Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(1):72-86. doi:10.7150/ijbs.13188 This issue Cite
Research Paper
Isovitexin Exerts Anti-Inflammatory and Anti-Oxidant Activities on Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting MAPK and NF-κB and Activating HO-1/Nrf2 Pathways
Hongming Lv1#, Zhenxiang Yu2#, Yuwei Zheng1, Lidong Wang1, Xiaofeng Qin1, Genhong Cheng1, Xinxin Ci1 ![]()
1. Institute of Translational Medicine, The First Hospital of Jilin University, College of Veterinary Medicine, Jilin University, Changchun, China
2. Department of Respiratory Medicine, The First Hospital of Jilin University, Changchun, China.
# These authors contributed equally to this work.
Received 2015-7-8; Accepted 2015-11-2; Published 2016-1-1
Abstract

Oxidative damage and inflammation are closely associated with the pathogenesis of acute lung injury (ALI). Thus, we explored the protective effect of isovitexin (IV), a glycosylflavonoid, in the context of ALI. To accomplish this, we created in vitro and in vivo models by respectively exposing macrophages to lipopolysaccharide (LPS) and using LPS to induce ALI in mice. In vitro, our results showed that IV treatment reduced LPS-induced pro-inflammatory cytokine secretion, iNOS and COX-2 expression and decreased the generation of ROS. Consistent findings were obtained in vivo. Additionally, IV inhibited H2O2-induced cytotoxicity and apoptosis. However, these effects were partially reversed following the use of an HO-1 inhibitor in vitro. Further studies revealed that IV significantly inhibited MAPK phosphorylation, reduced NF-κB nuclear translocation, and upregulated nuclear factor erythroid 2-related factor 2 (Nrf2) and heme oxygenase 1 (HO-1) expression in RAW 264.7 cells. In vivo, pretreatment with IV attenuated histopathological changes, infiltration of polymorphonuclear granulocytes and endothelial activation, decreased the expression of ICAM-1 and VCAM-1, reduced the levels of MPO and MDA, and increased the content of GSH and SOD in ALI. Furthermore, IV treatment effectively increased Nrf2 and HO-1 expression in lung tissues. Therefore, IV may offer a protective role against LPS-induced ALI by inhibiting MAPK and NF-κB and activating HO-1/Nrf2 pathways.
Keywords: Isovitexin, LPS, inflammation, oxidative stress, ALI
Introduction
Oxidative damage and inflammation are associated with the pathogenesis of various diseases, including acute lung injury (ALI) [1, 2]. Both ALI and its more serious form, acute respiratory distress syndrome (ARDS), are severe and acute respiratory dysfunctions. Both conditions present with overwhelming lung inflammation and cause high mortality rates worldwide [3, 4]. Furthermore, oxidative stress, which is present in many animal models of ALI and in ALI patients themselves, plays an essential role in regulating lung injury. For example, the antioxidant N-acetylcysteine attenuates pulmonary function in ARDS patients [5, 6]. Lipopolysaccharide (LPS), a pathogenic endotoxin present in the outer membranes of Gram-negative bacteria, enhances the formation of reactive oxygen species (ROS) and inflammatory mediators and is commonly used to induce pharmacological research models of ALI [7, 8].
Accumulating evidence suggests that LPS can activate nuclear factor-kappa B (NF-κB) and three mitogen-activated protein kinase (MAPK) pathways, inculding the c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 pathways, which result in the overexpression of inflammatory mediators such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), NO and prostaglandin (PG) E2. Cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) are responsible for the production of PGE2 and NO, respectively [9, 10]. On one hand, activation of inflammatory signaling resulted in increasing adhesion molecules expressions, including intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, which play significant roles in cell adhesion and migration [11, 12]. On the other hand, these pro-inflammatory cytokines lead to further damage of epithelial and endothelial cells in the lung. This damage leads to the activation of peripheral polymorphonuclear neutrophils (PMNs). Subsequently, over-activation of PMNs can regulate inflammatory responses and induce tissue damage through oxidative stress [13], the primary source of which is respiratory burst and degranulation. Oxidative stress can increase the production of ROS, myeloperoxidase (MPO) and malondialdehyde (MDA) and decrease the production of anti-oxidative enzymes, including reduced glutathione (GSH), superoxide dismutase (SOD), and heme oxygenase (HO)-1, which collectively protect lung tissues against oxidative damage in vivo [14]. These anti-oxidative enzymes are mediated by nuclear factor-erythroid 2-related factor 2 (Nrf2) [15, 16]. Nrf2 plays an essential role in protecting cells against various inflammatory- and oxidative stress-induced diseases, including age-related macular degeneration (AMD), chronic obstructive pulmonary disease (COPD) and asthma [17, 18]. Under unstressed conditions, Nrf2 is located in the cytoplasm in association with its repressor protein, Kelch-like ECH-associated protein 1 (Keap1). Under stress conditions, Nrf2 dissociates from Keap1, translocates into the nucleus and binds to the antioxidant response element (ARE). This interaction leads to the induction of phase II detoxifying enzymes and cytoprotective genes, such as HO-1 [19]. HO-1 is the major anti-inflammatory and anti-oxidative enzyme among the genes that are mediated by Nrf2 activation [20]. Several reports have suggested that the activation of HO-1 counteracts the inflammatory and oxidative damage that occurs in LPS-induced ALI [21]. Therefore, suppressing inflammation and oxidative stress may be an effective method of preventing and treating ALI.
Flavonoids exhibit a series of pharmaceutical activities, including anti-inflammatory and antioxidant activities, which may play a key role in the prevention of various diseases, including AMD, cancer and diabetes [22, 23]. The glycosylflavonoid isovitexin (IV) (Fig. 1A), extracted from rice hulls of Oryza sativa, possesses various biological activities, including anti-inflammatory and antioxidant activities [24]. However, the protective effect of IV against LPS-induced damage has not yet been demonstrated in vitro or in vivo. In the present study, we evaluated the protective effect of IV and the mechanisms related to inflammation and oxidative stress in RAW 264.7 cells exposed to LPS and in an LPS-induced ALI mouse model.
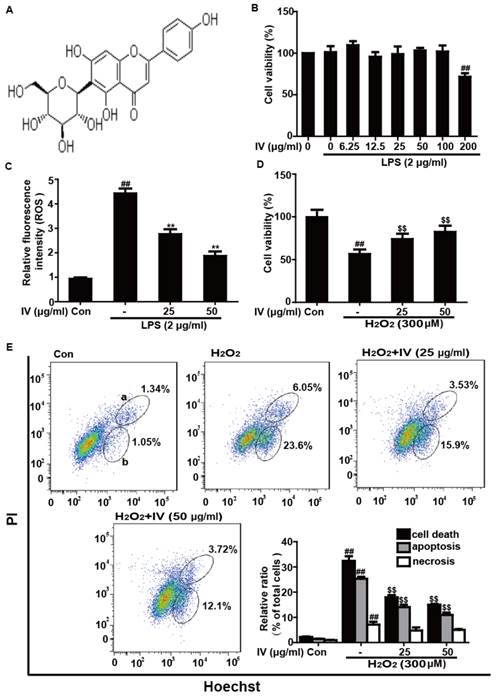
Effects of IV on LPS-induced and H2O2-stimulated damage in RAW 264.7 cells. (A) The chemical structure of isovitexin (IV). (B) Cells were pre-treated with various concentrations of IV for 1 h, and subsequently exposed to LPS (2 μg/ml) for an additional 24 h. Cell viability after LPS exposure was analyzed by an MTT assay. (C) Cells were exposed to IV (25 or 50 μg/ml) for 1 h and then subjected to LPS (2 μg/ml) for 24 h. Then, the LPS-stimulated ROS generation was measured as describes in the Experimental Section. Additionally, cells were exposed to IV (25 or 50 μg/ml) for 1 h and then subjected to H2O2 (300 μM) for 24 h. (D) Cell viability after H2O2 exposure was analyzed by an MTT assay. (E) The percentage of cell apoptosis and necrosis was determined using flow cytometry. “a” and “b” show necrosis and apoptosis, respectively. All data were expressed as means ± SEM of three independent experiments. ##p<0.01 vs the control group; **p<0.01 vs the LPS group; $$p<0.01 vs the H2O2 group.
