Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(2):219-234. doi:10.7150/ijbs.13062 This issue Cite
Research Paper
Long-term Stress with Hyperglucocorticoidemia-induced Hepatic Steatosis with VLDL Overproduction Is Dependent on both 5-HT2 Receptor and 5-HT Synthesis in Liver
Jihua Fu ![]() , Shaoxin Ma1#, Xin Li1#, Shanshan An1#, Tao Li1#, Keke Guo1#, Min Lin1, Wei Qu1, Shanshan Wang1, Xinyue Dong2, Xiaoyu Han2, Ting Fu2, Xinping Huang2, Tianying Wang2, Siyu He2
, Shaoxin Ma1#, Xin Li1#, Shanshan An1#, Tao Li1#, Keke Guo1#, Min Lin1, Wei Qu1, Shanshan Wang1, Xinyue Dong2, Xiaoyu Han2, Ting Fu2, Xinping Huang2, Tianying Wang2, Siyu He2
1. Postgraduates of China Pharmaceutical University, Nanjing, China
2. Undergraduates of China Pharmaceutical University, Nanjing, China
# These authors contributed equally to this work
Received 2015-6-27; Accepted 2015-10-23; Published 2016-1-1
Abstract

Hepatic triglycerides production and adipose lipolysis are pivotal for long-term stress (LTS) or hyperglucocorticoidemia-induced insulin resistance. 5-hydroxytryptamine (5-HT) has been demonstrated to induce hepatic lipid metabolic abnormality by activating mammalian target of rapamycin (mTOR). In present study, we explored whether 5-HT is involved in LTS effects in liver using restraint stress-exposed rats and cultured primary rat hepatocytes and HepG2 cells. LTS with hyperglucocorticoidemia induced hepatic 5-HT synthetic increase with tryptophan hydroxylase 1 (Tph1) up-regulation, and 5-HT2 receptor (5-HT2R, including 5-HT2A, 2B receptor) up-regulation in liver and visceral adipose, as well as hepatic mTOR activation with triglycerides and VLDL overproduction with steatosis, and visceral adipose lipolytic increase with high blood free fatty acids (FFAs) level. 5-HT exposure exhibited LTS-like effects in both tissues, and both LTS and 5-HT effects could be abolished significantly by blocking 5-HT2R. In HepG2 cells dexamethasone or palmitate-induced mTOR activation with triglycerides and VLDL overproduction were accompanied by up-regulations of 5-HT synthesis and 5-HT2R, which were significantly abolished by gene silencing Tph1 or 5-HT2R and were almost fully abolished by co-silencing of both, especially on VLDL overproduction. Chemical inhibition of Tph1 or/and 5-HT2R in both hepatocytes exhibited similar abolishment with genetic inhibition on dexamethason-induced effects. 5-HT-stimulated effects in both hepatocytes were fully abolished by blocking 5-HT2R, while 5-HT itself also up-regulated 5-HT2R. In conclusion, up-regulated hepatic 5-HT synthesis and 5-HT2R induced by both glucocorticoid and FFAs are crucial for LTS-induced hepatic steatosis with VLDL overproduction, while 5-HT by acting on 5-HT2R mediates mTOR activation in liver.
Keywords: Long-term stress, 5-hydroxytryptamine 2A, 2B receptor (5-HT2A, 2BRs), 5-hydroxytryptamine synthesis, Triglycerides (TGs) synthesis, Very low-density lipoprotein (VLDL) assembly
Introduction
5-hydroxytryptamine (5-HT), also known as serotonin, is a primal signaling molecule in the center and periphery, which is conserved across phyla that is implicated in the control of energy balance [1]. The majority of body's 5-HT presents in periphery, which is synthesized from L-tryptophan by the enzymes tryptophan hydroxylase 1 (Tph1) and aromatic amino acid decarboxylase (AADC), and regulates physiological functions in the hepato-gastrointestinal tract etc [2, 3]. Liver steatosis has been confirmed to involve 5-HT in several animal models, such as in a nonalcoholic steatohepatitis (NASH) model induced by a choline-methionine-deficient diet, Tph1-deficient mice showed reduced hepatocellular injury and less severe inflammation [4], lymphocytic choriomeningitis virus infection-induced liver steatosis is also serotonin-dependent [5] and more importantly, 5-HT mediates L-tryptophan feeding-caused exacerbation on hepatic steatosis induced by a high fat and high fructose diet through the activation of mammalian target of rapamycin (mTOR)-p70 S6 kinase (p70S6K) signaling pathway [6]. The various functions of 5-HT can be attributed to its diverse receptors, and seven major isoforms of 5-HT receptor (5-HTR), namely 5-HT1R to 5-HT7R, were found in the different types of cells. There are three subtypes of 5-HT2R, 5-HT2AR, 5-HT2BR and 5-HT2CR, among of which 5-HT2AR and 5-HT2BR are found in liver [7, 8]. In addition, 5-HT reuptake transporter (5-HTT), which helps 5-HT enter into the cell through plasma membrane, and other 5-HTRs are also found in the hepatocytes. It is showed that 5-HT effects on regulating hepatic energy metabolism are mediated by 5-HT2R. An in vitro study using 3T3-L1 adipocytes has demonstrated that 5-HT2AR involved in 5-HT-induced insulin resistance (IR) [9]. In an animal research using mouse, gut-derived 5-HT is upregulated during fasting, and the 5-HT signaling through 5-HT2BR in hepatocytes promotes gluconeogenesis and prevents glucose uptake in a glucose transporter 2 (GLUT2)-dependent manner, thereby further favoring maintenance of blood glucose levels [10]. These evidences suggest that 5-HT by acting on 5-HT2R is significant for controlling hepatic lipid and glucose metabolism.
Stress is an unavoidable state in daily life. Stress induces over-activity in the hypothalamic-pituitary-adrenocortical (HPA) axis with a resultant consequence of hyperglucocorticoidemia, and other increases in many hormones and cytokines levels in blood, such as adrenaline, growth hormone, serotonin, and proinflammatory cytokines [11, 12]. It is known that chronic over-secretion of stress mediators, particularly glucocorticoids (GCs), induce widely disorders, such as increased hepatic gluconeogenesis, decreased glucose uptake in peripheral tissues, hepatic steatosis, and suppressed immune responses [13]. Although the harmfulness of long-term stress (LTS) to health appears quite severe, the cellular or molecular mechanism involving in pathophysiology of stress remains largely unknown. In the present study, by rats exposed to LST or 5-HT with or without blocking 5-HT2A, 2BR, and primary cultured rat hepatocytes or HepG2 cells treated with 5-HT, dexamethasone (Dex), or palmitate (PA) with or without chemical or genetic inhibition of Tph1 or/and 5-HT2A, 2BR, we demonstrated that LTS with hyperglucocorticoidemia can stimulate 5-HT synthesis and 5-HT2A, 2BR expression in the hepatocytes in a direct and indirect manner, which is crucial for GC-induced abnormality of hepatic lipid metabolism by activating mTOR.
Materials and Methods
Materials and reagents
Male Sprague-Dawley rats were purchased from B&K Universal Group Limited (China). Sarpogrelate Hydrochloride (Sar) was from Mitsubishi Tanabe Pharma Corporation (Japan). 5-hydroxytryptamine hydrochloride (5-HT) was from Alfa Aesar (China). HepG2 and 293T cell lines were from American Type Culture Collection (ATCC, USA). DMEM, fetal bovine serum, and Trypsin-EDTA were from Gibco (USA). Dexamethasone (Dex) was from Cisen Pharmaceutical Ltd (China). Sodium palmitate (PA), BSA, collagenase IV, sodium heparin and trypan blue were from Sigma Chemical Co (USA). Rat tail tendon collagen Type I was from Shengyou Biotechnology Co., Ltd (China). Rapamycin (RAP) was from ApexBio Technology LLC (USA). Para-chlorophenylalanine (pCPA) was from Tokyo Chemical Industry Co. (Japan). GV248 vector was from GeneChem (China). Lipofectamine 2000 was from Invitrogen (USA). Glucose, TGs, low density lipoprotein cholesterol (LDL-c), alanine aminotransferase (ALT), glutamic-oxalacetic transaminase (AST), FFAs, and glycerol spectrophotometer kits for serum or adipose tissue detection were from Nanjing Jiancheng Bioengineering Institute (China). TGs enzyme-test kit for liver tissue and cells detection was from Applygen Technologies Inc (China). ELISA kits for tumor necrosis factor-α (TNF-α), corticosterone (Cort), growth hormone (GH), adrenline (Adr), insulin, VLDL and 5-HT were from Abcam (Hong Kong). RNAiso Plus Isolation Reagent was from Takara (Japan). Gene primers were from Baocheng Biology Co Ltd. (China). Cell lysis buffer and phosphatase inhibitor were from KeyGEN Boitech (China). Cytosol and Membrane Protein Extraction Kit was from Beyotime Institute of Biotechnology (China). Anti-5-HT2AR antibody was from GeneTex, Inc (USA). Anti-5-HT2BR antibody was from Becton and Dickinson Company Inc (USA). Anti-microsomal TGs transfer protein (MTTP) and anti-glycerin-3-phosphate acyltransferase 1 (GPAT1) antibodies were from Santa Cruz Biotechnology, Inc (CA). Anti-phospho-mTOR (Ser2448), anti-mTOR, anti-phospho-p70S6K (Thr389/412) and anti-p70S6K antibodies were from Signalway Antibody (USA). Anti-tryptophan hydroxylase-1 (Tph1) and anti-aromatic L-amino acid decarboxylase (AADC) antibodies were from Epitomics Abcam (HK). Anti-β-actin antibody was from Bioworld Technology, Inc (China). Horseradish peroxidase (HRP)-coupled secondary antibodies were from Santa Cruz Biotechnology, Inc (CA).
