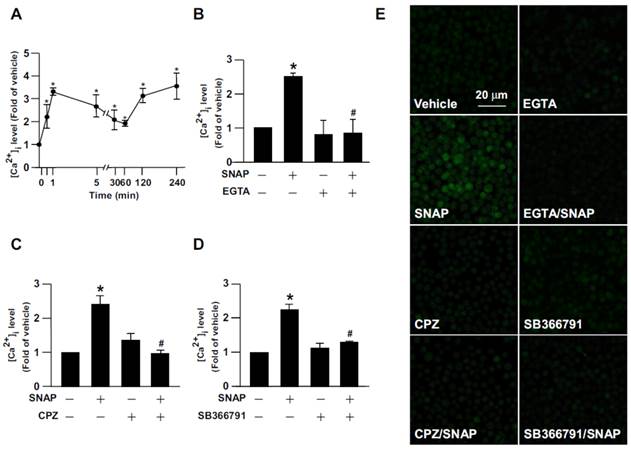
SNAP induces TRPV1-dependent Ca2+ influx in macrophages. (A) J774.A1 macrophages were treated with SNAP (200 μM) for up to 240 min. (B) Macrophages were pretreated with Ca2+ chelator EGTA (500 nM), (C) capsazepine (CPZ) (10 μM) or (D) SB366791 (10 μM) for 1 h followed by SNAP (200 μM) treatment for another 240 min. The intracellular level of Ca2+ was measured by Fluo-8 calcium assay. (E) Fluorescence images were photographed by fluorescence microscopy. Data are mean ± SEM from 4 independent experiments. *, P < 0.05 vs. vehicle-treated, #, P < 0.05 vs. SNAP-treated.
Role of TRPV1 in SNAP-increased intracellular Ca2+ content in HEK293 cells. (A and B) HEK293 cells were transiently transfected with vector or TRPV1-WT plasmid, then incubated with SNAP (200 μM) for up to 240 min. (C) After transfection with vector, TRPV1-WT, TRPV1-D mutant or TRPV1-K mutant plasmid, HEK293 cells were treated with SNAP (200 μM) for 240 min. (D-F) TRPV1-WT SNAP-treated group. After transfection with TRPV1-WT plasmid, HEK293 cells were pretreated with (D) Ca2+ chelator EGTA (500 μM), (E) CPZ (10 μM) or (F) SB366791 (10 μM) for 1 h followed by SNAP (200 μM) for 240 min. The intracellular level of Ca2+ was measured by Fluo-8 calcium assay. Fluorescence images were photographed by fluorescence microscopy. Data are mean ± SEM from 4 independent experiments. *, P < 0.05 vs. vector-transfected or vehicle-treated, #, P < 0.05 vs. TRPV-WT-transfected or SNAP-treated.
Activation of TRPV1 channels plays a central role in the detrimental effect of SNAP on calpain activation and LXRα-ABCA1-dependent cholesterol efflux
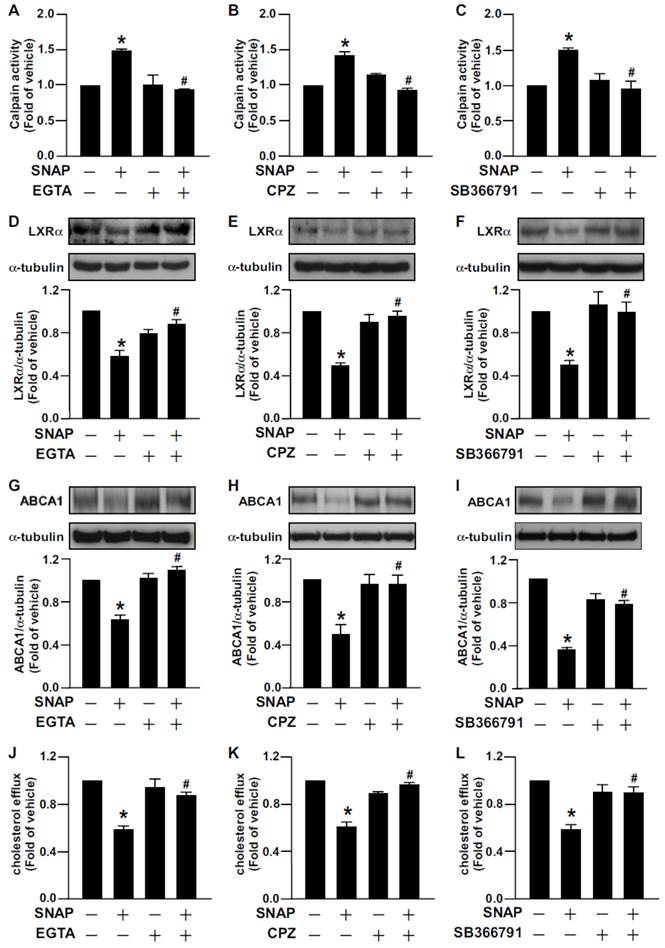
We next determined the significance of TRPV1 channels in SNAP-mediated cholesterol metabolism in macrophages. Treatment with EGTA or a TRPV1 antagonist prevented SNAP-induced calpain activity (Fig. 7A-C). The inhibitory effects of SNAP on LXRα and ABCA1 expression and cholesterol efflux capacity were also suppressed by EGTA (Fig. 7D, G, J), CPZ (Fig. 7E, H, K) and SB366791 (Fig. 7F, I, L). Thus, TRPV1-Ca2+ signaling may be required for SNAP-activated calpain and deregulation of cholesterol metabolism in macrophages (Fig. 8).
