Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(2):198-209. doi:10.7150/ijbs.13716 This issue Cite
Research Paper
Cyclosporine A Suppressed Glucose Oxidase Induced P53 Mitochondrial Translocation and Hepatic Cell Apoptosis through Blocking Mitochondrial Permeability Transition
Weihua Yu1, Xiaodi Zhang1, Jiangzheng Liu1, Xin Wang1, Shuang Li2, Rui Liu1, Nai Liao1, Tao Zhang1, Chunxu Hai1, ![]()
1. Department of Toxicology, the Ministry of Education Key Lab of Hazard Assessment and Control in Special Operational Environment, Shaanxi Provincial Key Lab of Free radical biology and medicine, School of Public Health, The Fourth Military Medical University, Xi'an, 710032, P. R. China
2. Department of Cardiology, Xijing Hospital, the Fourth Military Medical University, Xi'an, Shaanxi, 710032, China
Received 2015-9-1; Accepted 2015-11-9; Published 2016-1-1
Abstract

P53 is known as a transcription factor to control apoptotic cell death through regulating a series of target genes in nucleus. There is accumulating evidences show that p53 can directly induce cell apoptosis through transcription independent way at mitochondria. However, the mechanism by which p53 translocation into mitochondria in response to oxidative stress remains unclear. Here, glucose oxidase (GOX) was used to induce ROS generation in HepG2 cells and liver tissues of mice. The results showed that p53 was stabilized and translocated to mitochondria in a time and dose dependent manner after GOX exposure. Interestingly, as an inhibitor of mitochondrial permeability transition, cyclosporine A (CsA) was able to effectively reduce GOX mediated mitochondrial p53 distribution without influencing on the expression of p53 target genes including Bcl-2 and Bax. These indicated that CsA could just block p53 entering into mitochondria, but not affect p53-dependent transcription. Meanwhile, CsA failed to inhibit the ROS generation induced by GOX, which indicated that CsA had no antioxidant function. Moreover, GOX induced typical apoptosis characteristics including, mitochondrial dysfunction, accumulation of Bax and release of cytochrome C in mitochondria, accompanied with activation of caspase-9 and caspase-3. These processions were suppressed after pretreatment with CsA and pifithrin-μ (PFT-μ, a specific inhibitor of p53 mitochondrial translocation). In vivo, CsA was able to attenuate p53 mitochondrial distribution and protect mice liver against from GOX mediated apoptotic cell death. Taken together, these suggested that CsA could suppress ROS-mediated p53 mitochondrial distribution and cell apoptosis depended on its inhibition effect to mitochondrial permeability transition. It might be used to rescue the hepatic cell apoptosis in the patients with acute liver injury.
Keywords: Cyclosporine A, Reactive oxygen species, p53, mitochondrial permeability transition, liver disease, cell apoptosis
Introduction
ROS-induced hepatic cell apoptosis is the basis of multiple hepatopathies, such as alcoholic hepatitis (1), virus hepatitis (2), liver resection (3) and drug-induced liver disease (4). Accumulating evidence suggests that p53 plays a central role in the process of ROS-mediated cell apoptosis (5, 6). Under a normal state, p53 exists in the cytoplasm, and sustains a low protein level because of the ubiquitin-mediated protein degradation. But upon cellular stress, p53 is stabilized and induced cell apoptosis. Traditionally, p53 is considered to be a transcription factor and exert pro-apoptotic activity through regulating the transcription of target genes such as Puma, Bcl-2, Bax and Bak in nucleus (7, 8). These p53-dependent transcriptions can be suppressed by pifithrin-α (PFT-α), which is a small, water-soluble pharmacologic inhibitor of p53. In addition, recent reports indicate that p53 can directly activate cell apoptosis through migrating into mitochondria in a transcription-independent manner (9, 10). PFT-μ is able to suppress p53 binding to mitochondria and inducing apoptotic cell death. Meanwhile, it has demonstrated that mitochondrial p53 plays an essential role in the occurrence of various diseases, including ischemia-reperfusion injury, cerebral stroke and Parkinson's disease (10-12). However, the mechanism of p53 translocation into mitochondria under the stress of ROS is still unclear.
Mitochondria are essential for cell life, which affect multiple cellular progresses, including energy metabolism, calcium homeostasis, and signal transduction (13). As the targets and the resources of ROS, mitochondria play an important role in cell apoptosis (14). Upon apoptotic stress stimulation, excessive ROS generation will cause lipid peroxidation and biological dysfunction in mitochondria membrane. Mitochondrial permeability transition pore (MPTP) is a protein channel that spans the inner and outer membranes of mitochondria (15). In unstressed cells, MPTP is closed and has selective membrane permeability. Once under apoptotic stress, sudden opening of MPTP triggers massive ion influx and the collapse of mitochondrial membrane potential, which is called mitochondrial permeability transition (16). In this situation, protons and some apoptotic molecules, such as cytochrome C, are able to enter the mitochondria without restraint. Meanwhile, water and ion influx causes mitochondrial swelling (17). As mitochondrial permeability transition inhibitor, CsA and Sanglifehrin A (SfA) can potently and specifically prevent MPTP opening by binding to CypD (18-20). Therefore, we wonder that whether p53 mitochondrial translocation is related to the MPTP opening in response to ROS. And the mechanism that CsA protects against GOX-inducing hepatic cell apoptosis was also detected.
In present study, glucose oxidase was used to induce ROS generation and hepatic cell apoptosis in vivo and in vitro. GOX can mediate a slowly release of hydrogen peroxide (H2O2) through specifically catalyzing β-D-glucose reaction with oxygen. Since glucose and oxygen are abundant in hepatic tissues, GOX-system is considered as an ideal model for H2O2 releasing to induce oxidative stress (21, 22). Our findings demonstrated that p53 translocation into mitochondria was associated with the opening of MPTP. CsA protected hepatic cells against oxidative stress induced cell apoptosis through blocking p53 mitochondrial distribution and MPTP opening.
Materials and methods
Reagents
Glucose oxidase, Cyclosporine A, Sanglifehrin A, DCFH-DA, DAPI, calcein-AM and Rhodamine-123 were purchased from the Sigma Chemical Corp (St. Louis, USA). The molecular probes including MitoSOX™, Mito-Tracker-Red/Green were obtained from Invitrogen Corporation. (Carlsbad, USA). The Malondialdehyde assay kits were purchased from Beyotime Institute of Biotechnology (Nanjing, CHINA). The Annexin V-FITC/Propidium iodide apoptosis kit, TUNEL apoptosis detection kit, Mitochondria Fractionation Kit, and Nucleus Fractionation Kit was purchased from the Roche (Cambridge, USA). GAPDH, COX IV, Bax, Bcl-2, cytochrome C antibodies, Pifithrin-α, Pifithrin-μ and ionomycin were obtained from Santa Cruz Biotechnology (Santa Cruz, USA). P53 antibodies, Goat anti-Rabbit IgG-FITC, and Goat anti-Rabbit IgG-(H+L)-HRP were purchased from the abcam (Cambridge, USA). Caspase-9 and Caspase-3 Colorimetric Assay Kit was obtained from Keygen (Nanjing, China). All other chemicals and solvents were of analytical grade with highest purity commercially available.