Animal experiments
The experiments and procedures were approved by the Laboratory Animal Care Committee at China Pharmaceutical University (Protocol No. 20130905). The rats (8-week old) were housed in standard polypropylene cages, and were kept on a 12-h day/night cycle with free access to food and water in a temperature-controlled environment (24 ± 2 ◦C).
The rats were exposed to stress with or without sarpogrelate (Sar) treatment for 25 days. The stress was executed using restraint stress modified from Eunice Y. Yuen et al [14]. The rats were individually restrained in an air-accessible cylinder with a free movement banned, food and water-deprived manner for 8 hours daily (4 hours in the morning and afternoon, respectively), while the control rats were placed in their original cages undisturbed deprived to food and water. Sar were orally administrated with a dose of 30 mg/kg twice daily in the morning and afternoon, which were done at 30 min before executing the stress. Meanwhile, the control or stressed without drug-treated rats were orally administrated with 0.5 % CMC-Na, a vehicle of dissolving Sar. During the un-stressed period, all rats were returned to their home environment like control rats with free accessed to food and water. At the end of the study period, the rats were all fasted but accessed to water freely for 8 hours, after administration rats were then further deprived to food and water for 4 hours with that, except for control rats, stress-exposed rats were all stressed. Food intake and bodyweight were recorded per five days in the 25-day experimental period.
In another experiment, the rats were exposed to 5-HT with or without Sar treatment, or not for 25 days. 5-HT was subcutaneously administrated with 7.0 mg/kg twice daily in the morning and afternoon, while the control rats were subcutaneously administrated with normal saline. Sar was administrated like the Dex-exposed experiment. In the 25-day 5-HT-exposured period, food intake and bodyweight were measured, and serum 5-HT levels were also detected by collecting blood via tail bleeding under local ethocaine anaesthesia before 5-HT administration in the afternoon. At the end of the study period, the rats were all fasted but accessed to water freely for 12 hours, and 5-HT and/or Sar were administrated at 4 hour before killed.
The rats were anesthetized and humanely killed by withdrawal of blood to obtain serum by low speed centrifugation. The liver and intra-abdominal adipose (mesenteric, bilateral perirenal and epididymal) were immediately removed with washing in ice-cold phosphate-buffered saline (PBS) and weighed. Hepatic index (HI) and visceral obesity index (VOI) (Percentage of body weight) were calculated. Serum samples, liver and adipose tissue were stored at -80 °C until analysis.
Cell culture
HepG2 cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS) supplemented with penicillin and streptomycin and maintained at 37 °C in a 5% CO2 atmosphere, and were incubated for 24 h. Then, cells were washed twice with PBS, and the medium was changed to serum-free DMEM containing the antibiotics. After one-hour incubation, the cells were treated with drug, respectively, according to each experiment.
Preparation of primary hepatocytes
Male Rats (8-10 weeks old) were anesthetized, and then hepatocytes were isolated by a nonrecirculating in situ collagenase perfusion of livers cannulating through the hepatic portal vein as described previously [15] with minor modifications. Livers were first perfused in situ with 2.4 g/l HEPES containing calcium-free salt solution, followed by perfusion with calcium-Hank's balanced salt solution containing collagenase (0.05 g/100 ml). The livers were then gently minced on a Petri dish and filtered with 200 nm nylon mesh. Hepatocytes were washed three times with DMEM medium. Cell viability was consistently >90%, as determined by trypan blue exclusion. Isolated hepatocytes were incubated on 6-well plates (1×106 cells/well) coated with rat tail collagen type I in DMEM containing 20% FBS at 37 °C in a 5% CO2 atmosphere for 2-10 h before treatment. Then, cells were changed to serum-free DMEM. After one-hour incubation, the cells were treated with drug, respectively, according to each experiment.
RNA Interference
The target shRNAs against the human Tph1, 5-HT2AR and 5-HT2BR genes (Genbank access numbers: NM_004179, NM_000621, NM_000867, respectively) for RNA interference were designed as follows, Tph1: 5'-CCG GCC CAA GAA ATT GGC TTG GCT TCT CGA GAA GCC AAG CCA ATT TCT TGG GTT TTT-3', 5-HT2AR: 5'-CCG GGC CTA CAA GTC TAG CCA ACT TCT CGA GAA GTT GGC TAG ACT TGT AGG CTT TTT-3', 5-HT2BR: 5'-CCG GCC GAT ATA TCA CCT GCA ATT ACT CGA GTA ATT GCA GGT GAT ATA TCG GTT TTT-3', and the lentiviral vector GV248-negative (control siRNS): TTC TCC GAA CGT GTC ACG T without interference suppression on the expression of human gene. Target fragment was annealed by 3 'and 5' single strand and digested by AgeI and EcoRI restriction enzymes, then it was connected to the GV248 vector (GeneChem, China). After sequencing identification, except for GV248-vector, GV248-Tph1 alone or in a combination with GV248-5-HT2AR and GV248-5-HT2BR were used to transfect 293T packaging cells by Lipofectamine 2000. The supernatant was collected after 48 h and filtered through a 0.45 μm filtrate membrane, then Polybrene was add into it until the final concentration was 5 μg/mL. Supernatant was used to infect target cells for 12 h, and then placed into new medium for another 72 h. After infection, fluorescence of green fluorescent protein (GFP) was detected via fluorescence inversion microscope system (Olympus, Japan), and Tph1, 5-HT2AR and 5-HT2BR protein expressions were detected by western blot. Infected HepG2 cells were then transferred to DMEM supplemented with 10% FBS in a six-well plate, further incubation with Dex or sodium palmitate (PA) treatment as same as HepG2 cells culture.
Serum, liver tissue, and cells biochemical analysis
Liver or adipose tissue was homogenized in 0.02 M phosphate buffered solution (pH 7.4). Each parameter in serum and liver was measured according to the protocols of respective kits. ALT, AST, Glucose, TGs, FFAs, and low density lipoprotein cholesterol (LDL-c) level in the serum, and glycerol in adipose tissue were measured using spectrophotometer kit, respectively. TGs content in the liver tissue and cells was measured using a TGs enzyme-test kit. Tumor necrosis factor-α (TNF-α), corticosterone (Cort), growth hormone (GH), adrenline (Adr), insulin, VLDL and 5-HT level in the serum, liver tissue, or the cultured primary rat hepatocytes and HepG2 cells was measured using ELISA kit, respectively. The Homeostasis Model of Assessment-IR (HOMA-IR) index, a marker of IR, was calculated as serum glucose × serum insulin / 22.5 [16].
Oil-red O staining in the liver tissue
Liver tissue was placed in an optimal cutting temperature using a cryostat, and 6 μm sections were made. Tissue slices were rinsed with PBS, rinsed with 60% isopropanol, and stained with freshly prepared Oil-red O working solution for 15 min, then rinsed with 60% isopropanol again and counterstained with hematoxylin. For cells, primary rat hepatocytes or HepG2 cells were fixed with 4% paraformaldehyde and then stained with Oil-red O. The slides were mounted and examined under an optical microscope.
RNA isolation and gene expression analysis
Reverse transcription-polymerase chain reaction (RT-PCR) assay was used to analyze gene expression. Total RNA was isolated from frozen rat hepatic tissue or HepG2 cells using RNAiso Plus Isolation Reagent (TAKARA, Otsu, Shiga, Japan). Total RNA solution was first reserve transcribed and then immediately amplified in a GeneAmp PCR system (Eppendorf, Germany). Primers used in the RT-PCR were: Tph1 (forward-ACT GCG ACA TCA ACC GAG AA; reverse-GGC TAA CCC TGA CAG GAA AT), 5-HT1AR (forward-TGG GTA CTC TCA TTT TCT GC; reverse-GCG TCC TTT TGT TCA CAT AG), 5-HT1BR (forward-TGT AAT CGC TAC GGT GTA TC; reverse-TGT AAT CGC TAC GGT GTA TC), 5-HT2AR (forward-GGA TTT ACC TGG ATG TGC; reverse-TGG ATG GAC CGT TGG AAG), 5-HT2BR (forward-CAG CAG CAG AGG AAA TGA; reverse-ATC CAG GGA AAT GGC ACA), 5-HT2CR (forward-CTG AAG CAA TCA TGG TGA AC; reverse-CGC AGT TGA AAA TAG CAC AT), 5-HT3AR (forward- GCC TTT ATT CTA CGC AGT CA; reverse-TGC GAA ACA GCA GCC TGT C), 5-HT7R (forward-GCT CAA ACA CGA AAG GAA AA; reverse-AGA GCT TCC GGT TGA TAT TC), apolipoprotein B100 (ApoB100) (forward-AGT GCT GCC GTC TCT ACA GT; reverse-AGA GAC AGC GGT GGC TAA CT), adipose triglyceride lipase (ATGL) (forward-TTC AAG TTT CCT TGC AGA GT; reverse-CTC CCA AAC TGA CCC TTA AA), acetyl-CoA carboxylase (ACCase) (forward-GCC AGC AGA ATT TGT TAC TC; reverse-AGA CGA TGC AAT CTT ATC CC), and GAPDH (forward-TAT CGG ACG CCT GGT TAC; reverse-TGC TGA CAA TCT TGA GGG A) in the rat liver tissure, and ApoB100 (forward-ATC TCG TGT CTA GGA AAA GC; reverse-AAA ACA ACT CAG CTC TTG AC), ACCase (forward-TATC ACA GCA ACC AAG TAG T; reverse-CGA ACA AAG AAC CTG TAG TC), and GAPDH (forward-CAA CCG GGA AGG AAA TGA AT; reverse-TTG ATT TTG GAG GGA TCT CG) in HepG2 cells. PCR products were then subjected to agarose gel electrophoresis and analyzed with GeneGenius automatic gel imaging and analysis system (Tanon-3500, China). To exclude variations due to RNA quantity and quality, the data for all genes were adjusted to GAPDH.