Removal of Ca2+ or inhibition of TRPV1 channel function reversed SNAP-impaired cholesterol efflux and -downregulated LXRα and ABCA1. J774.A1 macrophages were treated with EGTA (500 μM), CPZ (10 μM) or SB366791 (10 μM) in the absence or the presence of SNAP (200 μM) for 12 h (A-C) or 18h (D-I). Western blot analysis of (A-C) calpain activity and (D-I) LXRα (D-F), ABCA1 (G-I) and α-tubulin protein levels. (J-L) Cholesterol efflux measured in macrophages treated with or without SNAP in the presence of EGTA, CPZ or SB366791 for 12 h after the equilibration of NBD-cholesterol (1 μg/ml) with apoAI (10 μg/ml) for an additional 6 h. Data are mean ± SEM from 4 independent experiments. *, P < 0.05 vs. vehicle-treated, #, P < 0.05 vs. SNAP-treated.
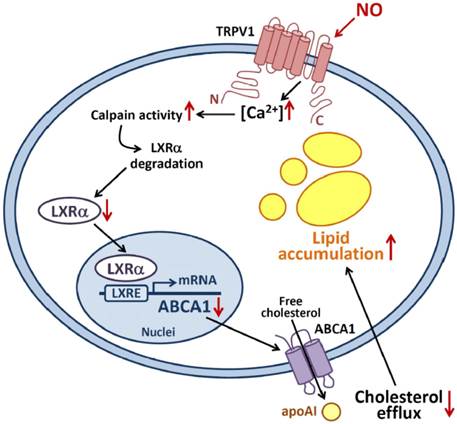
Proposed model of nitric oxide-induced TRPV1-Ca2+-calpain signaling that leads to proteolytic LXRα degradation and pathologic accumulation of lipids in macrophages. Activation of TRPV1 by excess NO increases the Ca2+ influx, which in turn increases calpain activity, promotes LXRα degradation and downregulates protein expression of LXRα and ABCA1, for impaired reverse cholesterol efflux and worsened lipid accumulation in macrophage-foam cells.
Discussion
In this study, we characterized a new regulatory mechanism underlying the posttranslational modification of LXRα in excess NO-deregulated cholesterol homeostasis of macrophage-foam cells. Exposing macrophages to the NO donor SNAP decreased the LXRα protein and mRNA levels by promoting LXRα degradation. In parallel with the enhanced proteolytic degradation of LXRα, SNAP increased calpain activity and promoted the formation of a calpain-LXRα complex. The PEST motif of LXRα was found to be the target site for calpain-dependent proteolysis. Treatment with a specific calpain pharmacological inhibitor showed that calpain plays an important role in SNAP-exacerbated lipid accumulation in foam cells. All these key events were prevented by removal of extracellular Ca2+ or inhibition of TRPV1-Ca2+ signaling. Activation of a TRPV1-Ca2+-calpain signaling cascade may be a key event in SNAP-mediated LXRα degradation and deregulated cholesterol metabolism in macrophage-foam cells.