Cell cultures and treatment
The human hepatoma cell lines HepG2 (p53++) were purchased from the ATCC (American Type Culture Collection, USA). Cells were cultured in RMPI 1640 medium with 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin at 37℃ in an atmosphere containing 5% CO2. Cells were exposed to GOX (0-50U) for 0-8 hours with or without PFT-α (20μM/L) (23), Pifithrin-μ (5μM/L) (24), CsA (10μM/L), Sanglifehrin A (20μM/L) (20) and NAC (5mM/L) (21) for 1 hour, respectively. After treatment, cells were collected and processed for further experiments.
Animal's experimental design
Eight-week-old male BabiC mice (23-25g) purchased from the Experimental Animal Center of the Fourth Military Medical University. They were maintained in a standard environmental condition and fed with a standard pellet diet and water ad libitum for 4 weeks. The mice were randomly allocated into four groups (n=6). And then these mice were treated as following: (A) saline + saline; (B) saline + GOX; (C) CsA + GOX; (D) CsA + saline. Animals were injected with CsA (10mg/kg) (25) or physiological saline 4 hours prior to GOX (400U/kg) in vena caudalis for 3 consecutive days. After treatment, the mice were anesthetized with intraperitoneal administration of butylenes (50mg/kg), and then liver tissues were collected. All procedures of animal work were approved by the Animal Welfare Committee of the Fourth Military Medical University in compliance with the relevant laws and institutional guidelines.
Apoptosis detection
HepG2 cells were harvested and suspension in 500 μl binding buffer, which mixed with FITC-labeled Annexin-V (10μl) and PI (10μl). Cells were incubated at room temperature for 30 min, and then detected by Accuri C6 Flow cytometer (BD Sciences, USA). The scatter parameters of the cells (104 cells) were analyzed using a Flow plus system.
Apoptosis was also assessed by TUNEL apoptosis detection kit. After treatment, the liver tissues of mice were staining with TUNEL according to the protocol. Cell nuclei were visualized under a Fluoview laser scanning confocal microscope (OLYMPUS, JAPAN).
Sub-cellular fractionation
Mitochondria were obtained from cultured cells and liver tissue by using Mitochondria/Cytosolic Fractionation Kit. After treatment, HepG2 cells or mice liver tissues were homogenized and centrifuged at 1000g for 10 minutes to remove intact cells and debris, then the supernatants were collected and then centrifuged with at 10,000g for 10 minutes. The pellets were collected as intact mitochondria. In order to collect mitochondrial protein, the pellets were re-suspending with 100 µl of mitochondrial extraction buffer mix containing DTT and protease inhibitors and saved as mitochondrial protein.
Nuclear protein was obtained from HepG2 cells and hepatic tissues using Nuclear Fractionation Kit. After treatment, cells and tissues washed with PBS twice and then homogenated with 1000 µl hypotonic buffer (150 mM NaCl, 10 mM HEPES-KOH (pH 7.9), 0.5 mM PMSF, 1 mM EDTA (pH 8.0), 0.6% NP-40). The homogenates were centrifuged at 2000g for 5 min. Then, the pellets resuspended in lysis buffer (420 mM NaCl, 20 mM HEPES-KOH (pH 7.9), 0.5 mM PMSF, 0.2 mM EDTA (pH 8.0), 0.5 mM dithiothreitol, 1.2 mM MgCl2, 25% glycerol, 0.5 µg/ml aprotinin), kept at 4℃ for 30 min with constant agitation, and centrifuged for 15 min at 10000 g. The supernatants containing the nuclear protein were collected and stored at -80℃.
Determination of ROS
ROS generation was analyzed by flow cytometry after staining with a non-fluorescent probe DCFH-DA. Once penetrated into the cells, DCFH-DA was rapidly oxidized to DCFH in presence of cellular ROS and showed a high green fluorescence. HepG2 cells were harvested and loaded with 500 μl DCFH-DA (10μM) for 30 min at 37◦C in the dark, then washed with PBS (pH=7.4) for three times. The fluorescence was analyzed by flow cytometry.
Measurement of MPTP opening
Calcein-AM was a non-fluorescent probe to detect MPTP activity. In living cells, calcein-AM was hydrolyzed to calcein and well-retained in mitochondria with a strong green fluorescent. Once MPTP opening, calcein was released from mitochondria and quenched by CoCl2 (quenching agent). HepG2 cells were incubated with calcein-Am for 30 min at 37◦C in the dark, and then washed twice with quenching agent CoCl2. Then, cells were analyzed by flow cytometry or confocal microscope with excitation at 488nm and emission at 525nm.
Mitochondrial membrane potential Assessment
Mitochondrial membrane potential was measured by staining with Rhodamine-123, a cationic membrane permeant fluorescent probe, which was accumulated rapidly and specifically in mitochondria. After washing three times with PBS (pH 7.4), the fluorescence intensity of Rhodamine-123 was tested with excitation at 488nm and emission at 525nm. The change of MMP level was indicated by fluorescence intensity of Rhodamine-123, which was analyzed using the Image-Pro plus 6 Software.
MDA content determination
The MDA content was analyzed according to the manufacturer's protocol by using the TBA method and Malondialdehyde (MDA) assay kits.
Mitochondrial swelling
Mitochondria isolated from HepG2 cells and mice liver tissues were incubated with mitochondrial store buffer. Mitochondrial swelling was measured at optical density of 540 nm with a microplate reader.
Determination of hepatic ATP level
ATP levels were measured in HepG2 cells and mice liver tissues using a luciferase's ATP determination kit. After treatment, the prepared cells and tissues were homogenized with the lysis buffer and centrifuged at 1000g for 15 minutes. 5 μl samples were mixed with 195 μl reacting buffer. Luminescence was monitored at 560 nm using a luminometer. The results were normalized to the protein content and ATP level was expressed as μmol/g protein.
Detection activities of caspase-9 and caspase-3
Caspase-9 and Caspase-3 Colorimetric Assay Kit was used to detect the caspase activation. In brief, the lysates of HepG2 cells was processed according to the manufacturer's protocol. The activation of caspase-9 and caspase-3 were measured at optical density of 450 nm and 405 nm, respectively.