Materials and Methods
Reagents and chemicals
Isovitexin (IV) with a purity >98% was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Jilin, China). LPS (Escherichia coli 055:B5) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against p-(ERK1/2), ERK1/2, p-(JNK1/2), JNK1/2, p-p38, p38, p65, IκB, p-IκB, Nrf2, HO-1, Lamin B and β-actin were purchased from Cell Signaling (Boston, MA, USA). SP600125, SB203580, and U0126 (specific inhibitors of the JNK1/2, p38, and ERK1/2, respectively) were purchased from Sigma-Aldrich (St. Louis, MO). ICAM-1 and VCAM-1 polyclonal antibody were purchased from ABclonal (MA, USA). The tin protoporphyrin IX (SnPP IX, HO-1 inhibitor) was purchased from Calbiochem (La Jolla, CA, USA). Control siRNA and Nrf2 siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). A Prime-Script RT-PCR kit was purchased from Takara (Dalian, China). In addition, GSH, MDA, SOD and MPO test kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Total ROS/Superoxide Detection Kit was purchased from Enzo Life Sciences Inc (Farmingdale, NY, USA). All other chemicals, unless specifically stated elsewhere, were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Male BALB/c mice, 6-8 weeks old and weighing approximately 18-20 g, were obtained from Liaoning Changsheng Technology Industrial Co., LTD (Certificate SCXK2010-0001; Liaoning, China). All animals were housed in a room kept at a temperature of 24 ± 1 °C under a 12 h light-dark cycle with a relative humidity of approximately 40-80%. The animals were allowed free access to tap water and normal food after feeding for several days. All studies were performed in accordance with the International Guiding Principles for Biomedical Research Involving Animals, which was published by the Council for the International Organizations of Medical Sciences.
Cell culture and MTT assay
The RAW 264.7 mouse macrophage cell line was purchased from the China Cell Line Bank (Beijing China). The RAW 264.7 cells were maintained in DMEM medium supplemented with 3 mM glutamine, 10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 U/ml of streptomycin at 37 °C in a humidified atmosphere containing 5% CO2.
Cell viability was determined by an MTT assay following the manufacturer's instructions. RAW 264.7 cells were plated in 96-well plates (1 × 104 cells/well) and incubated with various concentrations of IV (final concentration: 0-200 μg/ml) and LPS (2 μg/ml) for 24 h. In addition, the cells were pretreated with IV (25 or 50 μg/ml) for 1 h, followed by the addition of H2O2 (300 μM). After 24 h, MTT (5 mg/ml) was added to the cells, which were then incubated for another 4 h.
Endothelial cell culture studies
Human pulmonary artery endothelial cells (HPAECs) were obtained from ATCC and grown in complete endothelial cell growth media. Cells were pretreated for 1 h with IV at 0, 25, or 50 μg/ml. Following pretreatment, cells were exposed to either PBS or LPS at 100 ng/ml for 24 h, and then supernatant was collected for cytokine analysis.
Experimental protocol to create an in vivo ALI model
To create an ALI model, mice were randomly divided into six groups: control (saline), IV only (100 mg/kg, dissolved in 0.5% DMSO), LPS only (0.5 mg/kg, dissolved in saline), LPS (0.5 mg/kg) + IV (50 or 100 mg/kg), and LPS (0.5 mg/kg) + dexamethasone (Dex, 5 mg/kg dissolved in saline). IV (50 or 100 mg/kg) or Dex (5 mg/kg) were administered intraperitoneally. After exposure to IV or Dex for 1 h, the mice were anesthetized with diethyl ether, and LPS was administered intranasally (i.n.) to induce lung injury. After LPS administration for 12 h, the animals were euthanized. Accordingly, bronchoalveolar lavage fluid (BALF) and lung tissue samples were harvested to measure cytokine levels; ROS generation; SOD, GSH, MDA and MPO activity; and COX-2, iNOS, HO-1, and Nrf2 protein expression.
Detection of ROS levels
To measure ROS production, RAW 264.7 cells were grown in 24-well plates (1 × 105 cells/well) for 24 h and then in serum-free DMEM for 6 h. The cells were then pre-incubated with IV (25 or 50 μg/ml) for 1 h, after which LPS (2 μg/ml) was added to the treatment group of plates for an additional 24 h. Next, the cells were stained with 100 μl of ROS/Superoxide detection reagent for 1 h to detect ROS generation. Absorbance was measured in a multi-detection reader (Bio-Tek Instruments Inc.) at excitation and emission wavelengths of 488 nm and 520 nm, respectively.
Flow cytometry assay
H2O2, a type of ROS, can cause various types of cellular injury. Therefore, to further assess the role of IV pretreatment in ROS-induced cell damage, RAW 264.7 cells were grown in 12-well plates (5 × 105 cells/well) for 24 h and were then treated with different concentrations of IV for 1 h, after which H2O2 (300 μM) was added for an additional 24 h. The cells were then washed twice with ice-cold PBS and collected by centrifugation at 1,500 rpm/min for 5 min at 4 °C. Next, the cells were subjected to Hoechst 33342 and propidium iodide staining, and percentages of apoptosis and necrosis were determined using flow cytometry (LSR II Flow Cytometer; BD Biosciences, San Jose, CA, USA).
ELISA assay
RAW 264.7 cells were seeded into 24-well plates (2.5 × 105 cells/well) and then IV (25 or 50 μg/ml) plus LPS (2 μg/ml) were added to the treatment group of plates. Either IV (50 μg/ml) or LPS alone was added to the control group of plates. After stimulation with LPS for 24 h, cell-free supernatants were collected and assayed to determine TNF-α and IL-6 levels. In addition, each recovered BALF sample was centrifuged to detect TNF-α and IL-6 levels using an enzyme-linked immunosorbent assay (ELISA) kit in accordance with the manufacturer's instructions (BioLegend, Inc., CA, USA). The optical density of each well was read at 450 nm.
Total RNA extraction and qPCR
Total RNA was isolated from cells using Trizol reagent according to the procedure described by the manufacturer. After the concentration of RNA was determined using a spectrophotometer, 1 μg of RNA was transformed into cDNA using a Prime-Script RT-PCR kit (Takara). The following PCR primer sequences (forward and reverse, respectively) were used: iNOS: 5′-ACA TCG ACC CGT CCA CAG TAT-3′ and 5′-CAG AGG GGT AGG CTT GTC TC-3′; COX2: 5′-ACA CAC TCT ATC ACT GGC ACC-3′ and 5′-TTC AGG GCG AAG CGT TTGC-3′; β-actin: 5′-TCT GTG TGG ATT GTG GCT CTA-3′ and 5′-CTG CTT GCT GAT CCA CAT CTG-3′.
Isolation of nuclear and cytosolic fractions
Cytoplasmic and nuclear extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Pierce Biotechnology, Rockford, IL, USA), following the manufacturer's instructions. All steps were performed either on ice or at 4 °C.
Nrf2-siRNA transfection
For Nrf2-siRNA transfection, RAW 264.7 cells were grown in 6-well plates (2 × 105 cells/well) and allowed to reach 40%-60% confluence. The cells were then subjected to transient transfection with either Nrf2-negative control siRNA or Nrf2-siRNA using the siRNA transfection reagent LipofectamineTM 2000 following the manufacturer's protocol. After 6 h, the transfected cells were exposed to IV for 18 h, followed by lysis buffer for Western blot analysis.