The LXRα-ABCA1 pathway is the most important mechanism responsible for cholesterol efflux from macrophages to apoA1 and thus plays a central role in maintenance of cholesterol homeostasis of foam cells and progression of atherosclerosis [35-37]. Loss of or impaired function in the LXRα-ABCA1 pathway by inflammatory insults decreases the capacity for cholesterol efflux and leads to excessive cholesterol deposition in cells or peripheral tissues [31, 38]. Mice lacking LXRα show cholesterol accumulation in liver when fed a high-cholesterol diet [39]. In an LDL-receptor-deficient background, genetic deletion of LXRα worsens atherosclerosis [40]. Reactive oxygen species downregulate the expression of LXRα and ABCA1, thereby leading to lipid accumulation in foam cells [38]. In contrast, activation of LXRα by ligands such as GW3965 or TO901317 upregulates ABCA1 expression, promotes reverse cholesterol transport, inhibits foam-cell formation and retards atherosclerosis [41, 42]. Collectively, LXRα could be a pharmaceutical target for treatment of atherosclerosis and related metabolic diseases.
Most nuclear receptors such as LXRα function via ligand-induced conformational change and subsequent translocation into the nucleus to mediate the transcriptional activation of target genes. However, increasing evidence indicates that the transcriptional activity of nuclear receptors is also modulated by transcriptional or post-translational regulation [20-22, 43, 44]. For example, LXRα mRNA expression is directed by the autoregulation of LXRα itself or peroxisome proliferator-activated receptor γ [43, 44]. LXRα also undergoes a post-translational modification that includes phosphorylation, sumoylation and ubiquitination [20-22]. Protein kinase A phosphorylates LXRα at Ser195 and Ser196, which impairs DNA binding ability and thus transcriptional activity of LXRα [21]. Moreover, a ubiquitin E3-ligase protein complex can interact with LXRα and promote its degradation via ubiquitin-dependent proteolysis [22]. In this study, we demonstrated a novel mechanism in the regulation of LXRα protein expression. SNAP treatment activated the calpain-dependent proteolytic process, increased the formation of an LXRα-calpain complex, and resulted in LXRα degradation. Inhibition of calpain activity blunted the harmful effect of excess NO in cholesterol metabolism in macrophages. These results are consistent with previous evidence showing that suppressing calpain activation increased reverse cholesterol efflux and attenuated the cholesterol accumulation in foam cells [11, 45]. Because calpain protease has a detrimental role in the progression of atherosclerosis, the damaging effect of SNAP on LXRα-ABCA1-dependent cholesterol efflux from macrophages may be due to the increased calpain activity. Post-transcriptional regulation may be potential mechanism in regulating the LXRα-ABCA1 pathway.
Our observations of LXRα instability and the formation of an LXRα-calpain complex after SNAP treatment broaden the implication of calpain involvement in the protein stability of LXRα. In general, the protein amino acid sequence enriched in the PEST domain is rapidly degraded by calpain protease [4-6]. The PEST motif in ABCA1 is required for clapain-dependent degradation [11]. As well, the PEST sequence within IκB is necessary for μ-calpain-mediated degradation [46]. Indeed, we found a PEST-enriched motif in amino acids 69-80 of LXRα; deletion of this PEST domain prevented μ-calpain-mediated LXRα degradation in vitro, which suggests the potential role of calpain in regulating LXRα protein stability. This evidence suggests a new therapeutic trarget for maintaining the protein stability of LXRα. However, mutation of the PEST domain in cystic fibrosis transmembrane conductance regulator (CFTR) did not significantly alter the degradation rate of CFTR [47], so much remains to be learned about the exact mechanism of the PEST motif in calpain proteolysis.