Immunofluorescence
To evaluate the sub-cellular localization of p53, we used Confocal imaging of cells double labeled with Mitotraker Red CMX-Ros and p53 primary antibody. HepG2 cells incubated with 100 nM Mitotraker Red CMX-Ros for 30 min at 37 °C. Next, cells were washed with PBS and fixed with 4% paraformaldehyde at 37 °C for 30 min. Fixed cells were permeabilized with 0.1% Triton X-100 for 10 min at 4 °C. Then cells were incubated in blocking solution (5% BSA in PBS) for 1 hour and primary monoclonal p53 antibody (1:100) for 4h at room temperature. After washing, cells were incubated for 1 hour in PBS containing FITC-conjugated goat anti-rabbit antibody (1:100). After staining with DAPI (5μg/ml) for 20 min at 37 °C, images were obtained on a confocal microscope. The excitation and emission wavelengths for Mitotraker were 510/590 nm; for FITC-conjugate, they were 488/510 nm, respectively.
Western blot analysis
After quantifying protein concentrations, the whole, nuclear and mitochondrial protein lysates (30μg/sample) were analyzed by SDS-PAGE (10-15% gels) and blotted onto polyvinyl difluoride membranes. The blots were incubated with primary antibodies about 24h at 4°C and horseradish peroxidase-conjugated anti-IgG for 1 h at 37°C. After washing, the blots were developed with enhanced chemiluminescence and exposed to X-ray film.
Statistical analysis
All data were expressed as mean and standard error of the mean (SEM) from at least three independent experiments. Statistical analysis was carried out by 1-way analysis of variance (ANOVA) and unpaired t test. P<0.05 was considered significantly different.
Results
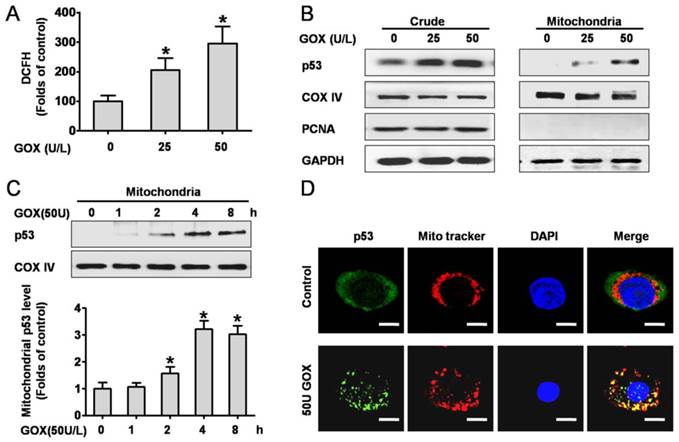
P53 protein was activated and translocated into mitochondria in response to ROS
We firstly investigated the intracellular ROS production when HepG2 cells exposed to GOX. Analysis of the DCFH fluorescence showed that ROS abundance increased approximately 2-3 folds after treated with 25 and 50 U GOX for 4 h (Figure 1A). To determine the effect of ROS on p53 expression and distribution, the crude and mitochondrial lysates were extracted after HepG2 cells exposure to GOX (0-50 U) for 0-8 hours. Western-blot results indicated that GOX caused obvious activation and stabilization of endogenous p53 protein in whole cell lysates (Figure 1B). Moreover, p53 protein in mitochondria lysates showed a robust accumulation and reached its peak after treatment with GOX (50U/L) for 4 hours (Figure 1C). Immunofluorescence staining also verified the above results. Compared with the untreated control cells, GOX induced significant p53 activation and translocation into mitochondria, as indicated by the increased fluorescent intensity and co-localization with Mitotraker red fluorescence. (Figure 1D).
P53 protein was activated and translocated into mitochondria in response to ROS. HepG2 cells were treated with 25 and 50 U GOX for 4 hours. (A) ROS generation was assessed by flow-cytometry method after DCFH-DA staining. (B) P53 protein expression in HepG2 whole-cell lysates and mitochondrial lysates were analyzed by Western-blot. In these experiments, GAPDH, COX IV and PCNA were employed as loading controls respectively. (C) Western blot was used to determined p53 protein in mitochondrial lysates after treatment with 50 U GOX for 0-8 hours. (D) Immunofluorescence staining was detected after cells were either untreated or exposed to GOX (50 U) for 4 hours. Confocal laser scanning microscope images showed the endogenous p53 (green), Mitotraker red fluorescence (red), DAPI staining (blue) and merged images (yellow sites indicated co-localization of p53 and Mitotraker) in HepG2 cells. Bar=10 μm (×300). Data of A and C were presented as the mean ± SD of triplicate independent experiments (n=3). *P<0.05, indicated a significant difference from control.
CsA was able to block oxidative stress induced p53 mitochondrial translocation due to inhibition of MPTP opening
Next, we attempted to find the potential mechanism of GOX-induced mitochondrial p53 translocation. It was well known that the increase of mitochondrial membrane permeability was an important event in cell apoptosis. Once MPTP opening, some apoptosis related molecules were able to flow across the mitochondria uncontrolled. On this basis, we asked whether p53 translocation into mitochondria was related to the opening of MPTP. As a specific fluorescence probe for mitochondrial permeability transition, Calcein-AM was used to evaluate the activity of MPTP. Once MPTP opening, the calcein could efflux from mitochondria and be quenched by CoCl2 in cytoplasm (18). Therefore, mitochondrial permeability transition of HepG2 cells could be reflected by quenched Calcein fluorescence. After treated with GOX, the green fluorescence of calcein decayed gradually in 2-8 hours (Figure 2A). This result indicated that GOX induced the mitochondrial membrane permeability increasing in a time dependent manner. Interestingly, these changes coincided with the accumulation of p53 protein in mitochondria lysates when HepG2 cells exposure to GOX for 0-8 hours (Figure 2B). Therefore, we considered that GOX-induced p53 translocation into mitochondria perhaps depended on MPTP opening.
CsA was able to block oxidative stress induced p53 mitochondrial translocation due to inhibition of MPTP opening. (A) After exposure to GOX (50 U) for 0-8 hours, HepG2 cells were stained with Calcein-AM and assessed by flow-cytometry. (B) The ratio of retained and quenched Calcein fluorescence within 8 hours was calculated relative to the retained Calcein fluorescence in control group. (C) Cells were pretreated with or without CsA (10 μM) for 1 hour, followed by GOX (50 U) and ionomycin (5 μM) stimulation for 4 hours. After treatment, cells stained with Calcein-AM and analyzed by confocal laser scanning microscope. (D) The retained Calcein fluorescence was assessed by using Image-Pro plus 6 Software, and the ratio of retained and quenched Calcein fluorescence was calculated. (E) Cells were pretreated with CsA (10 μM) and PFT-μ (5 μM) for 1 hour following by exposure to GOX (50 U) for 4 hours. The expression of p53 protein in mitochondrial lysates was detected by Western blot. (F) The relative protein abundance of mitochondrial p53 was normalized by banding to COX-IV protein. (G) P53 protein sub-cellular location was detected by immunofluorescence method. (H) Cells were exposed to GOX (50 U) for 4 hours in presence or absence of SfA (20μM) for 1 hour. And mitochondrial permeability transition was assessed by flow-cytometry method after staining with Calcein-AM. (I) P53 protein level in mitochondrial lysates was detected by Western blot. Data were expressed as the mean ± SD of triplicate independent experiments (n=3). *P<0.05, indicated a significant difference from control; #P<0.05, indicated a significant difference with the GOX-treated control.