Histopathological evaluation
The left lungs of the mice were excised at 12 h after the LPS challenge. Histopathological examinations were performed on the mice that were not subjected to BALF collection. The collected lung tissues were immersed in normal 10% neutral buffered formalin, fixed for 48 h, dehydrated in a series of graded ethanol, embedded in paraffin wax, and cut into 5-μm-thick sections. The paraffin-embedded sections were stained with hematoxylin and eosin (H & E) for pathological analysis.
Cell counting in BALF
After the administration of LPS for 12 h, all mice were euthanized prior to BALF collection. The BALF samples were centrifuged to pellet cells. The sedimented cells were resuspended in PBS to obtain total counts of cells, neutrophils and macrophages, which were counted using a hemocytometer. The Wright-Giemsa staining method was used for cytosine staining.
(ROS)/superoxide detection in BALF
Intracellular ROS levels were measured by Fluorence microplate reader or flow cytometry using the Total ROS/Superoxide Detection Kit (Enzo Life Sciences Inc., Farmingdale, NY, USA), according to the manufacturer's instructions. Moreover, BALF cells were washed with 200 μl of washing buffer and loaded with 100 μl of ROS/Superoxide detection reagent, prior to incubation at 37 °C in the dark for 30 min. Absorbance was read at a wavelength of 520 nm after excitation at 488 nm. ROS production was determined based on the increase of relative fluorescence intensity. Thus, in order to further measure ROS levels in alveolar macrophage, BALF cells were stained with SIGLEC-F, which be used for identification of alveolar macrophage in mice. After staining, the cells were washed with 200 μl of washing buffer and loaded with 100 μl of ROS/Superoxide detection reagent, prior to incubation at 37 °C in the dark for 30 min. Finally, the cells were resuspended with 0.4 ml of washing buffer and analyzed by flow cytometry (LSR II Flow Cytometer; BD Biosciences, San Jose, CA, USA).
Measurements of MPO, MDA, GSH and SOD and IL-6 in lung tissues
All of the mice were sacrificed using diethyl ether anesthesia, and their right lungs were excised after 12 h of LPS administration. The lung tissues were homogenized and dissolved in extraction buffer to analyze MPO, MDA, SOD, GSH and IL-6 levels. To examine the accumulation of neutrophils, level of lipid peroxidation and endothelial barrier function in the lung tissues, MPO, MDA and IL-6 content were assessed using commercially available assay kits in accordance with the manufacturer's instructions. Furthermore, to measure antioxidative enzyme activities in the lung tissue, SOD and GSH levels were detected according to the manufacturer's instructions.
Western blot analysis
Cells and lung tissue samples were lysed in RIPA with protease and phosphatase inhibitors for 30 min. Protein concentrations were measured using a BCA protein assay kit (Beyotime, China), and a 40 μg protein sample was electrophoretically transferred onto a PVDF membrane following separation on a 10% SDS-polyacrylamide gel. The membrane was blocked with blocking solution (5% (w/v) nonfat dry milk) for 2 h, followed by an overnight incubation at 4 °C with a specific primary antibody. On the following day, the membrane was incubated for an additional 1 h with HRP-conjugated secondary antibody (1:5000 dilution) at room temperature after thoroughly washing three times with TBST. Bands were detected by ECL (Amersham Pharmacia Biotech, Piscataway, NJ), and band intensities were quantified using Image J gel analysis software. All experiments were performed in triplicate.
Statistical analysis
All of the above-referenced data were expressed as mean ± SEM and analyzed using SPSS19.0 (IBM). Comparisons between experimental groups were conducted using one-way ANOVA, while multiple comparisons were made using the LSD method. Statistical significance was defined as p<0.05 or p<0.01.
Results
Effects of IV on cell viability and LPS-induced ROS generation in RAW 264.7 cells
The effects of different concentrations of IV and LPS (2 μg/ml) on cell viability were assessed using an MTT assay. The results indicated that LPS (2 μg/ml) and up to 100 μg/ml IV were not toxic to RAW 264.7 cells, but treatment with 200 μg/ml IV caused significant cytotoxicity (Fig. 1B). Additionally, because LPS enhances ROS production, leading to oxidative injury, we determined that IV protected against LPS-induced oxidative damage by suppressing intracellular ROS generation in RAW 264.7 cells. In the present study, LPS exposure significantly increased ROS production, which was suppressed by IV treatment (Fig. 1C). Furthermore, the addition of H2O2 markedly reduced cell viability and induced apoptosis, which were attenuated by IV exposure at 25 and 50 μg/ml (Fig. 1D-E).
Effects of IV treatment on LPS-induced increases in TNF-α, IL-6, iNOS, and COX-2 levels in RAW 264.7 cells
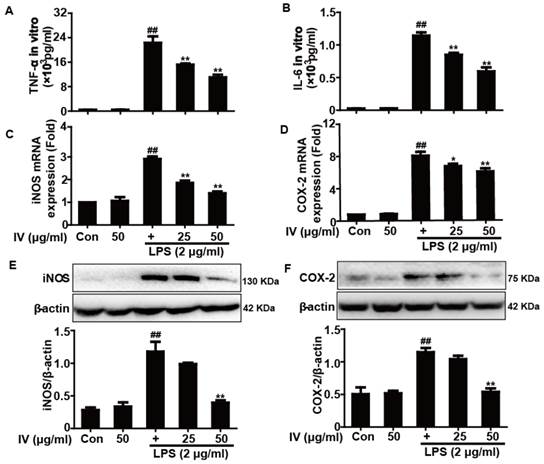
Treatment of RAW 264.7 cells with LPS alone significantly increased cytokine production, whereas IV treatment considerably inhibited production of the cytokines TNF-α and IL-6 (Fig. 2A-B). Moreover, LPS exposure clearly induced iNOS and COX-2 mRNA and protein expression, which were suppressed by IV treatment in RAW 264.7 cells (Fig. 2C-F).
Effects of IV treatment on LPS-induced the levels of TNF-α, IL-6, iNOS, and COX-2 in RAW 264.7 cells. RAW 264.7 cells were pretreated with or without IV for 1 h, and were then subjected to LPS for an additional 24 h. (A, B) The effect of IV on LPS-induced TNF-α and IL-6 production in RAW 264.7 cells. (C, D) Effects of IV on LPS-induced iNOS and COX-2 mRNA expression. Total RNA was extracted from RAW 264.7 cells and gene expression was quantified using real-time PCR. (E, F) Effects of IV on LPS-induced iNOS and COX-2 protein expression. All data were presented as means ± SEM of three independent experiments. ##p<0.01 vs the control group; *p<0.05 and **p<0.01 vs the LPS group.