Western blot analysis
Frozen liver tissues or cells were homogenized in lysis buffer with or without adding phosphatase inhibitors for detection of protein phosphorylation. Then samples were incubated on ice for 15 mins. Cytosol or plasma membrane protein was extracted by cytosol and membrane protein extraction kit, respectively. Debris was removed by centrifugation. Protein concentration of each sample was determined in order to calculate the loading amount. Samples were separated by SDS-PAGE gel and electrophoretically transferred to nitrocellulose membrane. Nonspecific binding sites were blocked with Trisbuffered saline containing 5% BSA for 12 h at 4°C. Membranes were first incubated with the primary antibodies, respectively. Then the membranes were incubated with the horseradish peroxidase (HRP)-coupled secondary antibodies. Immunopositive bands were visualized by a chemiluminescent method (ECL, Tanon-5200, China), and the protein bands were quantified by densitometry. Parallel blotting of β-actin was used as internal control.
Statistical analysis
Results were expressed as mean ± standard deviation (S.D.). Data differences between groups were tested for statistical significance using one-way analysis of variance (ANOVA) followed by LSD's multiple comparison test, while the differences of mRNA or protein levels of 5-HT1B, 2A, 2B, 7Rs between the LST or 5-HT group and Ctrl group were tested for using student's t-test. P < 0.05 was considered significant.
Results
LTS-induced hepatic steatosis with VLDL overproduction and adipose lipolysis in rat are associated with 5-HT 2 receptor
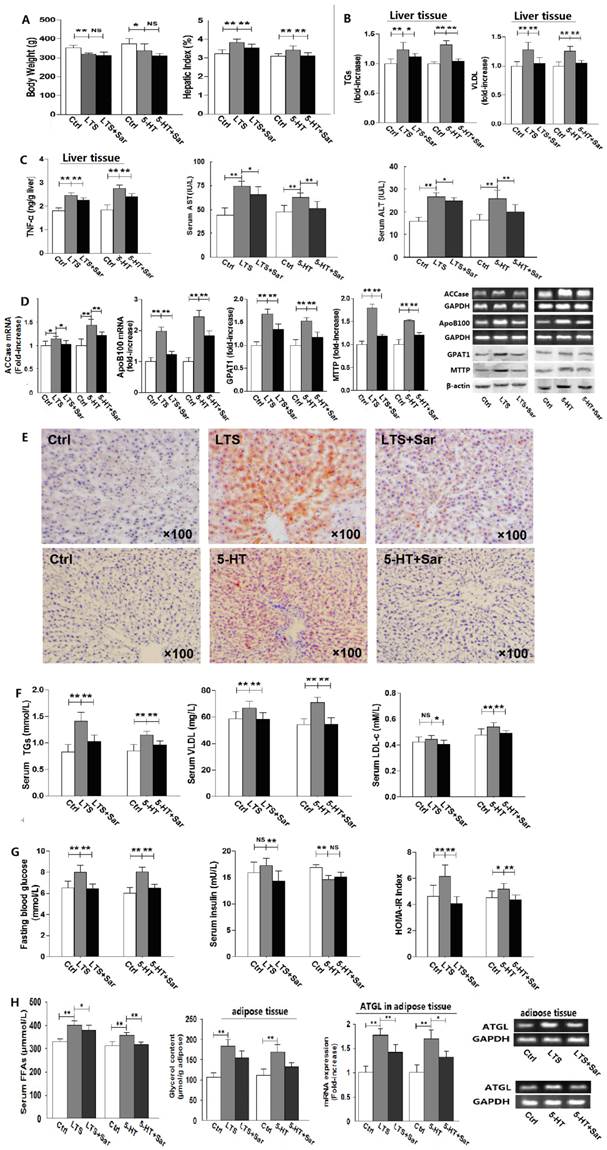
To examine whether or not 5-HT 2 receptor (5-HT2R) involves in long-term stress (LTS)-induced hepatic steatosis with VLDL assembly and adipose lipolysis, rats were exposed to stress treated with or without Sar, a broad-spectrum antagonist of various subtypes of 5-HT2R, for 25 days. And as a comparison, rats were also exposed to 5-HT with or without Sar treatment for 25 days. Both LTS and 5-HT exposure caused a decrease in body weight gain (Fig. 1A) and food intake (data not shown), while liver weight to body weight ratio, i.e. HI, increased (Fig. 1A). In addition, LTS or 5-HT exposure resulted in an attenuated tendency in visceral adipose weight, while the adipose weight to body weight ratio, i.e. VOI, was no marked difference between LTS or 5-HT-exposed and the control rats owing to significantly decreased bodyweight but slightly decreased visceral adipose weight by LTS or 5-HT versus the control (Data not shown). Sar treatment also merely showed an inhibited tendency on LTS or 5-HT-caused change in visceral adipose weight (Data not shown). Liver damage with increased levels of hepatic TNF-α, serum ALT and AST were observed but mildly in the LTS or 5-HT-exposed rats, which were attenuated by Sar (Fig. 1C). Both LTS and 5-HT induced an abnormality of hepatic fatty metabolism with increased TGs and VLDL contents (Fig. 1B) with steatosis (Fig. 1E), up-regulations of GPAT1 (a rate-limiting enzyme of TGs synthesis [17]) protein and ACCase (vital for fatty acids (FAs) synthesis[18]) gene (Fig. 1D), which suggest an up-regulated de novo lipogenesis (DNL), and up-regulations of MTTP (a rate-limiting factor of VLDL assembly [19]) protein and ApoB100 (a major apoprotein in VLDL particle [19]) gene (Fig. 1D), which suggest an increased VLDL assembly. In line with hepatic lipid metabolic disorder, LTS or 5-HT resulted in dyslipidemia with an increase or increased tendency in serum TGs, VLDL, and LDL-c level (Fig. 1F). We also detected LTS or 5-HT-stimulated lipolysis in visceral adipose tissue with increased serum FFAs level, up-regulated ATGL gene expression, a key enzyme of liposysis [20], and elevated glycerol content in the visceral adipose tissue (Fig. 1H). Though Sar treatment didn't affect LTS or 5-HT-caused decrease in body weight gain (Fig. 1A) and food intake (data not shown), it significantly attenuated LTS or 5-HT-induced abnormality of hepatic fatty metabolism, visceral adipose lipolytic increase, and dyslipidemia (Fig. 1, A-F and H). In addition, increased but slightly blood glucose and HOMA-IR index were detected in both LTS and 5-HT-exposed rats (Fig. 1G), suggesting a mild IR, while both had a different effect on serum insulin level with an increased tendency by LTS but markedly decreased by 5-HT (Fig. 1G). Sar treatment markedly attenuated LTS or 5-HT-induced elevation of blood glucose and HOMA-IR index, and LTS-induced increase in serum insulin, while it failed to markedly change 5-HT-caused decrease in serum insulin (Fig. 1G). These results suggest that, rat chronically exposed to LTS or 5-HT results in a similar consequence with abnormality of hepatic fatty metabolism, increased white adipose lipolysis, dyslipidemia, and hyperglycemia, all of which are associated with 5-HT2 receptor.
Long-term stress (LTS) or 5-HT exposure results in hepatic steatosis, VLDL overproduction, increased lipolysis in visceral adipose, and dyslipidemia in rats, which can be abolished significantly by blocking 5-HT 2 receptor. Except for control (Ctrl), rats were exposed to LTS (LTS) or 5-HT (5-HT) with or without sarpogrelate treatment (LTS+Sar or 5-HT+Sar) for 25 days. A, Body weight (Left) and hepatic index (Right) were detected. B, Hepatic content of TGs (Left) and VLDL (Right) were detected. C, Hepatic TNF-α content (Left), and serum AST (Middle) and ALT (Right) level were detected. D, Gene expression of hepatic ACCase and ApoB100 was detected by RT-PCR, while expression of hepatic GPAT1 and MTTP was detected by western blot. E, A representative image (original magnification, × 100) from all eight rats was assessed using Oil-red O staining in LTS-exposed (Up) or 5-HT-exposed (Down) rats. F, Serum concentration of TGs (Left), VLDL (Middle), and LDL-c (Right) was detected. G, Serum concentration of fasting blood glucose (Left) and insulin (Middle), HOMA-IR index (Right) were detected. H, Serum FFAs level (Left) and visceral adipose glycerol content (Middle) were detected, while gene expression of AGTL (Right) in the visceral adipose was detected by RT-PCR. Except for ApoB100, ACCase, GPAT1, MTTP, and ATGL from six of eight rats, other results from all eight rats are shown. Data are mean ± S.D., which are fold increase of gene or protein expression in control rats without LTS or 5-HT exposure, measured value, or calculated value (Hepatic index and HOMA-IR indexe). *, P < 0.05; **, P < 0.01. NS, not significant.
LTS-induced hyperglucocorticoidemia with increased hepatic 5-HT synthesis and 5-HT2R is associated with LTS-induced activation of mTOR―S6K pathway
Stress induces over-activity of HPA axis and also elevates many hormones levels in blood [12, 21]. To examine the effect of LTS on stress hormones, we measured serum corticosterone (Cort), 5-HT, GH, and Adr level, and hepatic 5-HT level. LTS significantly elevated serum Cort and 5-HT level, with slight elevation of serum Adr level without elevation of serum GH level (Fig. 2A).