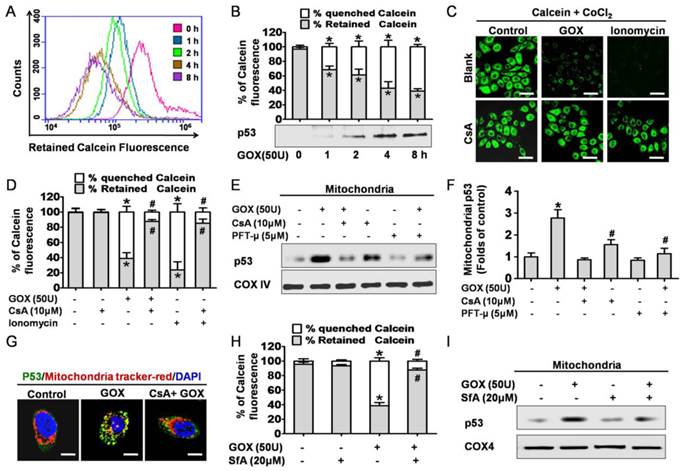
To further demonstrate this hypothesis, cyclosporine A was used to specifically prevent the MPTP opening. Compared with the control group, CsA was able to block GOX-induced increasing of quenched calcein (Figure 2C and 2D). Ionomycin acted as a positive control of mitochondrial permeability transition. As a potent and selective calcium ionophore, ionomycin could mobilize intracellular calcium stores and induce MPTP opening. Then, western blot was conducted to analyze the p53 protein in purified mitochondrial fractions. PFT-μ, a specific inhibitor of p53 mitochondrial translocation, was also used 1 hour prior to GOX exposure. The results showed that both CsA and PFT-μ were able to attenuate the GOX-mediated p53 protein accumulation in mitochondrial lysates (Figure 2E and 2F). Moreover, we also assessed the distribution of p53 protein through immunofluorescence staining method. The mitochondrial p53 protein indicated as the yellow sites, which formed by merging of the green fluorescence (p53 protein) and the red fluorescence (Mitotraker). Compared to GOX treatment group, CSA intervention was able to significantly reduce the p53 protein that anchoring to mitochondria (Figure 2G). Meanwhile, as pharmacological inhibitors of MPTP opening, SfA could effectively inhibit GOX-mediated Calcein fluorescence quench and p53 protein accumulation in mitochondria (Figure 2H and 2I). In conclusion, CsA was able to attenuate the p53 mitochondrial distribution through blocking the opening of MPTP.
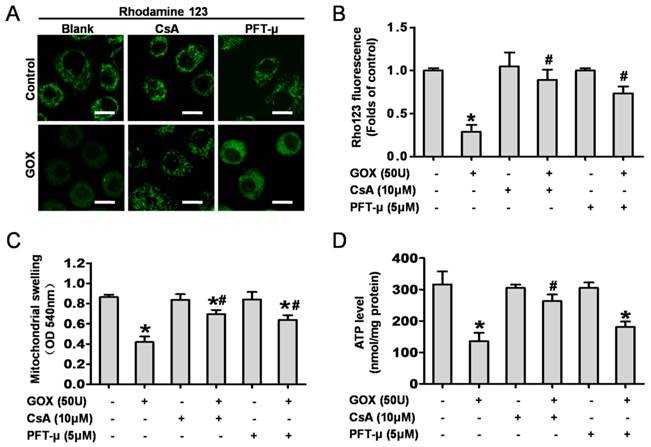
CsA and PFT-μ pretreatment ameliorated GOX exposure induced mitochondrial dysfunction
Then, we detected the role of mitochondrial p53 and the MPTP opening in GOX-mediated mitochondrial dysfunction. CsA and PFT-μ were administrated prior to GOX exposure. The results showed that both CsA and PFT-μ pretreatment were able to significantly attenuate the loss of mitochondrial membrane potential that induced by GOX (Figure 3A and 3B). Upon oxidative stress, sudden MPTP opening triggered massive ion and water influx which would cause mitochondrial swelling. As noted in Figure 3C, GOX induced mitochondrial swelling in a CsA dependent and PFT-μ dependent manner. In response to ROS, the damaged mitochondria would shut down the procession of oxidative phosphorylation and reduce the ATP production (10). Compared with control group, GOX caused an obvious decrease in ATP generation. However, CsA and PFT-μ pretreatment reversed the ATP level decreasing induced by GOX (Figure 3D). These indicated that inhibition of MPTP opening and p53 mitochondrial translocation improved the energy metabolism in mitochondria. Hence, GOX induced mitochondrial dysfunction was able to ameliorate by inhibition of p53 mitochondrial translocation and MPTP opening with CsA and PFT-μ.
CsA and PFT-μ pretreatment ameliorated GOX exposure induced mitochondrial dysfunction. In presence and absence of CsA (10 μM) or PFT-μ (5 μM) for 1 hour, HepG2 cells were exposure to GOX (50 U) for 4 hours. (A) The mitochondrial membrane potential was analyzed by flow cytometer after staining with rhodamine-123. (B) The fluorescence of rhodamine-123 was assessed by using Image-Pro plus 6 Software. Values were expressed as the folds of control. (C) Mitochondrial swelling was measured with the mitochondria isolated from HepG2 cells at optical density of 540 nm. (D) ATP level was measured by ATP Determination Kit. Data were expressed as the mean ± SD of triplicate independent experiments (n=3). *P<0.05, indicated a significant difference from control; #P<0.05, indicated a significant difference with the GOX-treated control.