Elevated [Ca2+]i level is thought to be associated with activation of calpain, which could be a key modulator in regulating downstream protein stability [1, 7]. Indeed, our observations suggested that SNAP-evoked Ca2+ influx was essential for calpain-mediated downregulation of LXRα and ABCA1. Additionally, SNAP-induced Ca2+ oscillation depended on TRPV1. Not unsurprisingly, TRP channels are activated by a variety of reactive chemicals [48-51]. NO activates TRP channels, including TRPV1, by inducing cysteine S-nitrosylation [31]. Moreover, treatment with an NO donor directly activated TRPV1- and TRPA1-dependent Ca2+ influx, thereby triggering peripheral nociceptive action [49]. Also, NO induced TRPV1-dependent apoptosis by modifying S-nitrosylation of TRPV1 in osteoblasts [52]. We also found that treatment with SNAP increased TRPV1-dependent Ca2+ influx. However, the exact molecular mechanism underlying excess NO-activation of TRPV1 channels in macrophage-foam cells remains for further investigation.
As well, both macrophage-mediated cholesterol clearance and pro-inflammatory mediator production are key steps in the initiation and progression of atherosclerosis [35-37]. Particularly, excessive cholesterol accumulation in macrophages may due to impaired LXRα-ABCA1-dependent cholesterol efflux induced by pro-inflammatory mediators in macrophages [23]. For example, oxLDL or pro-inflammatory cytokines induces lipid accumulation in macrophage-foam cells and inflammation, thereby inducing iNOS expression and excess NO production [53-55]. In addition, the activation of TRP channels by NOS-derived NO plays a crucial role in the regulation of several inflammatory diseases [49, 50, 56]. Deletion of TRPM2 diminishes iNOS-elicited signaling transduction and CXCL2 production in macrophages and microglia, thereby preventing inflammatory and neuropathic pain [56]. NO donors can induce TRPV1-dependent thermal hyperalgesia and nociceptive action [48, 49]. Indeed, we found that inhibiting TRPV1 activation ablated SNAP-activated calpain, downregulated LXRα and ABCA1, impaired cholesterol efflux and exacerbated oxLDL-induced lipid accumulation. TRPV1 has been implicated in calpain activation and pathogenesis of inflammatory diseases [57-59]. We also found that exposing macrophages to excess NO increased calpain activity in a TRPV1-Ca2+-dependent manner. Our previous study showed that excess NO augmented oxLDL-induced lipid accumulation in macrophages by deregulating LXRα-ABCA1-mediated cholesterol efflux; here, we provide advanced information for the link of TRPV1 channels to Ca2+-dependent calpain activity in excess NO-deregulated cholesterol metabolism.
In conclusion, we discovered a link between excess NO, TRPV1-Ca2+ signaling, calpain activity, LXRα protein stability and cholesterol metabolism in macrophages, which broads the cardiovascular disease-related significance of excess NO. Exposing macrophages to excess NO triggered TRPV1-dependent Ca2+ influx, increased calpain activity, and deregulated LXRα-ABCA1-dependent cholesterol efflux. These findings offer a new molecular mechanism underlying the pro-atherogenic property of excess NO in the pathogenesis of atherosclerosis and provide new insights into possible targets for therapeutic interventions in atherosclerosis and related cardiovascular diseases.
Supplementary Material
Supplementary Figure 1.
Acknowledgements
The authors thank Laura Smales for help in language editing.