Effects of IV treatment on LPS-induced activation of the MAPK and NF-κB signaling pathways in RAW 264.7 cells
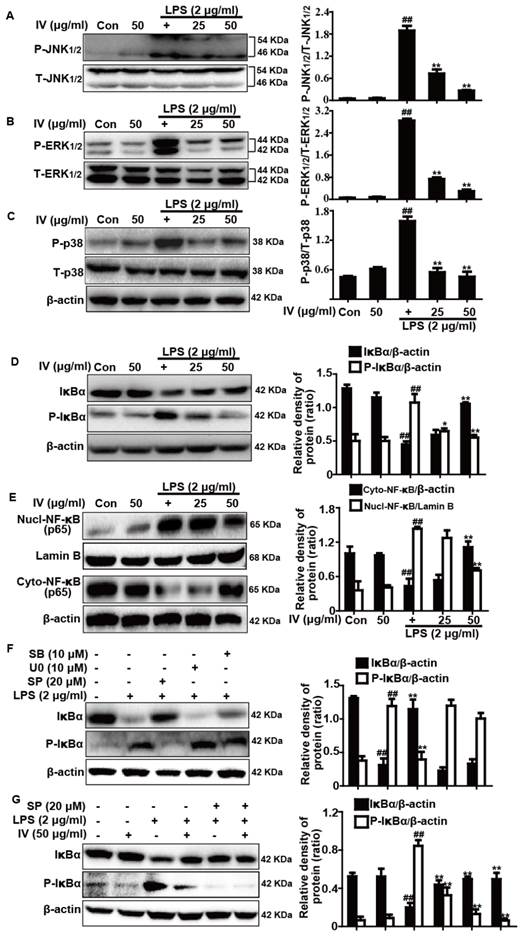
Because the MAPK and NF-κB signaling pathways are two typical inflammatory pathways, we analyzed the effects of IV treatment on LPS-stimulated MAPK and NF-κB activation. The results showed that the levels of JNK1/2, ERK1/2 and p38 phosphorylation dramatically increased in RAW 264.7 cells exposed to LPS, whereas the phosphorylation of these proteins clearly decreased following IV pretreatment (Fig. 3A-C). Furthermore, IV pretreatment effectively blocked IκBα phosphorylation and degradation, which was induced by LPS (Fig. 3D). NF-κB (p65) levels were further evaluated by Western blot analysis of cytoplasmic and nuclear extracts from the macrophages. The results suggested that pretreatment with 50 μg/ml IV significantly reduced nuclear levels of NF-κB (p65), whereas pretreatment with 25 μg/ml IV did not induce this effect (Fig. 3E). Because recent reports showed that MAPKs are responsible for the activation of NF-κB pathway, we treated cells with LPS and MAPK inhibitors (ERK1/2, p38 and JNK1/2 inhibitor) to investigate which inhibitor abrogated LPS-induced NF-κB pathway activation by evaluating IκBα and p-IκBα protein expression. The results suggested that pretreated with JNK1/2 inhibitor alone blocked IκBα phosphorylation and degradation, which was consistent with IV treatment (Fig. 3F-G).
Effects of IV treatment on LPS-activated MAPK and NF-κB signaling pathways in RAW 264.7 cells. Cells were pre-treated with IV (25 or 50 μg/ml) 18 h prior to exposure to LPS for 1 h. The protein levels were analyzed by Western blot. Quantification of relative expression of (A) P-JNK1/2/T-JNK1/2, (B) P-ERK1/2/T-ERK1/2, and (C) P-p38/T-p38. (D) Quantification of P-IκBα and IκBα expression were normalized to that of β-actin. Cells were pre-treated with IV (25 or 50 μg/ml) 18 h prior to expose LPS for 2 h, and the nuclear protein of NF-κB (p65) was analyzed by Western blot. (E) Quantification of NF-κB (p65) expression was normalized as Lamin B. Additionally, (F) Cells were pretreated with or without SP600125 (JNK1/2 inhibitor, 20 μM), U0126 (ERK1/2 inhibitor, 10 μM), and SB203580 (p38 inhibitor, 10 μM) for 18 h, and were then subjected to LPS for an additional 1 h; (G) Cells were pretreated with or without SP600125 (20 μM) for 1 h, and were then subjected to IV (50 μg/ml) for 18 h, and followed by exposure to LPS for an additional 1 h. Quantification of P-IκBα and IκBα expression were normalized to that of β-actin. Similar data were repeated in three independent experiments, and one of three representative experiments is shown. ##p<0.01 vs the control group; *p<0.05 and **p<0.01 vs the LPS group.
Effects of IV treatment on HO-1 protein expression and Nrf2 nuclear translocation in RAW 264.7 cells
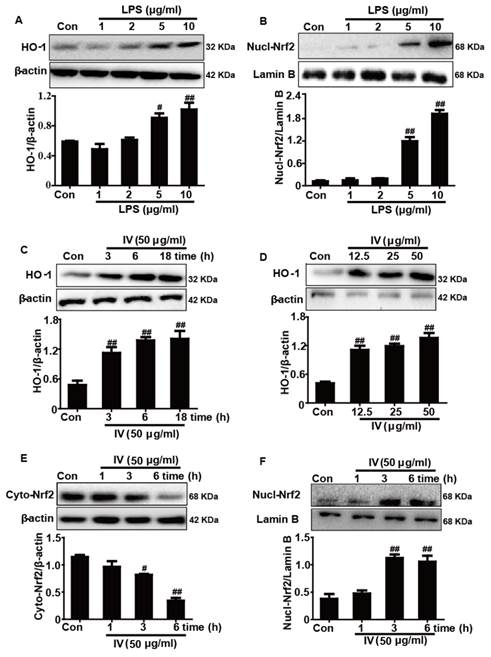
Because Nrf2 and HO-1 protein expression was upregulated by excessive accumulation of intracellular ROS, which was likely the result of high doses of LPS, we first investigated the effects of different concentrations of LPS on Nrf2 and HO-1 protein expression. Our results demonstrated that a 2 μg/ml dose of LPS alone has almost no effect on Nrf2 transcription and HO-1 protein expression in RAW264.7 cells (Fig. 4A-B). In addition, to examine IV-induced HO-1 protein expression, the cells were exposed to different concentrations of IV for varying periods of time. Our results showed that exposure to 50 μg/ml IV for 18 h markedly increased HO-1 protein expression in RAW 264.7 cells. Because Nrf2 is essential for the transcriptional regulation of HO-1 expression, we further determined whether IV treatment enhanced the involvement of Nrf2 activation in HO-1 up-regulation. The results showed that IV treatment increased nuclear levels of Nrf2 and concomitantly decreased cytoplasmic Nrf2 levels in RAW264.7 cells (Fig. 4C-F).
Effects of IV treatment on HO-1 protein expression and Nrf2 nuclear translocation in RAW 264.7 cells. (A, B) Cells were subjected to LPS (1, 2, 5 or 10 μg/ml) for 24 h, and then the total expressed HO-1 protein and the nuclear expressed Nrf2 protein were analyzed by Western blot. (C, D) Cells were treated with IV (50 μg/ml) for the indicated time periods, and subjected to various concentrations of IV (12.5, 25 or 50 μg/ml) for 18 h. The total protein was then analyzed by Western blot. (E, F) Cells were treated with IV (50 μg/ml) for the indicated time periods, and the nuclear and cytoplasmic levels of Nrf2 were analyzed by Western blot. The β-actin and Lamin B were acted as an internal control, respectively. All data were presented as means ± SEM of three independent experiments. #p<0.05 and ##p<0.01 vs the control group.
Effects of Nrf2-siRNA transfection and SnPP on IV-induced HO-1 protein expression in RAW 264.7 cells
To further investigate the role of Nrf2 in IV-induced HO-1 protein expression, we used siRNA to knockdown Nrf2 in RAW 264.7 cells. The cells were subjected to transient transfection with either control or Nrf2 siRNA. Western blot analysis indicated that the Nrf2 siRNA reduced total Nrf2 and HO-1 protein expression compared to the negative control (Fig. 5A). Moreover, we investigated whether Nrf2 siRNA transfection inhibited IV-induced HO-1 protein expression. Our experiments demonstrated that IV-mediated enhancements in HO-1 protein expression were markedly reduced in Nrf2 siRNA-transfected cells versus cells transfected with nonspecific control siRNA (Fig. 5B). These results provide further evidence that the transcriptional activator Nrf2 can modulate HO-1 gene expression. In addition, SnPP, a HO-1 inhibitor, significantly inhibited IV-enhanced HO-1 protein expression (Fig. 5C).
Effects of Nrf2-siRNA transfection and SnPP on IV-induced HO-1 protein expression in RAW 264.7 cells. (A) Cells were subjected to a transient transfection with Nrf2-negative control siRNA or Nrf2-siRNA. (B) Nrf2 mediates IV-enhanced HO-1 protein expression. Nrf2-siRNA or Nrf2-negative control siRNA were transfected into cells for 6 h followed by treatment with IV (50 μg/ml) for 18 h, and proteins were analyzed by Western blot. (C) Cells were exposed to SnPP (40 μM) for 1 h and then treated with IV (50 μg/ml) for 18 h, and proteins were analyzed by Western blot. All data were presented as means ± SEM of three independent experiments. ##p<0.01 vs the control group; ++p<0.01 vs the IV group.