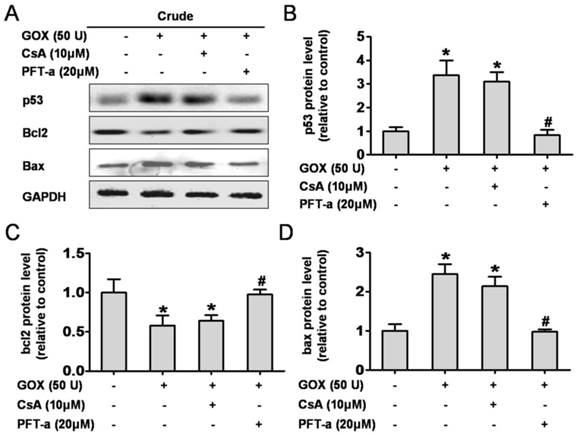
CsA failed to affect GOX-mediated p53 protein activation and transcription of p53 target genes, Bcl-2 and Bax
It is well known that p53 was able to control cell apoptosis through regulating the transcription of target genes such as Bcl-2 and Bax (5). Here, we assessed the role of CsA to protein activation and transcription when HepG2 cells exposed to GOX. Pifithrin-α is a water-soluble compound that could suppress p53 protein transcription. As shown in Figure 4A, PFT-α could suppress GOX-induced p53 protein increase in whole cell lysates, but CsA failed to show such an inhibition effect (Figure 4B). As an important anti-apoptotic Bcl-family member, Bcl-2 protein remained unaffected after CsA intervention compared with the GOX treated group. Notably, Pifithrin-α was able to block the GOX-induced Bcl-2 protein reduction (Figure 4C). Similarly, it was PFT-α rather than CsA that able to prevent the Bax increasing in whole cell lysates (Figure 4D). These data demonstrated that Cyclosporine A had no influence on activation and transcription of p53 target genes, including Bcl-2 and Bax.
CsA failed to affect p53 protein activation and transcription. Cells were treated with GOX (50 U) for 4 fours in presence and absence of CsA (10 μM) or PFT-α (20 μM). (A) Western blot analysis was performed to assess the expression of p53, Bcl-2 and Bax protein in whole cell lysates. (B-D) The relative protein levels were analyzed by densitometry. Data were expressed as the mean ± SD of triplicate independent experiments (n=3). *P<0.05, indicated a significant difference from control; #P<0.05, indicated a significant difference with the GOX-treated control.
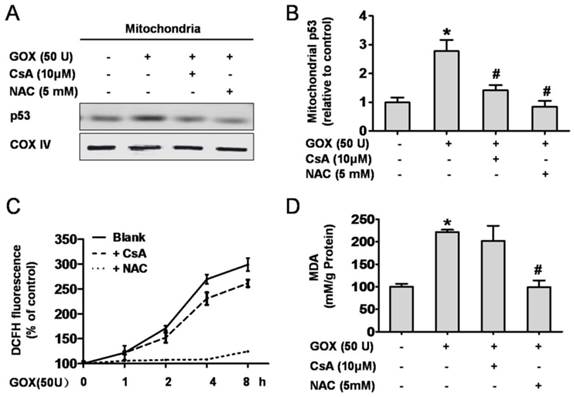
CsA suppressed GOX-mediated p53 mitochondrial translocation without affecting the intracellular redox state
GOX could catalyze β-D-glucose reacting with oxygen, and release of H2O2, which an important oxidant. Therefore, it was no doubt that GOX was able to induce oxidative stress damage in HepG2 cells. As an important antioxidant and free radical scavenger, NAC could effectively inhibit the oxidative stress (26, 27). As shown in 5A and 5B, both CsA and NAC pretreatment were able to suppress the increase of p53 protein abundance in mitochondrial lysates when HepG2 cells exposed to GOX. We wondered whether CsA blocked the p53 mitochondrial translocation through reducing the intracellular ROS generation. To verify this hypothesis, ROS and MDA levels were used to assess the intracellular redox state. After treatment with GOX for 0-8 h, ROS production increased gradually and NAC pretreatment could suppress this procession (Figure 5C). However, intervention with CsA failed to inhibit GOX inducing ROS generation. As a product of lipid peroxidation, MDA was raised about two folds after GOX treatment for 4 hours. Given NAC and CsA pretreatment respectively, we found that it was NAC rather than CsA that prevented the increase of MDA level (Figure 5D). These suggested that CsA failed to show the antioxidant effect, and its ability to suppress p53 mitochondrial distribution had nothing to do with the cellular redox state.
CsA suppressed GOX-mediated p53 mitochondrial translocation without affecting the intracellular redox state. Cells were treated with GOX (50 U) for 4 hours in presence and absence of CsA (10 μM) and NAC (5 mM). (A) Western blot analysis assessed p53 protein expression in mitochondrial lysates. (B) The relative mitochondrial p53 protein level was analyzed by densitometry. (C) ROS generation in different time points (0-8 hours) and (D) MDA level was assessed by DCFH-DA and TBA method respectively. Data were expressed as the mean ± SD of triplicate independent experiments (n=3). *P<0.05, indicated a significant difference from control; #P<0.05, indicated a significant difference with the GOX-treated control.
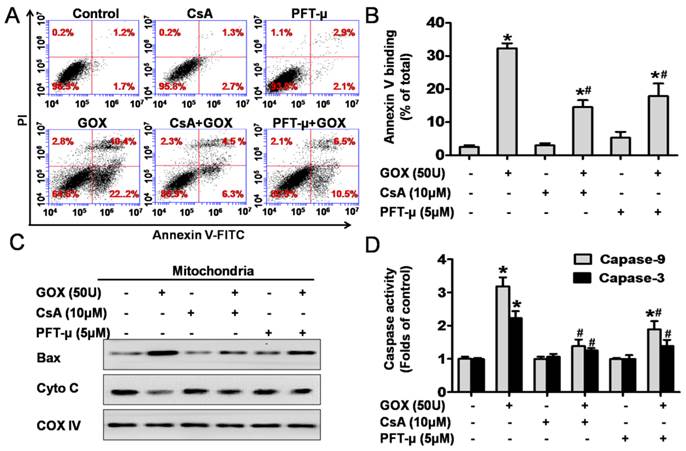
CsA could suppress GOX-mediated HepG2 cell apoptosis due to its inhibition effect to p53 mitochondrial translocation
To detect cell apoptosis, HepG2 cells were analyzed by flow cytometry method after staining with Annexin-V and PI (Figure 6A). The results revealed that the Annexin-V positive population increased from 3% to 32% after exposure to GOX (50U) for 4 hours. However, CsA and PFT-μ pretreatment abolished GOX-induced cell apoptosis to 10% and 17%, respectively (Figure 6B). It has been reported that Bax and cytochrome C played important role during the oxidative stress mediated cell apoptosis. Our previous experiment showed that pretreatment with CsA failed to inhibit Bax up-regulation induced by GOX in whole lysates. However, both CsA and PFT-μ were able to block the ROS-mediated the increasing of Bax protein in purified mitochondrial fractions (Figure 6C). Therefore, CsA did not affect the expression of Bax protein, but it inhibited the procession of Bax accumulation in mitochondria depended on mitochondrial p53. Meanwhile, CsA and PFT-μ treatment was able to prevent the releasing of cytochrome C from mitochondria, and subsequent activation of caspase-9 and caspase-3 in GOX treated HepG2 cells (Figure 6C and 6D). These results suggested that GOX-mediated apoptotic cell death was related to both p53 mitochondrial translocation and MPTP opening.