Besides up-regulating Tph1 and AADC expression with increasing 5-HT production, long-term stress (LTS) exposure similarly with long-term 5-HT exposure up-regulates 5-HT2AR and 5-HT2BR expression in the liver and visceral adipose with activation of mTOR-S6K pathway in the liver, as well as increased serum corticosterone (Cort) level. Except for control (Ctrl), rats were exposed to LTS (LTS) or 5-HT (5-HT) with or without sarpogrelate treatment (LTS+Sar or 5-HT+Sar) for 25 days. A, Serum level of Cort, growth hormone (GH), and adrenline (Adr), and 5-HT level in the serum and liver tissue of Ctrl, LTS, and LTS+Sar group was detected (Left and middle). Serum 5-HT levels in 25-day experimental period in the Ctrl, 5-HT, and 5-HT+Sar group were detected (Right). B, Gene expression of hepatic 5-HT1A, 1B, 2A, 2B, 2C, 3A, and 7 receptors, Tph1, and AADC from three rats was assessed by RT-PCR. C, Gene expressions of hepatic Tph1, AADC, 5-HT2AR, 5-HT2BR, 5-HT1BR, and 5-HT7R in the Ctrl and LTS group were detected by RT-PCR (Left), while gene expressions of 5-HT2AR and 5-HT2BR in the visceral adipose were also detected (Right). D, Expressions of hepatic 5-HT2AR, 5-HT2BR, Tph1, and AADC in the Ctrl and LTS group (Left), 5-HT2AR and 5-HT2BR expression in the liver (Middle) and their gene expressions in the visceral adipose (Right) in the Ctrl and 5-HT group were detected by western blot or RT-PCR. E, Expressions of hepatic phospho-mTOR (Serine 2448) (p-mTOR) and mTOR, phospho-p70S6K (Threonine 389/412) (p-S6K) and p70S6K (S6K) in either Ctrl, LTS, and LTS+Sar group or Ctrl, 5-HT, and 5-HT+Sar group were detected by western blot. Representative images from six of eight rats and results from six of eight rats (gene or protein expression) or eight rats are shown. Data are mean ± S.D., which are fold increase in control rats without LTS or 5-HT exposure, or measured value of serum 5-HT, Cort, GH, and Adr. *, P < 0.05; **, P < 0.01. NS, not significant.
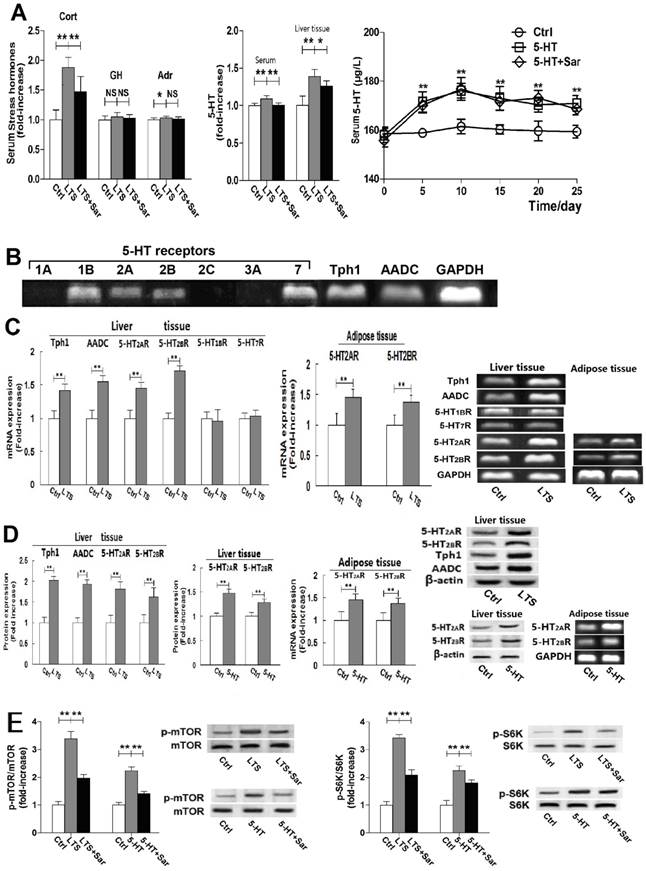
More importantly, a higher increase rate of 5-HT in the liver tissue than in the serum was detected (Fig. 2A), suggesting a non-blood-originated 5-HT production in the liver and activated by LTS. Sar treatment significantly attenuated LTS-caused elevation of serum Cort and 5-HT, but slightly attenuated hepatic 5-HT level (Fig. 2A). In addition, sustained high serum 5-HT levels during 25-day experiment were detected in the 5-HT-exposed rats, which wasn't changed by Sar treatment (Fig. 2A), suggesting that the Sar effects on 5-HT-induced disorders are not due to 5-HT level changed in the blood.
Expressions of 5-HT1BR, 5-HT2AR, 5-HT2BT, 5-HT7R, Tph1, and AADC gene without expressions of 5-HT1AR, 5-HT2CR, and 5-HT3AR gene in liver tissue were detected (Fig. 2B). We also detected protein expressions of 5-HT2AR, 5-HT2BT, Tph1, and AADC (Fig. 2D). LTS markedly up-regulated expressions of 5-HT2AR, 5-HT2BR, Tph1, and AADC, but failed to markedly affect 5-HT1BR and 5-HT7R expressions (Fig. 2, C and D). Long-term exogenic 5-HT exposure also induced up-regulation of 5-HT2AR and 5-HT2BR expression in the liver (Fig. 2D). In addition, gene expressions of 5-HT2AR and 5-HT2BR were also examined in the visceral adipose tissue, both of which were up-regulated by LTS or 5-HT exposure (Fig. 2, C and D). These results suggested that LTS can increase 5-HT synthesis with up-regulations of Tph1 and AADC expression in liver, resulting in a high 5-HT level in liver, and LTS like 5-HT exposure can also up-regulate 5-HT2AR and 5-HT2BR expression in the liver and visceral adipose tissues, which are associated with LTS or 5-HT-induced hepatic lipid metabolic abnormality and visceral adipose lipolytic increase.
It is reported that mTOR activation is the mechanism of 5-HT-induced hepatic steatosis and IR mediated by 5-HT2R in the liver and adipocytes [6, 22], so we examine LTS or 5-HT-indueced activation of hepatic mTOR and S6K, a downstream target of mTOR, by detecting Ser2448 phosphorylation of mTOR, and Thr389/412 phosphorylation of p70S6K. LTS or 5-HT didn't affect mTOR and p70S6K expression in the rat liver, but significantly up-regulated phosphorylation of mTOR and p70S6K (Fig. 2E). Sar treatment significantly abolished both-stimulated activation of mTOR and S6K (Fig. 3E). These results suggested that through activating mTOR-S6K pathway, both 5-HT and LTS induce lipid metabolic disorder in liver, which is associated with hepatic 5-HT2AR, 5-HT2BR, and 5-HT synthesis.
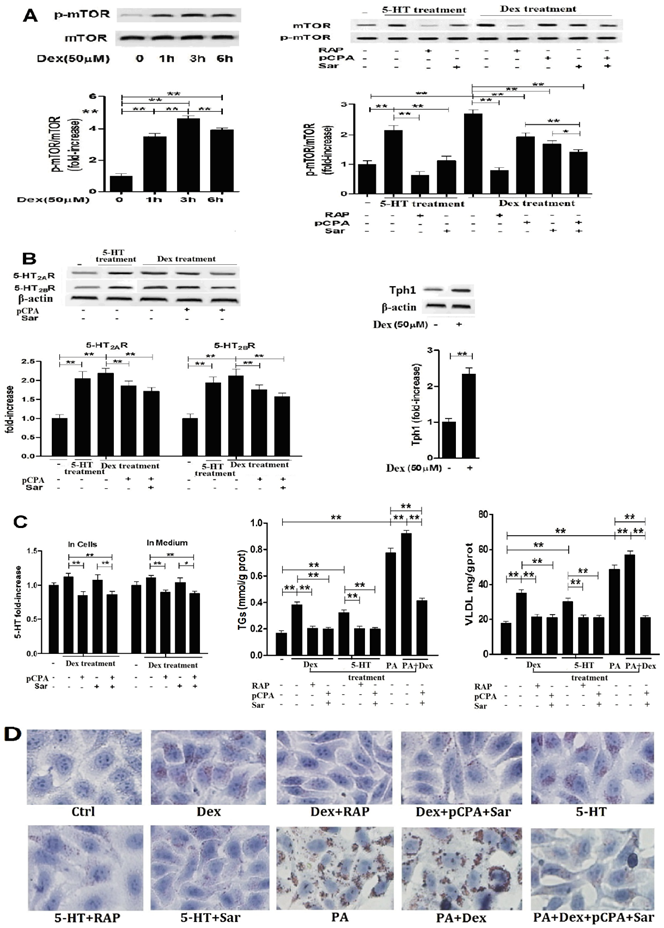
Dex-induced activation of mTOR with lipid droplet and VLDL overproduction in hepG2 cells is dependent on Dex-stimulated increases in 5-HT synthesis, 5-HT2R, while 5-HT-induced activation of mTOR is dependent on 5-HT2R. A, HepG2 cells were treated with 50 µM Dex for various periods, expression of phospho-mTOR (Serine 2448) and mTOR was then detected by western blot (Left). Cells were pretreated with vehicle (DMSO 10 µl/ml medium) (-), 100 nM rapamycin (RAP) (+), 30 µM para-chlorophenylalanine (pCPA) (+) or/and 30 µM Sarpogrelate (Sar) (+) for 30 min, respectively. After pretreatment, where indicated (+), except for one vehicle group, others were added with either 5-HT (in cells pretreated with vehicle, RAP, or Sar) or Dex (in cells pretreated with vehicle, RAP, pCPA, Sar, or pCPA+Sar) to the serum-free medium at a final concentration of 50 µM, respectively, and cells were incubated for an additional 3 h. Expression of phospho-mTOR (Serine 2448) and mTOR was detected (Right). B, Cells treated as described in A but only in the cells treated with vehicle, 5-HT, Dex, or Dex with pCPA or/and Sar. Expression of 5-HT2AR and 5-HT2BR (Left), Tph1 (Right) was detected by western blot. C, Cells treated as described in A. 5-HT levels inside HepG2 cells and in medium were measured using ELISA kit (Left). Cells were pretreated as described in A, then except for one vehicle group, others were added with 5-HT (in cells pretreated with vehicle, RAP, or Sar), Dex (in cells pretreated with vehicle, RAP, or pCPA+Sar), sodium palmitate (PA) (in cells pretreated with vehicle), or Dex + PA (in cells pretreated with vehicle, or pCPA+Sar) to the medium at a final concentration of 50 µM (Dex or 5-HT) or 400 µM (PA), respectively, and the cells were incubated for an additional 24 h. TGs (Middle) and VLDL (Right) level were measured using enzyme-test kit or ELISA kit, respectively. D, Cells treated as described in C with TGs and VLDL measurement. Lipid droplets were assessed by Oil Red O staining, a representative image (original magnification, × 400) is shown. Representative images or/and results from three independent experiments are shown. Data are mean ± S.D., which are fold increase from control cells without all-drug treatment, or measured value (TGs and VLDL). *, P < 0.05; **, P < 0.01.