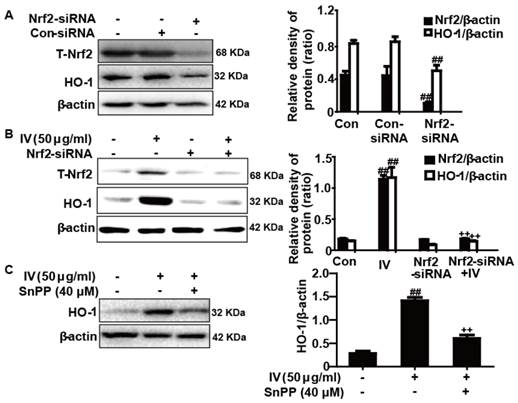
Effects of SnPP on IV-mediated decreases in ROS, TNF-α and IL-6 levels and on iNOS and COX-2 protein expression in RAW 264.7 cells
Increasing evidence has indicated that HO-1 plays an essential role in suppressing oxidative stress and inflammatory responses. We therefore utilized SnPP to determine whether the anti-oxidant and anti-inflammatory activities of IV were associated with increasing HO-1 expression. To accomplish this, RAW 264.7 cells were pretreated with either 25 or 50 μg/ml IV for 1 h with or without SnPP (40 μM), followed by LPS (2 μg/ml) for 24 h. Our results demonstrated SnPP partially counteracted the inhibitory effects of IV on LPS-induced ROS, TNF-α, and IL-6 production and iNOS and COX-2 protein expression (Fig. 6).
Effects of SnPP on IV-inhibited ROS, TNF-α and IL-6 levels as well as iNOS and COX-2 expression protein in RAW 264.7 cells. Cells were exposed to SnPP (40 μM) for 1 h followed by treatment with IV (50 μg/ml) or LPS (2 μg/ml) for 24 h. (A, B) Effects of SnPP on IV-inhibited TNF-α and IL-6 production. (C, D) Effects of SnPP on IV-inhibited iNOS and COX-2 protein expression. (E) The LPS-stimulated ROS generation was measured as described in the Experimental Section. All data were presented as means ± SEM of three independent experiments. ##p<0.01 vs the control group; **p<0.01 vs the LPS group; ++p<0.01 vs the IV group.
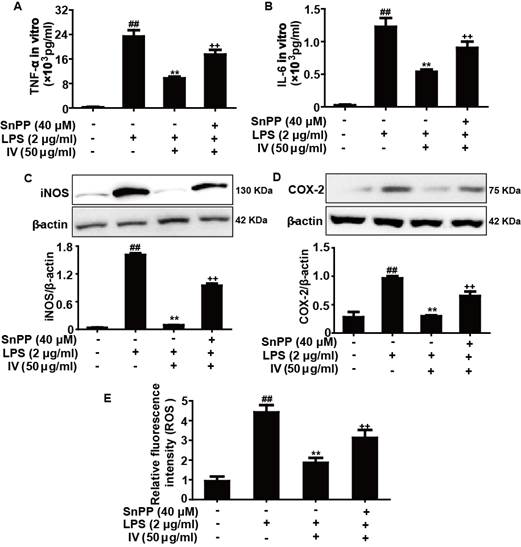
Effects of IV treatment on histological changes and inflammatory cell count in LPS-induced ALI mice
Normal pulmonary histology was observed in the control group (Fig. 7a). Conversely, the lung tissues of the mice administered LPS alone showed marked damage, including inflammatory cell infiltration and alveolar hemorrhage, whereas pretreatment with IV alone did not induce lung tissue damage (Fig. 7b). Furthermore, less severe histopathological changes were observed in the lung sections taken from the IV (50 and 100 mg/kg) (Fig. 7d-e) and Dex groups (Fig. 7f) than in those taken from the LPS group (Fig. 7c); these changes were evaluated by calculating a lung injury score, as shown in Fig. 7B. Additionally, as illustrated in Fig. 7C, the BALF taken from mice exposed to LPS contained more total cells, neutrophils and macrophages compared to the unchallenged group. Nevertheless, compared with LPS exposure, exposure to both IV and Dex significantly reduced the number of total cells, neutrophils and macrophages in BALF.
Effects of IV treatment on histological changes and inflammatory cell count in LPS-induced ALI mice. IV (50 or 100 mg/kg) or Dex (5 mg/kg) was administered intraperitoneally to mice 1 h before LPS pretreatment. (A) Lungs (n = 5) from each experimental group were processed for histological evaluation at 12 h after the LPS challenge: (a) control, (b) IV group, (c) LPS group, (d) LPS + IV (50 mg/kg), (e) LPS + IV (100 mg/kg), (f) LPS + Dex (5 mg/kg). (B) The lung injury score was determined following a five-point scale from 0 to 4 as follows: 0, l, 2, 3, and 4 represent no damage, mild damage, moderate damage, severe damage, and very severe damage, respectively. Representative histological sections of the lungs were stained with hematoxylin and eosin (H & E staining, magnification ×100). (C) The BALF was collected 12 h after LPS challenge to measure the number of total cells, neutrophils and macrophages. All data were presented as means ± SEM (n = 5 in each group). ##p<0.01 vs. control group, *p<0.05 and **p<0.01 vs LPS group.
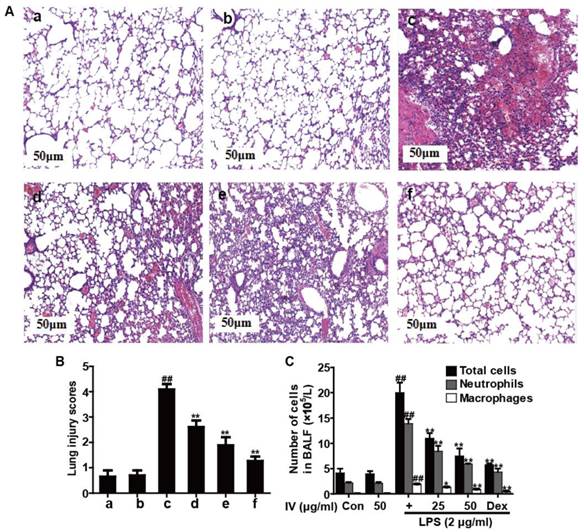
Effects of IV treatment on inflammation and oxidative damage in LPS-induced ALI mice
Based on the above outcome, we further investigated the potential protective effects of IV against inflammation and oxidative damage in LPS-induced ALI mice. Therefore, the levels of TNF-α and IL-6 in BALF were measured by ELISA; the levels ROS in BALF were detected by Fluorence microplate reader or flow cytometry. Additionally, analyses of MPO, MDA, SOD and GSH activities in the lung tissues were performed following manufacturer's instructions. iNOS and COX-2 protein expression in lung tissues was analyzed by Western blot. Our results indicated that IV pretreatment dramatically decreased TNF-α and IL-6 production, ROS generation, and MPO and MDA content, while it obviously increased SOD and GSH levels and effectively inhibited the protein expression of iNOS and COX-2, which play a key role in protecting against LPS-induced inflammation and oxidative stress in LPS-induced ALI mice (Fig. 8).
Effects of IV treatment on inflammation and oxidative damage in LPS-induced ALI mice. IV (50 or 100 mg/kg) or Dex (5 mg/kg) was administered intraperitoneally to mice 1 h before LPS pretreatment. (A, B) Effects of IV on the TNF-α and IL-6 production in the BALF. (C) Effects of IV on the iNOS and COX-2 protein expression in the lungs. (D, E, F and G) Effects of IV on the MPO, MDA, SOD and GSH activity in lung homogenates. (H) ROS generation in the BALF was measured by Fluorence microplate reader. (I) ROS production in alveolar macrophage was determined using flow cytometry. Images show the gated siglec-f high cell separated by ROS and superoxide. All data were presented as means ± SEM of three independent experiments. #p<0.05 and ##p<0.01 vs the control group; *p<0.05 and **p<0.01 vs the LPS group.