CsA could suppress GOX-mediated cell apoptosis due to its inhibition effect to p53 mitochondrial translocation. Cells were treated with GOX (50 U) for 4 hours in the presence and absence of CsA (10 μM) or PFT-μ (5 μM). (A) Cell apoptosis was assessed using flow cytometry with FITC-Annexin-V/PI double staining. (B) Apoptotic rate was represented by the percentage of Annexin-V positive cells in total cell population. (C) Western blot analysis was performed to assess the expression of apoptotic protein Bax and cytochrome C in mitochondrial lysates. (D) Colorimetric method was used to assay the activities of caspase-9 and caspase-3 at optical density of 450 nm and 405 nm, respectively. Data were expressed as the mean ± SD of triplicate independent experiments (n=3). *P<0.05, indicated a significant difference from control; #P<0.05, indicated a significant difference with the GOX-treated control.
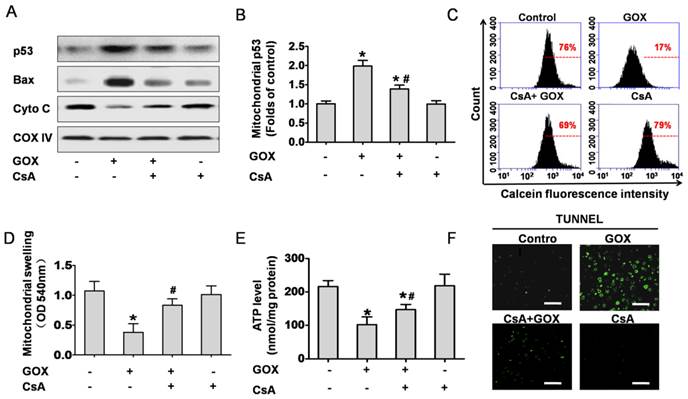
CsA prevented GOX-induced p53 mitochondrial translocation and cell apoptosis in the liver tissue of mouse
Based on the cell culture experiment, we investigated the role of CsA to hepatic tissues in vivo. Mitochondrial fractions in liver tissues were extracted to analyze protein expression. The results indicated that GOX induced p53 and Bax accumulation in mitochondria following by reduction of mitochondria cytochrome C, and CsA showed an obvious inhibition effect (Figure 7A and 7B). Meanwhile, mitochondria isolated from the liver tissues were stained with Calcein-AM and measured by flow cytometry. As shown in Figure 7C, CsA was able to suppress the quench of Calcein induced by GOX. Mitochondrial swelling was another classical hallmark of MPTP opening. Similarly, GOX caused dramatically increasing of mitochondrial swelling and CsA treatment reversed this procession (Figure 7D). As noted in Figure 7E, CsA was able to suppress the reduction of ATP after exposure to GOX. These indicated that CsA improved the energy metabolism of mitochondria. TUNEL staining was used to determine the cell apoptosis in hepatic tissues. GOX induced substantial TUNEL positive cells in the hepatic tissues, and CsA showed an obvious inhibition effect (Figure 7F). These results demonstrated that MPTP opening was crucial for oxidative stress induced p53 mitochondrial translocation and cell apoptosis in hepatic tissues of mice.
CsA prevented GOX-induced p53 mitochondrial translocation and cell apoptosis in the liver tissues of mice. Animals were injected with GOX (400 U/kg) with or without CsA (10 mg/kg) in vena caudalis for 3 consecutive days. (A) Western blot analyzed the expression of p53, Bax and Cyto C in the mitochondria of liver tissue. (B) The abundance of p53 protein was analyzed relative to COX-IV (n=3). (C) The mitochondria isolated from the mice liver were stained with Calcein-AM and measured by flow cytometry. (D) Mitochondrial swelling was measured at optical density of 540 nm (n=6). (E) Hepatic ATP level was detected by firefly luciferase ATP assay (n=6). (F) The liver tissue apoptosis was determined by TUNEL staining. Bar=50 μm (×60). Data were expressed as the mean ± SD. *P<0.05, indicated a significant difference from control; #P<0.05, indicated a significant difference with the GOX-treated control.
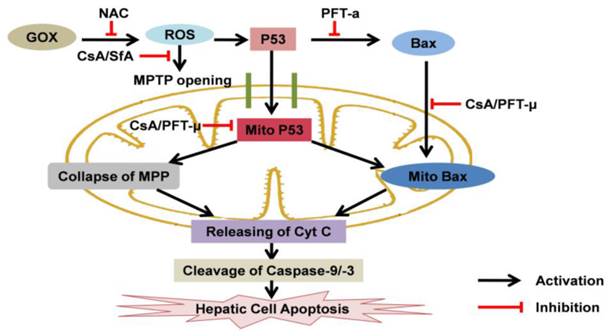
The effects of CsA to p53 protein and cell apoptosis when hepatic cells exposed to oxidative stress: CsA was able to suppress ROS-mediated MPTP opening, mitochondrial p53 accumulation and subsequent hepatic cell apoptosis. Mitochondrial p53 was crucial for GOX-mediated cell apoptosis, which associated with inducing collapse of MPP, Bax mitochondrial recruiting and mitochondrial cytochrome C releasing.
Discussion
In current study, GOX was used to induce H2O2 release and oxidative damage in HepG2 cells and liver tissues of mice (28). As a major stress sensor, p53 played an important role in ROS mediated cell apoptosis. After exposure to GOX, we observed a robust activation and mitochondrial translocation of p53 protein in vitro and in vivo. Meanwhile, GOX caused an obvious up-regulation of Bax protein in whole cell lysates, which indicated that GOX mediated apoptotic cell death in both p53 transcription-dependent and -independent manners. Here, we mainly focused on the p53 protein that located in mitochondria during ROS induced hepatic cell apoptosis. Interestingly, we found that the accumulation of mitochondrial p53 protein was synchronized with MPTP opening. MPTP was an important protein channel that regulated the mitochondrial membrane permeability. In physiological condition, it just allowed molecular masses less than 1.5 KD to flow across the mitochondria (15). Therefore, p53 protein mainly distributed in cytoplasm rather than mitochondria in unstressed hepatic cells, because of the closed MPTP. However, once the mitochondrial permeability transition occurred in response to ROS, substantial p53 protein was activated and accumulated in mitochondria. A further research demonstrated that both CsA and SfA were able to prevent ROS inducing mitochondrial p53 distribution through inhibiting the activity of MPTP. In conclusion, MPTP opening contributed to p53 translocation into mitochondria when hepatic cells exposed to GOX.