Dex-induced activation of mTOR pathway with lipid droplets and VLDL overproduction in the hepatocytes is dependent on enhancements in 5-HT synthesis, 5-HT2A and 2B receptor
To examine whether or not LTS-induced activation of mTOR pathway with abnormality of lipid metabolism in the hepatocytes is involved in 5-HT system through GC, we investigated the relationships between Dex-stimulated 5-HT synthesis, 5-HT2AR and 5-HT2BR expression and activation of mTOR in the cultured HepG2 cells and primary rat hepatocytes in vitro. As a comparison, we also investigated 5-HT effects on mTOR activation, 5-HT2AR and 5-HT2BR expression in both cells. Treatment with 50 μM Dex time-dependently increased the mTOR ser phosphorylation level in HepG2 cells (Fig. 3A, left), indicating Dex-evoked mTOR activation, while the most activation occurred at 3 h after Dex treatment. So, subsequent study for detecting mTOR activation in the cells would be treated with Dex or 5-HT for 3 h. Dex-induced mTOR activation was inhibited completely by rapamycin, a specific inhibitor of mTOR, in both hepatocytes (Fig. 3A, Fig. 4A). Dex-stimulated up-regulations of Tph1, 5-HT2AR, and 5-HT2BR expression with increased 5-HT levels in both cells and culture media, indicating that 5-HT has an autocrine in the hepatocytes, were simultaneous with mTOR activation (Fig. 3 and Fig. 4, A - C). More importantly, inhibiting Tph1 with pCPA, resulting in an abolishment to Dex-induced 5-HT production in the cell, or blocking 5-HT2AR and 5-HT2BR with Sar effectively attenuated Dex-induced mTOR activation (Fig. 3 and Fig. 4, A and C). Moreover, a co-treatment with Sar and pCPA exhibited most effective effect on abolishing Dex-caused mTOR activation (Fig. 3 and Fig. 4, A). In addition, pCPA also inhibited Dex-induced up-regulation of 5-HT2AR and 5-HT2BR expression in the Dex-treated cells with either pCPA or pCPA+Sar treatment (Fig. 3 and Fig.4, B). However, though pCPA completely inhibited Dex-induced production of 5-HT, it only resulted in a part inhibition to Dex-induced up-regulations of 5-HT2AR and 5-HT2BR (Fig. 3 and Fig. 4, B), suggesting that Dex presents a direct stimulation on 5-HT2AR and 5-HT2BR expression. As previous reports [6, 22], 5-HT treatment increased mTOR ser phosphorylation level, which was fully inhibited by either 100 nM rapamycin or 30 µM Sar treatment in both hepatocytes (Fig. 3 and Fig. 4, A). 5-HT per se also up-regulated 5-HT2AR and 5-HT2BR expression in both cells (Fig.3 and Fig.4, B).
Dex-induced activation of mTOR with lipid droplet and VLDL overproduction in primary cultured rat hepatocytes is also dependent on Dex-stimulated increases in 5-HT synthesis, 5-HT2AR, and 5-HT2BR. A, Primary cultured rat hepatocytes were pretreated with vehicle (DMSO 10 µl/ml medium) (-), 100 nM rapamycin (RAP) (+), 30 µM para-chlorophenylalanine (pCPA) (+) or/and 30 µM Sarpogrelate (Sar) (+) for 30 min, respectively. After pretreatment, where indicated (+), except for one vehicle group, others were added with either 5-HT (in cells pretreated with vehicle, RAP, or Sar) or Dex (in cells pretreated with vehicle, RAP, pCPA, Sar, or pCPA+Sar) to the medium at a final concentration of 50 µM, respectively, and cells were incubated for an additional 3 h. Expression of phospho-mTOR (Serine 2448) (p-Ser-mTOR) and mTOR was detected by western blot. B, Hepatocytes treated as described in A but only in the cells treated with vehicle, 5-HT, Dex, or Dex with pCPA or/and Sar. Expression of 5-HT2AR and 5-HT2BR (Left), Tph1 (Right) was detected by western blot. C, cells treated as described in B. 5-HT levels inside hepatocytes and in medium were measured using ELISA kit (Left). The hepatocyts were pretreated as described in A, then except for one vehicle group, others were added with 5-HT (in cells pretreated with vehicle, RAP, or Sar), Dex (in cells pretreated with vehicle, RAP, or pCPA+Sar), sodium palmitate (PA) (in cells pretreated with vehicle), or Dex + PA (in cells pretreated with vehicle, or pCPA+Sar) to the medium at a final concentration of 50 µM (Dex or 5-HT) or 200 µM (PA), respectively, and the cells were incubated for an additional 10 h. TGs (Middle) and VLDL (Right) level were measured using enzyme-test kit or ELISA kit, respectively. D, Cells treated as described in C with TGs and VLDL measurement. Lipid droplets were assessed by Oil Red O staining, a representative image (original magnification, × 400) is shown. Representative images or/and results from three independent experiments are shown. Data are mean ± S.D., which are fold increase from control cells without all-drug treatment, or measured value (TGs and VLDL). *, P < 0.05; **, P < 0.01.
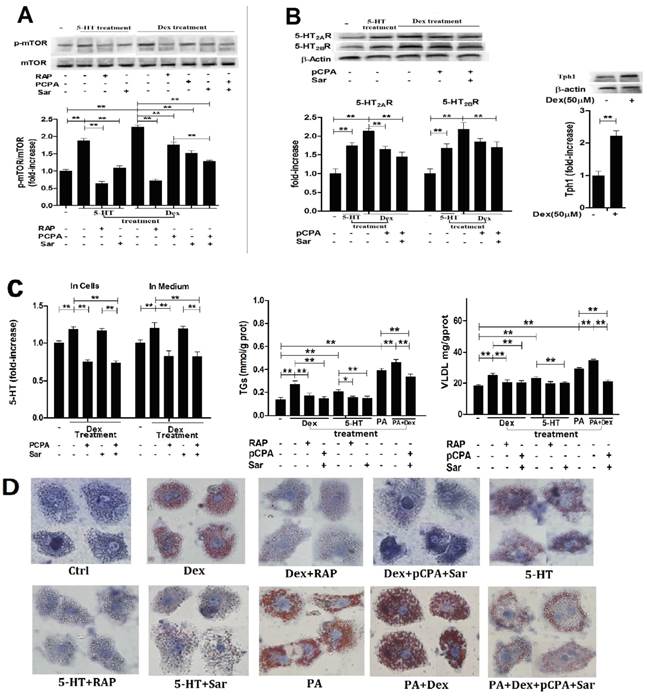
Subsequently, we detected Dex or 5-HT-induced lipid droplet formation, VLDL production. Treatment with Dex or 5-HT resulted in a visible accumulation of lipid droplets with increased TGs and VLDL levels in both cells, which could be abolished almost completely by rapamycin, Sar with pCPA (in the Dex-treated cells), or Sar (in the 5-HT-treated cells) (Fig. 3 and Fig. 4, C and D). In order to obtain severe accumulation of lipid droplets in the hepatocytes, we treated to cells with PA, a saturated fatty acid, or PA and Dex. PA treatment resulted in a severe accumulation in lipid droplets with increased TGs and VLDL contents in both cells, while Dex treatment exacerbated the PA effects (Fig. 3 and Fig. 4, C and D). Co-treatment with pCPA and Sar significantly inhibited above effects induced by co-exposure with PA and Dex (Fig. 3 and Fig. 4, C and D). Interestingly, the co-treatment showed a strong inhibition on the lipid droplet formation and increased TGs content that were mainly induced by PA in the PA and Dex-exposed cells, while the inhibition on the increased VLDL content was almost complete (Fig. 3 and Fig. 4, C).