This study was supported by grants from the Ministry of Science and Technology (102-2628-B-010-001-MY3, 103-2628-B-010-040-MY3, 103-2811-B-010-029 and 104-2811-B-010-020), and Ministry of Education, Aim for the Top University Plan, Taiwan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Goll DE, Thompson VF, Li H. et al. The calpain system. Physiol Rev. 2003;83:731-801
2. Khorchid A, Ikura M. How calpain is activated by calcium. Nat Struct Biol. 2002;9:239-41
3. Storr SJ, Carragher NO, Frame MC. et al. The calpain system and cancer. Nat Rev Cancer. 2011;11:364-74
4. Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267-71
5. Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364-8
6. Rechsteiner M. Regulation of enzyme levels by proteolysis: the role of pest regions. Adv Enzyme Regul. 1988;27:135-51
7. Miyazaki T, Koya T, Kigawa Y. et al. Calpain and atherosclerosis. J Atheroscler Thromb. 2013;20:228-37
8. Chen B, Zhao Q, Ni R. et al. Inhibition of calpain reduces oxidative stress and attenuates endothelial dysfunction in diabetes. Cardiovasc Diabetol. 2014;13:88
9. Letavernier E, Perez J, Bellocq A. et al. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res. 2008;102:720-8
10. Ariyoshi H, Okahara K, Sakon M. et al. Possible involvement of m-calpain in vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol. 1998;18:493-8
11. Wang N, Chen W, Linsel-Nitschke P. et al. A PEST sequence in ABCA1 regulates degradation by calpain protease and stabilization of ABCA1 by apoA-I. J Clin Invest. 2003;111:99-107
12. Hori N, Hayashi H, Sugiyama Y. Calpain-mediated cleavage negatively regulates the expression level of ABCG1. Atherosclerosis. 2011;215:383-91
13. Chen W, Wang N, Tall AR. A PEST deletion mutant of ABCA1 shows impaired internalization and defective cholesterol efflux from late endosomes. J Biol Chem. 2005;280:29277-81
14. Makishima M. Nuclear receptors as targets for drug development: regulation of cholesterol and bile acid metabolism by nuclear receptors. J Pharmacol Sci. 2005;97:177-83
15. Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. 2003;17:985-93
16. Joseph SB, Castrillo A, Laffitte BA. et al. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213-9
17. Repa JJ, Mangelsdorf DJ. The liver X receptor gene team: potential new players in atherosclerosis. Nat Med. 2002;8:1243-8
18. Schmitz G, Langmann T. Transcriptional regulatory networks in lipid metabolism control ABCA1 expression. Biochim Biophys Acta. 2005;1735:1-19
19. Kohro T, Nakajima T, Wada Y. et al. Genomic structure and mapping of human orphan receptor LXR alpha: upregulation of LXRα mRNA during monocyte to macrophage differentiation. J Atheroscler Thromb. 2000;7:145-51
20. Pascual G, Fong AL, Ogawa S. et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature. 2005;437:759-63
21. Yamamoto T, Shimano H, Inoue N. et al. Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver. J Biol Chem. 2007;282:11687-95
22. Kim KH, Yoon JM, Choi AH. et al. Liver X receptor ligands suppress ubiquitination and degradation of LXRalpha by displacing BARD1/BRCA1. Mol Endocrinol. 2009;23:466-74
23. Zhao JF, Shyue SK, Lin SJ. et al. Excess nitric oxide impairs LXR(α)-ABCA1-dependent cholesterol efflux in macrophage foam cells. J Cell Physiol. 2014;229:117-25
24. Caterina MJ, Schumacher MA, Tominaga M. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816-24
25. Caterina MJ, Rosen TA, Tominaga M. et al. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature. 1999;398:436-41
26. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387-417
27. Yang D, Luo Z, Ma S. et al. Activation of TRPV1 by dietary capsaicin improves endothelium-dependent vasorelaxation and prevents hypertension. Cell Metab. 2010;12:130-41
28. Ching LC, Kou YR, Shyue SK. et al. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc Res. 2011;91:492-501
29. Wei J, Ching LC, Zhao JF. et al. Essential role of transient receptor potential vanilloid type 1 in evodiamine-mediated protection against atherosclerosis. Acta Physiol. 2013;207:299-307
30. Wang L, Wang DH. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation. 2005;112:3617-23
31. Zhao JF, Ching LC, Kou YR. et al. Activation of TRPV1 prevents oxLDL-induced lipid accumulation and TNF-α-induced inflammation in macrophages: Role of liver X receptor α. Mediators Inflamm. 2013:925171