As a typical immunosuppressant, CsA was given to organ transplant patients and the ones with autoimmune diseases in clinical (29, 30). It works mainly depended on suppression the T cells activation and proliferation (31). Moreover, it was also known as a specific inhibitor to suppress mitochondrial permeability transition when cells encounter to apoptotic stress (19). MPTP opening was considered as a hall mark in mitochondrial pathway cell apoptosis. Due to MPTP opening, massive ion influx would trigger collapse of mitochondrial membrane potential and shut down oxidative phosphorylation and ATP production (32, 33). Subsequently, cytochrome C released from mitochondria into cytoplasm and binding with procaspase-9 and Apaf-1 in the presence of ATP (34). The cleaved caspase-9 triggered a further activation of caspase-3, which would execute the cell apoptosis (35). In the present study, GOX-induced obvious apoptosis features including collapse of mitochondrial membrane potential, ATP depletion, mitochondrial swelling, mitochondrial cytochrome C releasing which followed by activation of caspase-9 and caspase-3. Interestingly, these processions were suppressed due to closing the MPTP with CsA and inhibiting p53 mitochondrial translocation with PFT-μ. The results demonstrated that CsA attenuated GOX-mediated apoptotic cell death, which depended on the inhibition effect of p53 mitochondrial translocation. Although Bcl-2 and Bax might participate in the GOX-mediated cell apoptosis, we found that CsA didn't influence the Bcl-2 and Bax protein expression in whole cell lysates. However, GOX induced Bax accumulation in mitochondria which could be inhibited by PFT-μ and CsA. These indicated that mitochondrial p53 was involved in Bax mitochondrial translocation in GOX-induced apoptosis. Meanwhile, it had reported that collapse of MPP and mitochondrial Bax accumulations were able to induce the procession of cytochrome C release from mitochondria into cytoplasm (36-38). To sum up, CsA was able to protect oxidative stress induced mitochondrial pathway cell apoptosis through inhibiting p53 mitochondrial translocation and MPTP opening.
In previous study, we demonstrated that MPTP opening and p53 mitochondrial distribution were two important events in response to ROS. GOX was used to catalyze β-D-glucose reacting with oxygen, which could mediated slowly releasing of H2O2. Hence, it was no doubt that GOX induced an obvious increase of ROS and MDA. Although both NAC and CsA were able to suppress the p53 mitochondrial distribution in GOX treated HepG2 cells, the mechanisms that they worked were different. As a specific free-radical scavenger, NAC prevented p53 mitochondrial translocation through reducing the ROS abundance. However, CsA failed to reduce the intracellular oxidative stress induced by GOX, which suggested that CsA didn't have antioxidant ability. As an inhibitor of mitochondrial permeability transition, SfA was also able to block the p53 accumulation in mitochondria. Therefore, CsA suppressed p53 protein mitochondrial distribution was probably due to block the mitochondrial permeability transition.
ROS induced hepatic cell apoptosis was an important nosogenesis of multiple hepatic diseases, and there was no effective emergency treatment drug (39, 40). Here, we conferred one potential mechanism for oxidative stress induced p53 mitochondrial distribution, which was related to MPTP opening. P53 mitochondrial distribution and MPTP opening were crucial for ROS-mediated cell apoptosis, and CsA showed obvious inhibition effect for two events. Therefore, CsA might be a therapeutic strategy to rescue the hepatic cell apoptosis in the patients with acute liver injury.
Abbreviations
CypD: cyclophilin D; CsA: cyclosporine A; COX IV: Cytochrome c Oxidase IV; Cyt C: cytochrome C; DCFH-DA: 2',7'-dichlorofluorescin diacetate; DTNB: 5,5'-dithio bis-(2-nitrobenzoic acid); dUTP: 2'-deoxyuridine 5'-triphosphate; GOX: glucose oxidase; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; MPP: mitochondrial membrane potential; MPTP: mitochondrial permeability transition pore; MDA: malondialdehyde; PCNA: proliferating cell nuclear antigen; PFT-α: pifithrin-α; PFT-μ: pifithrin-μ; PMSF: phenylmethyl-sulfonyl fluoride; ROS: reactive oxygen species; Rho123: Rhodamine 123; SfA: Sanglifehrin A; TBA: Thiobarbituric acid; TUNEL: terminal-deoxynucleoitidyl transferase mediated nick end labeling.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81473010 and No. 31400724). We are grateful to Wen Song for providing language assistance and the article writing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jampana S C, Khan R. Pathogenesis of alcoholic hepatitis: Role of inflammatory signaling and oxidative stress. World J Hepatol. 2011;3(5):114-117
2. Blackham S, Baillie A, Al-Hababi F. et al. Gene Expression Profiling Indicates the Roles of Host Oxidative Stress, Apoptosis, Lipid Metabolism, and Intracellular Transport Genes in the Replication of Hepatitis C Virus. Journal of Virology. 2010;84(10):5404-5414
3. Oguz S, Kanter M, Erboga M. et al. Effects of Urtica dioica on oxidative stress, proliferation and apoptosis after partial hepatectomy in rats. Toxicology and Industrial Health. 2013
4. Jaeschke H, Mcgill M R, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metabolism Reviews. 2012;44(1):88-106
5. Ye Y, Liu J, Xu J. et al. Nano-SiO2 induces apoptosis via activation of p53 and Bax mediated by oxidative stress in human hepatic cell line. Toxicology in Vitro. 2010;24(3):751-758
6. Narayanan A, Amaya M, Voss K. et al. Reactive oxygen species activate NFκB (p65) and p53 and induce apoptosis in RVFV infected liver cells. Virology. 2014;449:270-286
7. Mondal S, Bhattacharya K, Mallick A. et al. Bak Compensated for Bax in p53-null Cells to Release Cytochrome c for the Initiation of Mitochondrial Signaling during Withanolide D-Induced Apoptosis. PLoS ONE. 2012;7(3):e34277
8. Nakajima W T N. Synergistic induction of apoptosis by p53-inducible Bcl-2 family proteins Noxa and Puma. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2007;74(2):148-157
9. Lee S K, Kim Y C, Song S B. et al. Stabilization and translocation of p53 to mitochondria is linked to Bax translocation to mitochondria in simvastatin-induced apoptosis. Biochemical and Biophysical Research Communications. 2010;391(4):1592-1597