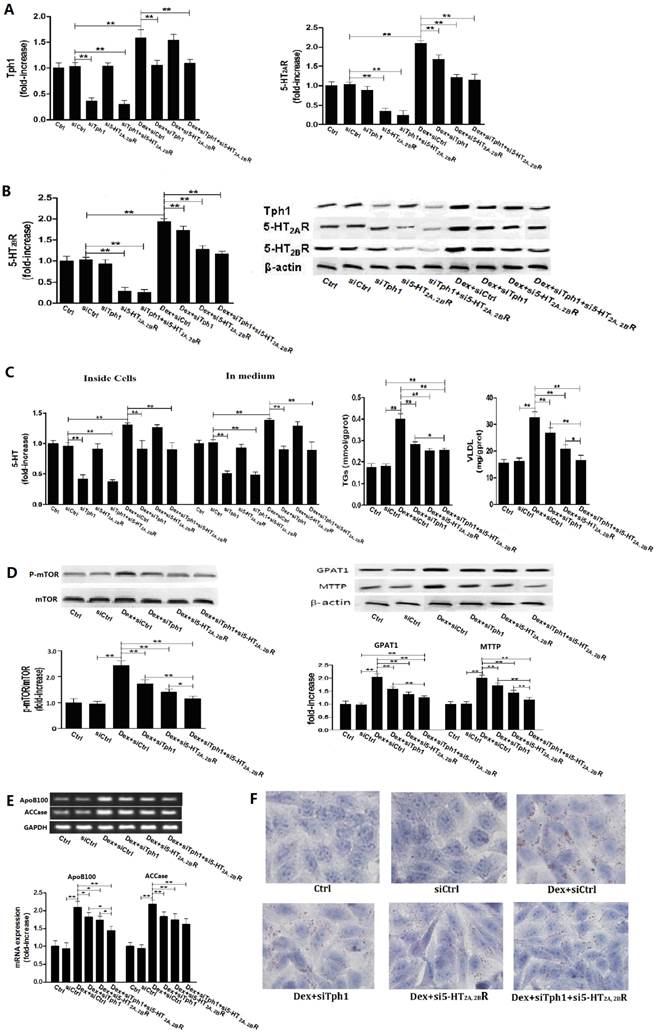
5-HT synthesis and 5-HT2 receptor are also crucial for Dex or PA-induced mTOR activation with production of lipid droplets and VLDL in HepG2 cells with silencing Tph1 or/and 5-HT2A, 2BR
To further confirm whether or not 5-HT synthesis and 5-HT2R are important roles in GC or FA-induced lipid metabolic disorder in hepatocytes, we investigated the Dex and PA effect on HepG2 cells under genetic inhibition of Tph1 (siTph1), 5-HT2A, 2BR (si5-HT2A, 2BR), alone or in a combination. siTph1, or/and si5-HT2A, 2BR reduced expression of Tph1 or 5-HT2A, 2BR by approximately 70% (Fig. 5, A and B), respectively, with reduction of 5-HT levels in the Tph1-silenced cells and culture medium by approximately 55%, respectively (Fig. 5C). Gene silencing Tph1 strongly abolished Dex or PA-induced up-regulation of Tph1 with increased 5-HT level in the cells and culture medium, while gene silencing 5-HT2A, 2BR strongly abolished Dex or PA-induced up-regulation of 5-HT2A, 2BR (Fig. 5 and Fig. 6, A - C). However, Dex or PA-induced up-regulation of 5-HT2A, 2BR was abolished slightly by gene silencing Tph1 (In Dex or PA+siTph1 group, Fig. 5A and 6B), indicating a direct effect of Dex or PA and an effect of 5-HT per se on up-regulating 5-HT2A, 2BR expression. As chemical inhibition of Tph1 or/and 5-HT2A, 2BR, genetic inhibition of them also significantly abolished Dex or PA-induced activation of mTOR (Fig. 5D, 6C). We found that Dex-induced up-regulations of GPAT1 and MTTP (protein expression) (Fig. 5D), and up-regulations of ApoB100 and ACCase (gene expression) (Fig. 5E) with increased TGs and VLDL level (Fig. 5C) and lipid droplet formation (Fig. 5F) were abolished significantly by gene silencing Tph1 or/and 5-HT2A, 2BR. We also found that PA-induced increases in TGs and VLDL level (Fig. 6D) and lipid droplets (Fig. 6E) were also abolished significantly by the gene silencing. The most effective abolishment to Dex or PA effects presented in the co-silencing Tph1 and 5-HT2A, 2BR, resulting in a strong abolishment on mTOR activation, full inhibition on increased VLDL content, and un-full inhibition on increased TGs with lipid droplets. And, the co-silencing exhibited a similar effect with silencing 5-HT2A, 2BR alone on Dex-induced up-regulations of GPAT1 and ACCase with increased TGs content, and on PA-induced increase in TGs content, but exhibited a more significant abolishment on Dex-induced up-regulations of MTTP and ApoB100 with increased VLDL content, and on PA-induced increase in VLDL content.
Gene silencing Tph1 or/and 5-HT2A, 2BR in HepG2 cells markedly abolishes Dex-induced mTOR activation, lipid droplets and VLDL overproduction. The gene silencing method was described in Material and Methods. Except for control (Ctrl), HepG2 cells with gene silencing control (siCtrl), Tph1 (siTph1), 5-HT2AR and 5-HT2BR (si5-HT2A, 2BR), and Tph1 with 5-HT2AR and 5-HT2BR (siTph1+si5-HT2A, 2BR) were treated with or without 50 µM Dex (Dex+siCtrl, Dex+siTph1, Dex+si5-HT2A, 2BR, Dex+siTph1+si5-HT2A, 2BR, respectively) for an additional 3 h (examining p-mTOR and mTOR expression) or 24 h (examining others agents) in serum-free culture medium. A and B, Expressions of Tph1 (A, left), 5-HT2AR (A, right) and 5-HT2BR (B, left), and representative image (B, right) were detected by western blot. C - E, 5-HT levels inside cells and in medium were measured using ELISA kit (C, left). Except for siTph1, si5-HT2A, 2BR, and siTph1+si5-HT2A, 2BR group, TGs (C, middle) and VLDL (C, right) level in other groups were measured using enzyme-test kit or ELISA kit, expressions of phospho-mTOR (Serine 2448) (p-mTOR) and mTOR (D, left), GPAT1 and MTTP (D, right) were detected by western blot, and gene expressions of ApoB100 and ACCase were detected by RT-PCR (E). F, Lipid droplets were assessed by Oil Red O staining, a representative image (original magnification, × 400) is shown. Representative images or/and results from three independent experiments are shown. Data are mean ± S.D., which are fold increase from Ctrl cells without Dex treatment, or measured value (TGs and VLDL). *, P < 0.05; **, P < 0.01.
Gene silencing Tph1 or/and 5-HT2A, 2BR in HepG2 cells also markedly abolishes palmitic acid (PA)-induced mTOR activation, lipid droplets and VLDL overproduction. The gene silencing method was described in Material and Methods. Except for control (Ctrl) and gene silencing control (siCtrl), 400 µM PA were treated to the cells with gene silencing control (PA+siCtrl), Tph1 (PA+siTph1), 5-HT2AR and 5-HT2BR (PA+si5-HT2A, 2BR), and Tph1 with 5-HT2AR and 5-HT2BR (PA+siTph1+si5-HT2A, 2BR) for an additional 3 h (examining expression of p-mTOR, mTOR, Tph1, 5-HT2AR, and 5-HT2BR) or 24 h (examining TGs and VLDL content, lipid droplets) in serum-free culture medium. A and B, Expressions of Tph1 (A, left), 5-HT2AR (A, right) and 5-HT2BR (B, left), and representative image (B, right) were detected by western blot. C, 5-HT levels inside cells were measured using ELISA kit (left). Expressions of phospho-mTOR (Serine 2448) (p-mTOR) and mTOR (Reft) were detected by western blot (Right). D, TGs (Left) and VLDL (Right) level were measured using enzyme-test kit or ELISA kit. E, Lipid droplets were assessed by Oil Red O staining, a representative image (original magnification, × 400) is shown. Representative images or/and results from three independent experiments are shown. Data are mean ± S.D., which are fold increase from Ctrl cells without PA treatment, or measured value (TGs and VLDL). *, P < 0.05; **, P < 0.01.
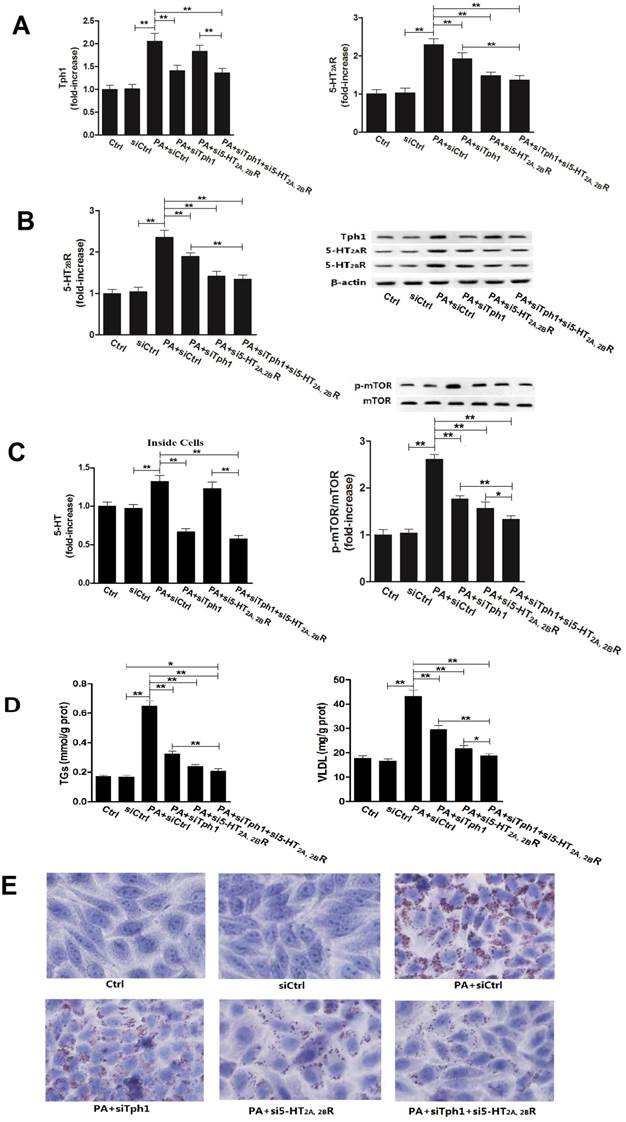
Together with above results from the primary rat hepatocytes and HepG2 cells experiments, these suggested that: 1) Dex-induced mTOR activation with TGs DNL, lipid droplet formation, and VLDL production in the hepatocytes is dependent on up-regulations of 5-HT synthesis, 5-HT2AR and 5-HT2BR; 2) PA-stimulated mTOR activation with TGs and VLDL overproduction, and lipid droplet formation in the hepatocytes is also closely associated with increased 5-HT synthesis and 5-HT2A, 2BR expression; 3) Though 5-HT mediates both TGs and VLDL syntheses in the hepatocytes, 5-HT action on mediating VLDL production is more significant than on mediating lipid droplet formation when the cells are stimulated by Dex or PA; 4) 5-HT2AR and 5-HT2BR expression can be up-regulated not only by Dex or PA but also by 5-HT per se in hepatocytes.