32. Mohapatra DP, Wang SY, Wang GK. et al. A tyrosine residue in TM6 of the Vanilloid Receptor TRPV1 involved in desensitization and calcium permeability of capsaicin-activated currents. Mol Cell Neurosci. 2003;23:314-24
33. Iwamoto N, Lu R, Tanaka N. et al. Calmodulin interacts with ATP binding cassette transporter A1 to protect from calpain-mediated degradation and upregulates high-density lipoprotein generation. Arterioscler Thromb Vasc Biol. 2010;30:1446-52
34. WWW PESTfind Analysis. European Molecular Biology Network. http://at.embnet.org/embnet/tools/bio/PESTfind/
35. Li AC, Glass CK. The macrophage foam cell as a target for the therapeutic intervention. Nat Med. 2002;8:1235-42
36. Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503-16
37. Lusis AJ. Atherosclerosis. Nature. 2000;407:233-41
38. Schmitz G, Grandl M. Role of redox regulation and lipid rafts in macrophages during Ox-LDL-mediated foam cell formation. Antioxid Redox Signal. 2007;9:1499-518
39. Peet DJ, Turley SD, Ma W. et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693-704
40. Bischoff ED, Daige CL, Petrowski M. et al. Non-redundant roles for LXRalpha and LXRbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J Lipid Res. 2010;51:900-6
41. Verschuren L, de Vries-van der Weij J, Zadelaar S. et al. LXR agonist suppresses atherosclerotic lesion growth and promotes lesion regression in apoE*3Leiden mice: time course and mechanisms. J Lipid Res. 2009;50:301-11
42. Joseph SB, McKilligin E, Pei L. et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002;99:7604-9
43. Laffitte BA, Joseph SB, Walczak R. et al. Autoregulation of the human liver X receptor alpha promoter. Mol Cell Biol. 2001;21:7558-68
44. Guan JZ, Tamasawa N, Murakami H. et al. Clofibrate, a peroxisome-proliferator, enhances reverse cholesterol transport through cytochrome P450 activation and oxysterol generation. Tohoku J Exp Med. 2003;201:251-9
45. Lin CY, Lee TS, Chen CC. et al. Endothelin-1 exacerbates lipid accumulation by increasing the protein degradation of the ATP-binding cassette transporter G1 in macrophages. J Cell Physiol. 2011;226:2198-205
46. Shumway SD, Maki M, Miyamoto S. The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mu-calpain. J Biol Chem. 1999;274:30874-81
47. Chen EY, Clarke DM. The PEST sequence does not contribute to the stability of the cystic fibrosis transmembrane conductance regulator. BMC Biochem. 2002;3:29
48. Yoshida T, Inoue R, Morii T. et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596-607
49. Miyamoto T, Dubin AE, Petrus MJ. et al. TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS One. 2009;4:e7596
50. Takahashi N, Mizuno Y, Kozai D. et al. Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels (Austin). 2008;2:287-98
51. Trevisan G, Hoffmeister C, Rossato MF. et al. TRPA1 receptor stimulation by hydrogen peroxide is critical to trigger hyperalgesia and inflammation in a model of acute gout. Free Radic Biol Med. 2014;72C:200-9
52. Pan L, Song K, Hu F. et al. Nitric oxide induces apoptosis associated with TRPV1 channel-mediated Ca(2+) entry via S-nitrosylation in osteoblasts. Eur J Pharmacol. 2013;715:280-5
53. Nathan C, Xie QW. Nitric oxide synthases: Roles, tolls, and controls. Cell. 1994;78:915-8
54. Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am J Physiol. 1996;271:C1424-37
55. Kuhlencordt PJ, Chen J, Han F. et al. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation. 2001;103:3099-104
56. Haraguchi K, Kawamoto A, Isami K. et al. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci. 2012;32:3931-41
57. Hong S, Agresta L, Guo C. et al. The TRPV1 receptor is associated with preferential stress in large dorsal root ganglion neurons in early diabetic sensory neuropathy. J Neurochem. 2008;105:1212-22
58. Lau JK, Brown KC, Dom AM. et al. Capsaicin induces apoptosis in human small cell lung cancer via the TRPV6 receptor and the calpain pathway. Apoptosis. 2014;19:1190-201
59. De Jong PR, Takahashi N, Harris AR. et al. Ion channel TRPV1-dependent activation of PTP1B suppresses EGFR-associated intestinal tumorigenesis. J Clin Invest. 2014;124:3793-806
Author contact
![]() Corresponding author: Dr. Tzong-Shyuan Lee, DVM, Ph.D, Department of Physiology, National Yang-Ming University, Taipei 11221, Taiwan; Tel: +886-2-2826-7365; Fax: +886-2-2826-4049; E-mail: tsleeedu.tw
Corresponding author: Dr. Tzong-Shyuan Lee, DVM, Ph.D, Department of Physiology, National Yang-Ming University, Taipei 11221, Taiwan; Tel: +886-2-2826-7365; Fax: +886-2-2826-4049; E-mail: tsleeedu.tw