10. Vaseva A V, Marchenko N D, Ji K. et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149(7):1536-1548
11. Lan R, Zhang Y, Xiang J. et al. Xiao-Xu-Ming decoction preserves mitochondrial integrity and reduces apoptosis after focal cerebral ischemia and reperfusion via the mitochondrial p53 pathway. J Ethnopharmacol. 2014;151(1):307-316
12. Ottolini D, Cali T, Negro A. et al. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet. 2013;22(11):2152-2168
13. Folmes C D L, Dzeja P P, Nelson T J. et al. Mitochondria in Control of Cell Fate. Circulation Research. 2012;110(4):526-529
14. Sousa C A, Soares E V. Mitochondria are the main source and one of the targets of Pb (lead)-induced oxidative stress in the yeast Saccharomyces cerevisiae. Applied Microbiology and Biotechnology. 2014;98(11):5153-5160
15. Bernardi P, Forte M. The mitochondrial permeability transition pore. Novartis Found Symp. 2007;287:157-164 164-169
16. Halestrap A P, Richardson A P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. 2014
17. Toledo F D, Pérez L M, Basiglio C L. et al. The Ca2+-calmodulin-Ca2+/calmodulin-dependent protein kinase II signaling pathway is involved in oxidative stress-induced mitochondrial permeability transition and apoptosis in isolated rat hepatocytes. Archives of Toxicology. 2014
18. Cao X H, Zhao S S, Liu D Y. et al. ROS-Ca(2+) is associated with mitochondria permeability transition pore involved in surfactin-induced MCF-7 cells apoptosis. Chem Biol Interact. 2011;190(1):16-27
19. Sharov V G, Todor A, Khanal S. et al. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. Journal of Molecular and Cellular Cardiology. 2007;42(1):150-158
20. Qin L S, Jia P F, Zhang Z Q. et al. ROS-p53-cyclophilin-D signaling mediates salinomycin-induced glioma cell necrosis. J Exp Clin Cancer Res. 2015;34:57
21. Kumar S, Sitasawad S L. N-acetylcysteine prevents glucose/glucose oxidase-induced oxidative stress, mitochondrial damage and apoptosis in H9c2 cells. Life Sci. 2009;84(11-12):328-336
22. Rost D, Welker A, Welker J. et al. Liver-homing of purified glucose oxidase: A novel in vivo model of physiological hepatic oxidative stress (H2O2). Journal of Hepatology. 2007;46(3):482-491
23. Kanno S, Kurauchi K, Tomizawa A. et al. Pifithrin-alpha has a p53-independent cytoprotective effect on docosahexaenoic acid-induced cytotoxicity in human hepatocellular carcinoma HepG2 cells. Toxicol Lett. 2015;232(2):393-402
24. Krukowski K, Nijboer C H, Huo X. et al. Prevention of chemotherapy-induced peripheral neuropathy by the small-molecule inhibitor pifithrin-mu. Pain. 2015;156(11):2184-2192
25. Nijboer C H, Heijnen C J, van der Kooij M A. et al. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann Neurol. 2011;70(2):255-264
26. Lee Y H, Su S B, Huang C C. et al. N-acetylcysteine attenuates hexavalent chromium-induced hypersensitivity through inhibition of cell death, ROS-related signaling and cytokine expression. PLoS One. 2014;9(9):e108317
27. Wang C, Chen K, Xia Y. et al. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One. 2014;9(9):e108855
28. Milton R D, Giroud F, Thumser A E. et al. Hydrogen peroxide produced by glucose oxidase affects the performance of laccase cathodes in glucose/oxygen fuel cells: FAD-dependent glucose dehydrogenase as a replacement. Phys Chem Chem Phys. 2013;15(44):19371-19379
29. Tourneur E, Ben M S, Chassin C. et al. Cyclosporine A impairs nucleotide binding oligomerization domain (Nod1)-mediated innate antibacterial renal defenses in mice and human transplant recipients. PLoS Pathog. 2013;9(1):e1003152
30. Macleod H, Goodwin D G, Damphousse C. et al. The DTH effector response and IL-2 are unaffected by cyclosporine A in autoimmune B6D2F1 mice. Cell Immunol. 2010;266(1):14-23
31. Wang X, Zhang D, Wang L. et al. Cyclosporine treatment of angioimmunoblastic T-cell lymphoma relapsed after an autologous hematopoietic stem cell transplant. Exp Clin Transplant. 2015;13(2):203-205
32. Bonora M, Wieckowsk M R, Chinopoulos C. et al. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene. 2015;34(12):1608
33. Mukherjee R, Mareninova O A, Odinokova I V. et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut. 2015
34. Matias A C, Manieri T M, Cipriano S S. et al. Diethyldithiocarbamate induces apoptosis in neuroblastoma cells by raising the intracellular copper level, triggering cytochrome c release and caspase activation. Toxicol In Vitro. 2013;27(1):349-357
35. Kanno T, Nishizaki T. Sphingosine induces apoptosis in hippocampal neurons and astrocytes by activating caspase-3/-9 via a mitochondrial pathway linked to SDK/14-3-3 protein/Bax/cytochrome c. J Cell Physiol. 2011;226(9):2329-2337
36. Gomez-Crisostomo N P, Lopez-Marure R, Zapata E. et al. Bax induces cytochrome c release by multiple mechanisms in mitochondria from MCF7 cells. J Bioenerg Biomembr. 2013;45(5):441-448
37. Li T, Brustovetsky T, Antonsson B. et al. Oligomeric BAX induces mitochondrial permeability transition and complete cytochrome c release without oxidative stress. Biochim Biophys Acta. 2008;1777(11):1409-1421
38. Gottlieb E, Armour S M, Harris M H. et al. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10(6):709-717
39. Dey A, Lakshmanan J. The role of antioxidants and other agents in alleviating hyperglycemia mediated oxidative stress and injury in liver. Food Funct. 2013;4(8):1148-1184
40. Zhu W J, Li M, Liu C. et al. Avermectin induced liver injury in pigeon: mechanisms of apoptosis and oxidative stress. Ecotoxicol Environ Saf. 2013;98:74-81
Author contact
![]() Corresponding author: Department of Toxicology, the Ministry of Education Key Lab of Hazard Assessment and Control in Special Operational Environment, Shaanxi Provincial Key Lab of Free radical biology and medicine, School of Public Health, The Fourth Military Medical University, Xi'an, 710032, Shaanxi, P. R. China. Tel: +86 02983374879. Fax: +86 02984774879. Email: cx-haiedu.cn
Corresponding author: Department of Toxicology, the Ministry of Education Key Lab of Hazard Assessment and Control in Special Operational Environment, Shaanxi Provincial Key Lab of Free radical biology and medicine, School of Public Health, The Fourth Military Medical University, Xi'an, 710032, Shaanxi, P. R. China. Tel: +86 02983374879. Fax: +86 02984774879. Email: cx-haiedu.cn