Discussion
Chronic GC exposure, such as in the setting of chronic stress-induced over-activation of HPA axis and Cushing syndrome, is well known to result in whole-body insulin resistance (IR). It has also been found to produce a persistent state of chronic low-grade inflammation in Cushing syndrome patients [23]. GCs result in abnormality of lipid metabolism in body is an important reason for GC-induced IR. Though GCs effect on adipose tissue lipolysis is controversial, multiple studies have demonstrated that GCs promote white adipose tissue lipolysis, such as the trials of rats exposed to GC in vivo and adipocytes exposed to GC in vitro [24-26], and the detection of Cushing syndrome patients through microdialysis [27]. Lipid metabolic disorder with steatosis in liver have been shown to be associated with IR and obesity, which represent an important marker of cardiovascular risk, possibly even more so than visceral fat [28, 29]. The development of hepatic steatosis has been recognized as a sequela of chronic excess GC exposure in humans [30-32], as well as GCs have found to increase VLDL production and secretion [33]. Besides GCs can directly enhance FA synthase to promote FA synthesis in liver [34, 35], indirect GC effects on hepatic lipogenesis are to induce adipose tissue lipolysis, which results in high FFAs level in the blood and delivering FFAs to liver, and systemic hyperinsulinemia and hyperglycemia, all of which drive hepatic DNL [36]. For the impact of GCs on 5-HT system, it is found that GC treatment can increase density of serotonin 5-HT2A receptors in humans [37], and induces a tissue-specific regulation of Tph mRNA levels in the pineal gland [38].
However, the correlation of GC-induced IR with peripheral 5-HT system is not investigated carefully to date according to our literature searching, while peripheral 5-HT has been reported to be an important factor in the high-fat diet-induced obesity and non-alcoholic fatty liver disease [39]. By studies in the rats, primary cultured rat hepatocytes and HepG2 cells treated with 5-HT, Dex, or PA under the chemical or genetic inhibition of Tph1 or/and 5-HT2AR and 5-HT2BR, present study has demonstrated that LST with hyperglucocorticoidemia-induced hepatic steatosis with VLDL overproduction is closely associated with enhancement of 5-HT synthesis and up-regulations of 5-HT2AR and 5-HT2BR in the liver. GC can directly induce 5-HT synthesis with up-regulating Tph1 and AADC expression, and 5-HT2AR and 5-HT2BR expression in hepatocytes, and indirectly up-regulate them through promoting lipolysis in visceral adipose tissue to result in high FFAs level in blood, while FFAs have a similar effect with GC on hepatic 5-HT synthesis and 5-HT2AR and 5-HT2BR expression. We found that increased 5-HT synthesis, 5-HT2AR and 5-HT2BR expression are crucial for GC-induced DNL with a rate-limiting enzyme of FA synthesis ACCase [18] up-regulation, and for GC or FA-induced TGs overproduction with a rate-limiting enzyme of TGs synthesis GPAT1 [17] up-regulation, and VLDL overproduction with a key factor of VLDL assembly MTTP (19) and VLDL precursor ApoB100 [19, 40] up-regulation in the hepatocytes. However, 5-HT-induced TGs-enriched VLDL production may have priority over lipid droplets formation in the hepatocytes according to present study, since inhibition of 5-HT synthesis and 5-HT2R simultaneously using chemical or genetic method in the cultured rat primary hepatocytes and HepG2 cells, could fully abolish Dex or/and PA-induced production of VLDL but un-fully abolish to formation of lipid droplets with TGs and FA synthesis. Additionally, we found that LTS-induced slightly hepatic damage with slightly increases in hepatic TNF-α, and serum AST and ALT levels is also associated with 5-HT2 receptor in liver, while LTS-induced hepatic lipid metabolic disorders seem to be prior to its action on evoking hyperglycemia and hyperinsulinemia. LTS-induced lipid metabolic disorders and Sar-treated effects in rats are not owing to their impact to body weight or food intake, because LTS exposure, like long-term 5-HT exposure, resulted in a decrease rather than an increase in bodyweight gain and food intake in rats, which agrees with previous studies [41, 42], while Sar treatment didn't have a marked influence on bodyweight and food intake in LTS-exposed rats.
The mTOR is a serine/threonine kinase and forms protein complexes that induce lipogenic gene expression and is found to be activated in the livers of obese rats fed a high fat and high sucrose diet [43, 44]. The mTOR complex is important in the stimulation of hepatic lipogenesis [45], and the mTOR kinase inhibitor rapamycin (RAP) reduces hepatic steatosis induced by a high fat diet [46]. 5-HT2R mediating hepatic steatosis and IR has been demonstrated in several researches [9], [47]. More importantly, mTOR activation by 5-HT has been demonstrated to be the mechanism by which L-tryptophan exacerbates hepatic steatosis in the fat and fructose-enriched diet-fed mice [6], and activation of mTOR is also the mechanism of 5-HT-induced IR by 5-HT2AR in the adipocytes by inhibition of insulin-stimulated activation of IRS-1-Akt signaling pathway and glucose uptake [22]. In present study, we have examined multiple expressions of 5-HT receptor isoform, and 5-HT synthesis with Tph1 and AADC expression in the rat liver, while LTS-induced activation of mTOR-S6K pathway with hepatic steatosis and VLDL overproduction is closely associated with up-regulations of 5-HT synthesis, 5-HT2AR and 5-HT2BR expression in liver, which is confirmed in the cultured HepG2 cells and primary rat hepatocytes. We found that Dex directly stimulated DNL with FA and TGs syntheses, lipid droplet formation, and VLDL overproduction, which was mediated by increased 5-HT acting on 5-HT2R to activate mTOR-S6K pathway in the hepatocytes. Surprisingly, PA-induced TGs and VLDL overproduction with severe lipid droplet accumulation in HepG2 cells was also mediated by 5-HT, 5-HT2R to mTOR pathway. Chemical or genetic inhibition of 5-HT synthesis or/and 5-HT2A, 2BR significantly abolished Dex and PA effects, and co-inhibition to 5-HT synthesis and 5-HT2A, 2BR almost fully abolished these effects. So, we have revealed a causal relationship between GC or FA-stimulated increases in 5-HT synthesis, 5-HT2AR and 5-HT2BR with abnormality of lipid metabolism in hepatocytes.
5-HT2AR, as a Gq/11-coupled receptor, is recognized to be a key role when 5-HT induces mTOR activation in the adipocytes [22]. We also detected that 5-HT-induced mTOR activation in the hepatocytes was fully abolished by Sar treatment, suggesting 5-HT action by 5-HT2R. However, our study detected that both 5-HT2AR and 5-HT2BR rather than 5-HT2AR alone were up-regulated by LTS in the rat liver, Dex or PA in the cultured hepatocytes. We also found that 5-HT itself also up-regulated 5-HT2AR and 5-HT2BR expression in both the rat liver and cultured hepatocytes. These results suggested that both 5-HT2AR and 5-HT2BR not alone work when mediating 5-HT, GC, or PA-induced mTOR activation in hepatocytes, both of which are up-regulated. So, we speculated that GC or FA-induced enhancement of 5-HT synthesis in the hepatocytes leads to two consequences that one is up-regulating 5-HT2AR and 5-HT2BR expression, and another is that 5-HT by autocrine to act on 5-HT2R activates mTOR-S6K pathway. We also infer that there are multiple inducements to up-regulating hepatic 5-HT2AR and 5-HT2BR expression owing to GC, 5-HT, and blood FFAs, respectively, when animal exposed to LTS or GC. Additionally, 5-HT2AR and 5-HT2BR may also play an important role in GC-induced adipose tissue lipolysis, since LTS or 5-HT exposure induced 5-HT2AR and 5-HT2BR up-regulation and lipolysis in the visceral adipose tissue of rats with a lipolytic marker of increased serum FFAs level, glycerol content and ATGL expression in the visceral adipose tissue [20], all of which were significantly inhibited by Sar treatment.
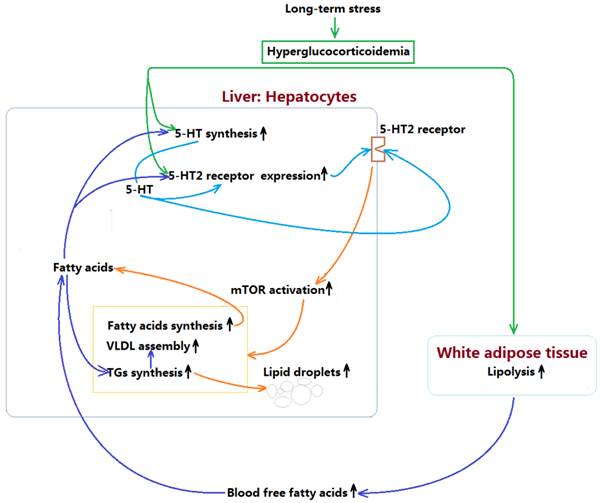
In summary, present study demonstrates that LTS with hyperglucocorticoidemia-induced hepatic steatosis with VLDL overproduction and dyslipidemia presents a causal relationship with enhancements of hepatic 5-HT2AR and 5-HT2BR expression and 5-HT synthesis. GC directly stimulates up-regulations of 5-HT synthesis, 5-HT2AR, and 5-HT2BR in the hepatocytes, which also has a indirect effects by inducing lipolysis in visceral adipose tissue to result in a high FFAs level in blood, while FFAs delivered to liver also up-regulates 5-HT synthesis, 5-HT2AR and 5-HT2BR expression. Further, increased intrahepatocellular 5-HT by autocrine to act on the 5-HT2R activates mTOR pathway, ultimately promotes FAs and TGs synthesis, and VLDL assembly, resulting in hepatic steatosis and VLDL overproduction with a closely related dyslipidemia. The mechanism process was shown in figure 7. In addition, GC effect on promoting lipolysis in visceral adipose tissue may also involve in 5-HT2 receptor according to present study. As GC effects on mediating lipid metabolic abnormality must be muzzled by 5-HT and 5-HT2 receptor in periphery, we guess that conflicting results in different studies implicating GC effects on energy metabolism may, at least in part, owe to whether or not peripheral 5-HT system altered by GC.