Effects of IV treatment on endothelial activation, neutrophils adhesion and endothelial barrier functions
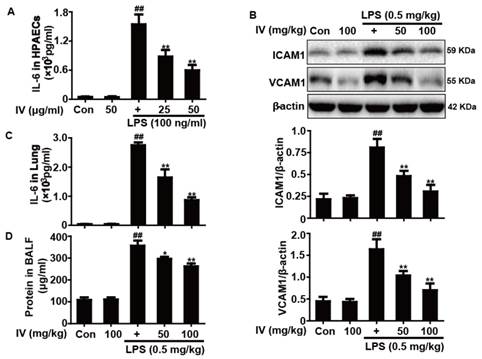
To evaluate IV's ability to suppress endothelial cell activation of LPS, HPAECs were pretreated with IV (25 or 50 μg/ml) for 24 h. The state of endothelial cell activation was determined by measuring IL-6 concentration in supernatants after exposure to LPS for 24 h. IV suppressed LPS-induced IL-6 production in lung endothelial cells in a dose-dependent manner (Fig. 9A). LPS activated inflammatory signaling and led to increased expression of adhesion molecules ICAM1 and VCAM1, which were suppressed by IV (Fig. 9B). In addition, lung IL-6 concentration and BALF protein concentration decreased in IV-treated mice, which suggested that IV can improve endothelial barrier function (Fig. 9C-D).
Effects of IV treatment on endothelial activation, neutrophils adhesion and endothelial barrier functions. HPAECs were pretreated with or without IV for 1 h, and were then subjected to LPS for an additional 24 h. (A) Supernatant was collected for measurement of IL-6 concentration using ELISA. IV (50 or 100 mg/kg) was administered intraperitoneally to mice 1 h before LPS pretreatment. The homogenates of whole and nuclear lung tissues were analyzed by Western blot. (B) Effects of IV on ICAM1 and VCAM1 protein expression in the homogenates of whole lung tissues from LPS-induced ALI mice. (C) Effects of IV on IL-6 concentration in the homogenates of whole lung tissues from LPS-induced ALI mice. The protein concentrations in BALF were measured using a protein assay kit. (D) Effects of IV on protein concentrations in BALF from LPS-induced ALI mice. All data were presented as means ± SEM of three independent experiments. ##p<0.01 vs the control group; *p<0.05 and **p<0.01 vs the LPS group.
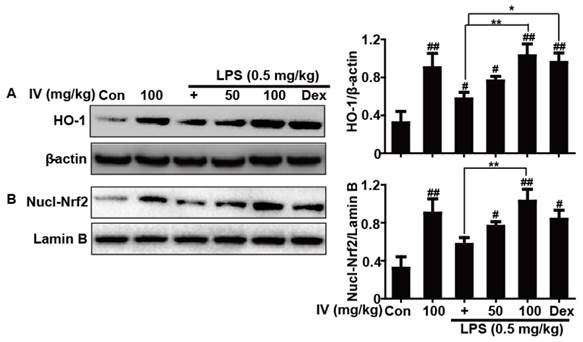
Effects of IV treatment on HO-1 protein expression and Nrf2 nuclear translocation in lung tissues of mice with LPS-induced ALI
Several previous studies have suggested that up-regulation of Nrf2/HO-1 plays a protective role in the devolvement and progression of ALI. In the present study, we found that IV alone clearly increased HO-1 total protein levels and Nrf2 nuclear protein levels, while reducing its cytoplasmic levels, in mice with LPS-induced ALI. These results were consistent with those obtained following IV treatment of RAW 246.7 cells. In addition, while LPS exposure remarkably induced HO-1 expression, IV further increased LPS-induced HO-1 expression compared with that observed in the control group (Fig. 10). Together, these findings suggest that the ability of IV to inhibit inflammation and oxidative stress in mice with LPS-induced ALI may be associated with the up-regulation of HO-1 protein expression and activation of Nrf2 nuclear translocation.
Effects of IV treatment on the HO-1 protein expression and Nrf2 nuclear translocation in the lung tissues of mice with LPS-induced ALI. IV (50 or 100 mg/kg) or Dex (5 mg/kg) was administered intraperitoneally to mice 1 h before LPS pretreatment. The homogenates of whole and nuclear lung tissues were analyzed by Western blot. (A) Effects of IV on HO-1 protein expression in the homogenates of whole lung tissues from LPS-induced ALI mice. (B) Effects of IV on Nrf2 nuclear translocation in the homogenates of nuclear lung tissues with LPS-induced ALI mice. All data were presented as means ± SEM of three independent experiments. #p<0.05 and ##p<0.01 vs the control group; *p<0.05 and **p<0.01 vs the LPS group.
Discussion and conclusion
Inflammation stimulates the generation of ROS, and oxidative stress also induces inflammatory responses [17]. Oxidative stress and inflammation are recognized as interrelated biological events that together are involved in the pathogenesis of ALI [25, 26]. Previous reports have suggested that LPS administration could trigger the release of excessive cytokines, chemokines and ROS, thus inducing an ALI model with similar pathological features to ALI in humans [27, 28]. Moreover, increasing evidence has shown that macrophages play an essential role in the regulation of inflammation responses and that activated macrophages mostly enhance oxygen depletion, which causes the overproduction of ROS [29, 30]. Therefore, any approach that inhibits oxidative stress and inflammation in vitro and in vivo may potentially have an effect on the prevention or treatment of ALI. IV has been shown to possess a variety of biological properties, including antioxidant and anti-inflammatory activities [31, 32]. However, it remains unclear whether these activities of IV serve a mechanistic role in LPS-induced macrophages and ALI mice.
Previous reports have shown that the duration of sustained inflammation is positively and directly associated with the risk of developing inflammatory diseases, including ALI [33, 34]. TNF-α and IL-6, both early inflammatory cytokines, are associated with lung injury severity [35]. Moreover, In contrast, in many inflammatory conditions, inhibition of mediators of NO and PGE2, regulated by iNOS and COX-2, respectively, may have a crucial effect on the prevention of inflammatory responses [36]. In our studies, we found that pretreatment with IV significantly decreased TNF-α and IL-6 levels and inhibited COX-2 and iNOS expression in LPS-induced RAW cells and ALI mice. Importantly, NF-κB, an essential transcription factor, is a key molecule in inflammation. Under normal physiological conditions, NF-κB remains in the cytoplasm linked to the inhibitor of alpha (IκBα) protein, but specific stimulants, such as LPS, lead to the release of NF-κB through phosphorylation and degradation of IκBα [37, 38]. IκB detaches from NF-κB, enabling NF-κB dimers to enter the nucleus and induce the expression of various inflammatory genes, such as those encoding inflammatory cytokines, iNOS, COX-2, and chemokines [39]. Moreover, MAPKs, including JNK1/2, ERK1/2 and p38, also regulate the expression of iNOS, COX-2 and inflammatory cytokines in various cells [40, 41]. The present study demonstrated that IV exposure clearly suppressed MAPK phosphorylation, and blocked IκBα phosphorylation and degradation, which resulted in a decrease in NF-κB (p65) nuclear translocation in LPS-induced RAW 264.7 cells. Intriguingly, recent reports showed that MAPKs are responsible for the activation of NF-κB pathway [42]. In our study, we found that IV suppressed the activation of NF-κB pathway via inhibiting JNK1/2 phosphorylation. Therefore, the IV-inhibited JNK1/2-NF-κB and MAPKs activation may potentially serve as therapeutic targets in inflammatory diseases.
Numerous reports have shown that instillation of LPS into the lungs can lead to overproduction of ROS. This overproduction may exacerbate inflammation and contribute to the pathogenesis of ALI/ARDS [43]. Moreover, H2O2, a type of ROS, can cause various types of cellular injury [44]. Hence, in the present study, we found that LPS treatment significantly increased ROS generation in RAW 264.7 cells and ALI mice; this was effectively counteracted with IV pretreatment. Furthermore, the addition of H2O2 clearly reduced cell viability and induced cell apoptosis, whereas IV ameliorated H2O2-induced cytotoxicity and cell apoptosis. These observations indicated that IV is capable of inhibiting LPS-stimulated ROS production in vitro and in vivo and can counteract ROS-induced oxidative damage in RAW 264.7 cells. Interestingly, several recent reports have suggested that HO-1 commonly acts to protect against inflammation and oxidative tissue injury. HO-1 exerts its protective effect by inhibiting cytokine production, iNOS and COX-2 expression, and ROS generation [45, 46]. Thus, we hypothesized that IV inhibits the induction of TNF-α, IL-6, iNOS, COX-2, and ROS through IV-mediated HO-1 expression. Our observations suggested that IV had an inhibitory effect on the production of the above molecules in RAW 264.7 cells; this effect was partially reversed by SnPP, an HO-1 inhibitor. Nrf2, a critical transcription factor, plays a key role in transcriptionally activating HO-1 gene expression [47]. However, a recent report showed that high-dose LPS exposure could activate HO-1 and Nrf2 expression [30]. Therefore, it is possible that LPS induced Nrf2 and HO-1 expression in our experiment. The results showed that LPS had no effect on the expression levels of Nrf2 or HO-1. In addition, we found that IV treatment remarkably increased HO-1 expression and reduced Nrf2 expression in the cytoplasm, which was directly associated with an increased Nrf2 level in the nucleus. However, transfection of Nrf2 siRNA partially abolished IV-induced HO-1 expression. This result indicated that IV induces HO-1 protein expression via Nrf2 activation in RAW 264.7 cells. In general, IV could play an essential role in inhibiting LPS-induced inflammation and oxidative stress via the up-regulation of Nrf2/HO-1 signaling in cases where LPS does not activate Nrf2 and HO-1 expression.
Based on the above outcome, we further investigated whether the role played by IV in mice with LPS-induced ALI was protective. ALI, or ARDS, is characterized by inflammatory mediator release, lung edema, endothelial and epithelial cell integrity loss, and extensive neutrophil infiltration [48]. To assess histological changes after IV pretreatment in LPS-induced mice, lung sections were evaluated using hematoxylin and eosin staining. We determined that both IV and Dex could ameliorate LPS-induced damage in lung tissues, including inflammatory cell infiltration and alveolar hemorrhage. On the other hand, recent report has suggested that the decreased BAL protein concentration and lung IL-6 secretion was related to the improvement of endothelial barrier function in LPS-induced ALI mice, and that the suppression of endothelial cell activation was associated with the inhibition of IL-6 in LPS-induced HPAECs [49]. Moreover, expression of ICAM-1 and VCAM-1, which are adhesion molecules, are essential for neutrophil adhesion, migration and infiltration [50]. In this study, we demonstrated that IV reduced BAL protein concentration and lung IL-6 secretion, inhibited the LPS-induced IL-6 secretion in HPAECs and mice, and decreased the ICAM-1 and VCAM-1 protein expression in the lung. Taken together, these findings illustrated that IV is a potential therapeutic role in ameliorating neutrophils adhesion and endothelial activation and endothelial barrier functions during ALI. Additionally, ALI leads to excessive macrophage activation and massive neutrophil accumulation in the lungs; together, these responses can promote inflammatory cytokine release and enhance ROS generation [27, 51]. In the present study, IV and Dex both effectively decreased the numbers of total cells, neutrophils and macrophages in BALF. MPO, an enzyme that exists in azurophilic granules of naive PMNs, produces excess oxidants, resulting in tissue damage [52]. Furthermore, LPS instillation into the lungs can induce massive accumulation of ROS, increasing MDA levels. MDA is an end-product of lipid oxidation and therefore can serve to indicate cell membrane damage and destruction [53]. Our data illustrated that IV markedly decreased MPO and MDA levels in lung tissues exposed to LPS. Under unstressed conditions, this amelioration of oxidative injury is derived from the activities of antioxidative enzymes, including SOD and GSH. SOD can convert superoxide anions into hydrogen peroxide, which is then metabolized to water by CAT or GSH [54]. In our experiments, IV increased the activities of SOD and GSH in mice with LPS-induced ALI. Importantly, many recent studies have suggested that Nrf2 is essential for regulating oxidative stress and inflammation in mice with LPS-induced ALI [55]. Moreover, HO-1, an antioxidative enzyme that is regulated by the Nrf2 pathway, also plays a protective role in LPS-induced ALI rats [56]. Our findings demonstrated that IV enhanced Nrf2 and HO-1 expression in LPS-induced ALI mice. These observations indicated that IV can improve severe lung damage by up-regulating the Nrf2/HO-1 signaling pathway.
In conclusion, IV effectively protected against LPS-induced damage, oxidative stress and inflammation in vitro and in vivo. These beneficial effects may be the result of suppression of ROS generation, decreased the TNF-α and IL-6 secretion, and inhibition of COX-2 and iNOS expression, reduced ICAM-1 and VCAM-1 protein expression. Moreover, IV significantly decreased MPO and MDA levels and increased GSH and SOD content in LPS-induced ALI mice. The underlying mechanisms of these findings may be closely associated with inhibition of MAPK and NF-κB and activation of the HO-1/Nrf2 pathways. The components of these pathways may therefore represent potential targets for the prevention and treatment of ALI.