Mechanism by which long-term stress with hyperglucocorticoidemia induces hepatic steatosis with VLDL overproduction through up-regulating 5-HT synthesis and 5-HT2 receptor in liver.
Acknowledgements
The authors are grateful to B Jun Yang, a physician of pathology department in Nanjing Drum Tower Hospital, China, for his contribution in histopathological examination.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81570720) and the Students' Innovation and Entrepreneurship Training Program in China Pharmaceutical University (No. J1030830, SY12108, SZ14122) and Huahai Pharmaceuticals Graduate Innovation Fund (No. CX13S-003HH).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tecott LH. Serotonin and the orchestration of energy balance. Cell metabolism. 2007;6:352-61
2. Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annual review of medicine. 2009;60:355-66
3. Lesurtel M, Soll C, Graf R, Clavien PA. Role of serotonin in the hepato-gastroIntestinal tract: an old molecule for new perspectives. Cellular and molecular life sciences: CMLS. 2008;65:940-52
4. Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M. et al. Serotonin mediates oxidative stress and mitochondrial toxicity in a murine model of nonalcoholic steatohepatitis. Gastroenterology. 2007;133:608-18
5. Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M. et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nature medicine. 2008;14:756-61
6. Osawa Y, Kanamori H, Seki E, Hoshi M, Ohtaki H, Yasuda Y. et al. L-tryptophan-mediated enhancement of susceptibility to nonalcoholic fatty liver disease is dependent on the mammalian target of rapamycin. The Journal of biological chemistry. 2011;286:34800-8
7. Hoyer D, Hannon JP, Martin GR. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacology, biochemistry, and behavior. 2002;71:533-54
8. Kroeze WK, Kristiansen K, Roth BL. Molecular biology of serotonin receptors structure and function at the molecular level. Current topics in medicinal chemistry. 2002;2:507-28
9. Li Q, Hosaka T, Harada N, Nakaya Y, Funaki M. Activation of Akt through 5-HT2A receptor ameliorates serotonin-induced degradation of insulin receptor substrate-1 in adipocytes. Molecular and cellular endocrinology. 2013;365:25-35
10. Sumara G, Sumara O, Kim JK, Karsenty G. Gut-derived serotonin is a multifunctional determinant to fasting adaptation. Cell metabolism. 2012;16:588-600
11. Mo B, Feng N, Renner K, Forster G. Restraint stress increases serotonin release in the central nucleus of the amygdala via activation of corticotropin-releasing factor receptors. Brain research bulletin. 2008;76:493-8
12. McEwen BS. The neurobiology of stress: from serendipity to clinical relevance. Brain research. 2000;886:172-89
13. Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30:1-10
14. Yuen EY, Zhong P, Li X, Wei J, Yan Z. Restoration of glutamatergic transmission by dopamine D4 receptors in stressed animals. The Journal of biological chemistry. 2013;288:26112-20
15. Seglen PO. Preparation of isolated rat liver cells. Methods in cell biology. 1976;13:29-83
16. Haffner SM, Kennedy E, Gonzalez C, Stern MP, Miettinen H. A prospective analysis of the HOMA model. The Mexico City Diabetes Study. Diabetes care. 1996;19:1138-41
17. Wendel AA, Cooper DE, Ilkayeva OR, Muoio DM, Coleman RA. Glycerol-3-phosphate acyltransferase (GPAT)-1, but not GPAT4, incorporates newly synthesized fatty acids into triacylglycerol and diminishes fatty acid oxidation. The Journal of biological chemistry. 2013;288:27299-306
18. Foufelle F, Ferre P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J. 2002;366:377-91
19. Fisher EA, Pan M, Chen X, Wu X, Wang H, Jamil H. et al. The triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative pathways. The Journal of biological chemistry. 2001;276:27855-63
20. Geer EB, Islam J, Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: focus on adipose tissue function and lipid metabolism. Endocrinology and metabolism clinics of North America. 2014;43:75-102
21. Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, Maier SF. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nature neuroscience. 2005;8:365-71
22. Li Q, Hosaka T, Shikama Y, Bando Y, Kosugi C, Kataoka N. et al. Heparin-binding EGF-like growth factor (HB-EGF) mediates 5-HT-induced insulin resistance through activation of EGF receptor-ERK1/2-mTOR pathway. Endocrinology. 2012;153:56-68
23. Barahona M-J, Sucunza N, Resmini E, Fernandez-Real J-M, Ricart W, Moreno-Navarrete J-Ma. et al. Persistent body fat mass and inflammatory marker increases after long-term cure of Cushing's syndrome. The Journal of Clinical Endocrinology & Metabolism. 2009;94:3365-71
24. Campbell JE, Peckett AJ, D'souza AM, Hawke TJ, Riddell MC. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. American Journal of Physiology-Cell Physiology. 2011;300:C198-C209
25. Slavin B, Ong J, Kern P. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. Journal of lipid research. 1994;35:1535-41
26. Villena JA, Roy S, Sarkadi-Nagy E, Kim K-H, Sul HS. Desnutrin, an Adipocyte Gene Encoding a Novel Patatin Domain-containing Protein, Is Induced by Fasting and Glucocorticoids ECTOPIC EXPRESSION OF DESNUTRIN INCREASES TRIGLYCERIDE HYDROLYSIS. Journal of Biological Chemistry. 2004;279:47066-75
27. Krsek M, Rosicka M, Nedvidkova J, Hana V, Marek J, Haluzík M. et al. Increased lipolysis of subcutaneous abdominal adipose tissue and altered noradrenergic activity in patients with Cushings syndrome: an in vivo microdialysis study. Physiological research. 2006;55:421
28. Lim S, Son KR, Song IC, Park HS, Jin CJ, Jang HC. et al. Fat in liver/muscle correlates more strongly with insulin sensitivity in rats than abdominal fat. Obesity (Silver Spring, Md). 2009;17:188-95
29. Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW. et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15430-5
30. Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP. et al. Hepatic steatosis in Cushing's syndrome: a radiological assessment using computed tomography. European journal of endocrinology / European Federation of Endocrine Societies. 2003;149:543-8
31. Dourakis SP, Sevastianos VA, Kaliopi P. Acute severe steatohepatitis related to prednisolone therapy. The American journal of gastroenterology. 2002;97:1074-5
32. Nanki T, Koike R, Miyasaka N. Subacute severe steatohepatitis during prednisolone therapy for systemic lupus erythematosis. The American journal of gastroenterology. 1999;94:3379
33. Dolinsky VW, Douglas DN, Lehner R, Vance DE. Regulation of the enzymes of hepatic microsomal triacylglycerol lipolysis and re-esterification by the glucocorticoid dexamethasone. The Biochemical journal. 2004;378:967-74
34. Amatruda JM, Danahy SA, Chang CL. The effects of glucocorticoids on insulin-stimulated lipogenesis in primary cultures of rat hepatocytes. Biochem J. 1983;212:135-41
35. Zhao LF, Iwasaki Y, Zhe W, Nishiyama M, Taguchi T, Tsugita M. et al. Hormonal regulation of acetyl-CoA carboxylase isoenzyme gene transcription. Endocrine journal. 2010;57:317-24
36. Schwarz J-M, Linfoot P, Dare D, Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. The American Journal of Clinical Nutrition. 2003;77:43-50
37. Kling A, Mjörndal T, Rantapää-Dahlqvist S. Glucocorticoid treatment increases density of serotonin 5-HT2 A receptors in humans. Psychoneuroendocrinology. 2013;38:1014-20
38. Clark MS, Russo AF. Tissue-specific glucocorticoid regulation of tryptophan hydroxylase mRNA levels. Molecular brain research. 1997;48:346-54
39. Crane JD, Palanivel R, Mottillo EP, Bujak AL, Wang H, Ford RJ. et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nature medicine. 2015;21:166-72
40. Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S. et al. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. The Journal of clinical investigation. 2008;118:2347-64
41. Armario A. The hypothalamic-pituitary-adrenal axis: what can it tell us about stressors? CNS & neurological disorders drug targets. 2006;5:485-501
42. Blundell JE, Lawton CL, Halford JC. Serotonin, eating behavior, and fat intake. Obesity research. 1995;3(Suppl 4):471S-6S
43. Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Current biology: CB. 2009;19:R1046-52
44. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473-81
45. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3441-6
46. Chang GR, Chiu YS, Wu YY, Chen WY, Liao JW, Chao TH. et al. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. Journal of pharmacological sciences. 2009;109:496-503
47. Takishita E, Takahashi A, Harada N, Yamato M, Yoshizumi M, Nakaya Y. Effect of sarpogrelate hydrochloride, a 5-HT2 blocker, on insulin resistance in Otsuka Long-Evans Tokushima fatty rats (OLETF rats), a type 2 diabetic rat model. Journal of cardiovascular pharmacology. 2004;43:266-70
Author contact
![]() Corresponding author: Prof. Jihua Fu, Department of Physiology, China Pharmaceutical University, 639 Long Mian Road, 211198, Nanjing, Jiangsu Province, China. Tel.: +86 025 8618 52 26; Fax: + 86 025 8618 52 26; E-mail address: jihua_fuedu.cn
Corresponding author: Prof. Jihua Fu, Department of Physiology, China Pharmaceutical University, 639 Long Mian Road, 211198, Nanjing, Jiangsu Province, China. Tel.: +86 025 8618 52 26; Fax: + 86 025 8618 52 26; E-mail address: jihua_fuedu.cn