Acknowledgements
This work was supported by a grant from the Natural Science Foundation of Jilin (No. 20150520050JH).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349-61
2. Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal. 2008;10:739-53
3. Avecillas JF, Freire AX, Arroliga AC. Clinical epidemiology of acute lung injury and acute respiratory distress syndrome: incidence, diagnosis, and outcomes. Clin Chest Med. 2006;27:549-57 abstract vii
4. Rubenfeld GD, Caldwell E, Peabody E. et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685-93
5. Bernard GR. N-acetylcysteine in experimental and clinical acute lung injury. Am J Med. 1991;91:54S-59S
6. Bernard GR, Wheeler AP, Arons MM. et al. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest. 1997;112:164-72
7. Matsuzawa A, Saegusa K, Noguchi T. et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587-92
8. Chen X, Yang X, Liu T, Guan M. et al. Kaempferol regulates MAPKs and NF-kappaB signaling pathways to attenuate LPS-induced acute lung injury in mice. Int Immunopharmacol. 2012;14:209-16
9. Becker S, Mundandhara S, Devlin RB. et al. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol Appl Pharmacol. 2005;207:269-75
10. Kim JB, Han AR, Park EY. et al. Inhibition of LPS-induced iNOS, COX-2 and cytokines expression by poncirin through the NF-kappaB inactivation in RAW 264.7 macrophage cells. Biol Pharm Bull. 2007;30:2345-51
11. Lin CC, Pan CS, Wang CY. et al. Tumor necrosis factor-alpha induces VCAM-1-mediated inflammation via c-Src-dependent transactivation of EGF receptors in human cardiac fibroblasts. J Biomed Sci. 2015;22:53
12. Sans E, Delachanal E, Duperray A. Analysis of the roles of ICAM-1 in neutrophil transmigration using a reconstituted mammalian cell expression model: implication of ICAM-1 cytoplasmic domain and Rho-dependent signaling pathway. J Immunol. 2001;166:544-51
13. Korkmaz A, Kolankaya D. Protective effect of rutin on the ischemia/reperfusion induced damage in rat kidney. J Surg Res. 2010;164:309-15
14. Kuo MY, Liao MF, Chen FL. et al. Luteolin attenuates the pulmonary inflammatory response involves abilities of antioxidation and inhibition of MAPK and NFkappaB pathways in mice with endotoxin-induced acute lung injury. Food Chem Toxicol. 2011;49:2660-6
15. Giudice A, Montella M. Activation of the Nrf2-ARE signaling pathway: a promising strategy in cancer prevention. Bioessays. 2006;28:169-81
16. Zhang Y, Gordon GB. A strategy for cancer prevention: stimulation of the Nrf2-ARE signaling pathway. Mol Cancer Ther. 2004;3:885-93
17. Bian Q, Gao S, Zhou J. et al. Lutein and zeaxanthin supplementation reduces photooxidative damage and modulates the expression of inflammation-related genes in retinal pigment epithelial cells. Free Radic Biol Med. 2012;53:1298-307
18. Malhotra D, Thimmulappa RK, Mercado N. et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 2011;121:4289-302
19. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89-116
20. Nguyen T, Sherratt PJ, Huang HC. et al. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J Biol Chem. 2003;278:4536-41
21. Yeh CH, Yang JJ, Yang ML. et al. Rutin decreases lipopolysaccharide-induced acute lung injury via inhibition of oxidative stress and the MAPK-NF-kappaB pathway. Free Radic Biol Med. 2014;69:249-57
22. Sheu SJ, Liu NC, Ou CC. et al. Resveratrol stimulates mitochondrial bioenergetics to protect retinal pigment epithelial cells from oxidative damage. Invest Ophthalmol Vis Sci. 2013;54:6426-38
23. Havsteen BH. The biochemistry and medical significance of the flavonoids. Pharmacol Ther. 2002;96:67-202
24. Kang J, Xie C, Li Z, Nagarajan S. et al. Flavonoids from acai (Euterpe oleracea Mart.) pulp and their antioxidant and anti-inflammatory activities. Food Chem. 2011;128:152-7
25. Nicholls SJ. The complex intersection of inflammation and oxidation: implications for atheroprotection. J Am Coll Cardiol. 2008;52:1379-80
26. Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal. 2007;9:1991-2002
27. Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7:1-7
28. Rojas M, Woods CR, Mora AL. et al. Endotoxin-induced lung injury in mice: structural, functional, and biochemical responses. Am J Physiol Lung Cell Mol Physiol. 2005;288:L333-41
29. Pierce GF. Macrophages: important physiologic and pathologic sources of polypeptide growth factors. Am J Respir Cell Mol Biol. 1990;2:233-4
30. Ye M, Wang Q, Zhang W. et al. Oroxylin A exerts anti-inflammatory activity on lipopolysaccharide-induced mouse macrophage via Nrf2/ARE activation. Biochem Cell Biol. 2014;92:337-48
31. Lin CM, Chen CT, Lee HH, Lin JK. Prevention of cellular ROS damage by isovitexin and related flavonoids. Planta Med. 2002;68:365-7
32. Huang ST, Chen CT, Chieng KT. et al. Inhibitory effects of a rice hull constituent on tumor necrosis factor alpha, prostaglandin E2, and cyclooxygenase-2 production in lipopolysaccharide-activated mouse macrophages. Ann N Y Acad Sci. 2005;1042:387-95
33. Reuter S, Gupta SC, Chaturvedi MM. et al. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603-16
34. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334-49
35. Giebelen IA, van Westerloo DJ, LaRosa GJ. et al. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock. 2007;28:700-3
36. Xia Z, Triffitt JT. A review on macrophage responses to biomaterials. Biomed Mater. 2006;1:R1-9
37. Kang SR, Han DY, Park KI. et al. Suppressive Effect on Lipopolysaccharide-Induced Proinflammatory Mediators by Citrus aurantium L. in Macrophage RAW 264.7 Cells via NF-kappaB Signal Pathway. Evid Based Complement Alternat Med. 2011. 2011
38. de Martin R, Vanhove B, Cheng Q. et al. Cytokine-inducible expression in endothelial cells of an I kappa B alpha-like gene is regulated by NF kappa B. EMBO J. 1993;12:2773-9
39. Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13-20
40. Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol. 2007;292:R18-36
41. Yoon WJ, Lee NH, Hyun CG. Limonene suppresses lipopolysaccharide-induced production of nitric oxide, prostaglandin E2, and pro-inflammatory cytokines in RAW 264.7 macrophages. J Oleo Sci. 2010;59:415-21
42. Maulik N, Sato M, Price BD. et al. An essential role of NFkappaB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1998;429:365-9
43. Blackwell TS, Blackwell TR, Holden EP. et al. In vivo antioxidant treatment suppresses nuclear factor-kappa B activation and neutrophilic lung inflammation. J Immunol. 1996;157:1630-7
44. Mates JM, Sanchez-Jimenez FM. Role of reactive oxygen species in apoptosis: implications for cancer therapy. Int J Biochem Cell Biol. 2000;32:157-70
45. Chakraborty P, Saraswat G, Kabir SN. alpha-Dihydroxychalcone-glycoside (alpha-DHC) isolated from the heartwood of Pterocarpus marsupium inhibits LPS induced MAPK activation and up regulates HO-1 expression in murine RAW 264.7 macrophage. Toxicol Appl Pharmacol. 2014;277:95-107
46. Song YS, Park CM. Luteolin and luteolin-7-O-glucoside strengthen antioxidative potential through the modulation of Nrf2/MAPK mediated HO-1 signaling cascade in RAW 264.7 cells. Food Chem Toxicol. 2014;65:70-5
47. Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol. 1996;15:9-19
48. Zhang X, Song K, Xiong H. et al. Protective effect of florfenicol on acute lung injury induced by lipopolysaccharide in mice. Int Immunopharmacol. 2009;9:1525-9
49. Konter JM, Parker JL, Baez E. et al. Adiponectin attenuates lipopolysaccharide-induced acute lung injury through suppression of endothelial cell activation. J Immunol. 2012;188:854-63
50. Koh DJ, Ahn HS, Chung HS. et al. Inhibitory effects of casticin on migration of eosinophil and expression of chemokines and adhesion molecules in A549 lung epithelial cells via NF-kappaB inactivation. J Ethnopharmacol. 2011;136:399-405
51. Steinberg KP, Milberg JA, Martin TR. et al. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150:113-22
52. Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598-625
53. Victor VM, Rocha M, Esplugues JV. et al. Role of free radicals in sepsis: antioxidant therapy. Curr Pharm Des. 2005;11:3141-58
54. Annapurna A, Ansari MA, Manjunath PM. Partial role of multiple pathways in infarct size limiting effect of quercetin and rutin against cerebral ischemia-reperfusion injury in rats. Eur Rev Med Pharmacol Sci. 2013;17:491-500
55. Kim KH, Kim DH, Jeong N. et al. Therapeutic Effect of Chung-Pae, an Experimental Herbal Formula, on Acute Lung Inflammation Is Associated with Suppression of NF- kappa B and Activation of Nrf2. Evid Based Complement Alternat Med. 2013;2013:659459
56. Tsai CL, Lin YC, Wang HM. et al. Baicalein, an active component of Scutellaria baicalensis, protects against lipopolysaccharide-induced acute lung injury in rats. J Ethnopharmacol. 2014;153:197-206
Author contact
![]() Corresponding author: X.-X.Ci, Institute of Translational Medicine, The First Hospital, Jilin University, Changchun, 130001, China. Tel.: +86 431 88783044; E-mail addresses: xinxincicom (X.-X.Ci.)
Corresponding author: X.-X.Ci, Institute of Translational Medicine, The First Hospital, Jilin University, Changchun, 130001, China. Tel.: +86 431 88783044; E-mail addresses: xinxincicom (X.-X.Ci.)