Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(2):235-245. doi:10.7150/ijbs.13814 This issue Cite
Research Paper
Selective Regulation of MAPK Signaling Mediates RANKL-dependent Osteoclast Differentiation
Kyunghee Lee1, Yeoun Ho Chung1, Heejin Ahn1, Hyunsoo Kim1, Jaerang Rho2, Daewon Jeong1 ![]()
1. Department of Microbiology, Laboratory of Bone Metabolism and Control, Yeungnam University College of Medicine, Daegu 705-717, Korea
2. Department of Microbiology and Molecular Biology, Chungnam National University, Daejeon 305-764, Korea
Received 2015-9-10; Accepted 2015-11-21; Published 2016-1-1
Abstract

Different stimuli often activate the same intracellular signaling molecules but trigger distinct cell responses. We explored whether or not MAPK signaling induced by macrophage colony-stimulating factor (M-CSF), which is responsible for osteoclast proliferation, differs from that induced by receptor activator of NF-κB ligand (RANKL), which is essential for inducing osteoclast differentiation. The activation of MAPKs by M-CSF or RANKL differed in terms of the extent and duration of ERK, p38, and JNK phosphorylation as well as the isoform specificity of JNK phosphorylation. In particular, RANKL induced a second wave of MAPK activation coincident with the onset of osteoclast differentiation, whereas M-CSF triggered only a monophasic response. M-CSF was also able to trigger a full MAPK response on restimulation of cells earlier than was RANKL, representing that MAPK resensitization by M-CSF differs from that by RANKL. Furthermore, the adapter protein TRAF6 recruitment to the cytoplasmic tail of RANK in a submembrane compartment is specifically required for RANKL-induced activation of p38 MAPK, expression of osteoclastogenic transcription factors, and osteoclast differentiation, indicating that the switch from proliferation to differentiation in osteoclast precursors is dependent on p38 activation via the RANKL-RANK-TRAF6 axis. Our results suggest that selective control of MAPK signaling induced by M-CSF and by RANKL mediates the proliferation versus differentiation decision in osteoclast precursors.
Keywords: M-CSF, RANKL, MAPK signaling, osteoclast differentiation
Introduction
Numerous processes including the immune response and metabolism are regulated by overlapping and intertwined signaling networks that are activated by various triggers. A central issue in signal transduction therefore concerns how the activation of common signaling mediators in response to different stimuli leads to distinct outcomes in a given cell type. Such stimuli in mammals include hormones, growth factors, and inflammatory cytokines, many of which activate MAPK signaling pathways through interaction with different receptors and intracellular couplers, resulting in diverse outcomes in terms of regulation of gene expression and cellular functions [1-4]. For instance, epidermal growth factor and nerve growth factor each activate the canonical MAPK signaling pathway Raf-MEK-ERK in the neuroendocrine cell line PC12, but they induce cell proliferation or differentiation into sympathetic neuron-like cells, respectively [5, 6]. These distinct outcomes of Raf-MEK-ERK signaling are thought to result from transient and more persistent activation of ERK by epidermal growth factor and nerve growth factor, respectively [7, 8]. Sustained ERK activation by epidermal growth factor in PC12 cells overexpressing the epidermal growth factor receptor resulted in cell differentiation rather than proliferation, indicating that alteration of ERK activity can induce reversible changes in cell fate progression [9].
Activation of ERK signaling by fibroblast growth factor forces naive embryonic stem cells to exit the self-renewal program as well as promotes their transition to neural or mesodermal lineage commitment by altering the expression of key markers of multipotency such as Oct4, Nanog, and Rex1 [10]. Furthermore, differentiation of mesenchymal stem cells into the osteogenic lineage is regulated by sustained activation of ERK at an early stage of the differentiation process as well as by activation of the MAPK JNK (c-Jun NH2-terminal kinase) at a later stage during extracellular matrix protein synthesis and calcium deposition [11]. Blockade of ERK activation after induction of osteogenic differentiation results in a transition from commitment to the osteogenic lineage to adipogenic differentiation [11]. Although these various observations have revealed a versatile role for MAPKs in the regulation of cellular functions as a result of their selective activation and their differential control of gene expression, the precise spatiotemporal regulation of these enzymes in the context of cell fate determination has remained unclear.
Bone undergoes remodeling in a cooperative manner at the levels of bone matrix formation and mineral deposition by osteoblasts and of bone destruction by osteoclasts [12]. Osteoclast differentiation is coordinated by the binding of macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL) to their respective receptors c-Fms and RANK on the surface of osteoclast precursors [13]. Activation of MAPKs (ERK, JNK, and p38) and Akt signaling by M-CSF is implicated in regulation of osteoclast precursor proliferation [14, 15], whereas that of MAPKs, NF-κB (nuclear factor-κB), and NFATc1 (nuclear factor of activated T cells c1) by RANKL is thought to contribute to osteoclast differentiation [14, 16]. Compared with cells treated with M-CSF alone, the proliferation of osteoclast precursors was found to be retarded 2 days after exposure to M-CSF and RANKL, corresponding to the onset of osteoclast fusion and differentiation [17]. Osteoclast differentiation in the presence of M-CSF and RANKL is accompanied by cell cycle arrest resulting from down-regulation of cell cycle regulators such as cyclins and cyclin-dependent kinases as well as up-regulation of the cyclin-dependent kinases inhibitor p27KIP1 [17]. Furthermore, cell cycle arrest induced by treatment of osteoclast precursors with the DNA replication inhibitor hydroxyurea promoted osteoclast formation [18]. These observations thus indicate that arrest of the M-CSF-dependent proliferation of osteoclast precursors plays a key role in osteoclast differentiation. Moreover, treatment of confluent osteoclast precursors manifesting growth inhibition as a result of cell-cell contact with an ERK signaling inhibitor accelerated osteoclast formation through activation of p38, reflecting a seesaw-like balance between ERK and p38 phosphorylation [19]. Activation of MAPKs by M-CSF and RANKL may thus determine the balance between cell proliferation and differentiation in osteoclast precursors, with inhibition of cell proliferation being required for the shift in cell fate toward differentiation.
Given that interaction of M-CSF with c-Fms and that of RANKL with RANK in osteoclast precursors each result in the activation of MAPKs, it is unclear whether or not MAPK activation by RANKL differs from that by M-CSF, and there exists a distinct mechanism before and after MAPK activation during osteoclast precursor proliferation and differentiation toward bone-resorbing osteoclasts. We now show that differences in the extent, duration, isoform specificity, and resensitization of MAPK activation give rise to distinct fates in osteoclast precursors.
Materials and Methods
Cell isolation and culture
Bone marrow-derived mononuclear osteoclast precursors were isolated from the tibia and femur of 6-week-old male C57BL/6J mice (Central Lab Animals, Seoul, Korea) by flushing of the bone marrow cavity. Erythrocytes were removed by treatment of the cell preparation with hypotonic buffer (0.15 mM NH4Cl, 1 mM KCO3, and 0.1 mM EDTA at pH 7.4), and the remaining bone marrow cells were incubated under a humidified atmosphere of 5% CO2 at 37°C for 12 hours in α-minimum essential medium (α-MEM; Invitrogen) supplemented with antibiotics, 10% fetal bovine serum, and recombinant human M-CSF (5 ng/mL). Nonadherent cells were collected and cultured for 3 days in α-MEM supplemented with M-CSF (30 ng/mL) before experiments. All animal procedures were approved by the Institutional Review Board of Yeungnam University College of Medicine and were in accordance with the Guide for the Care and Use of Laboratory Animals.
Assay of cell proliferation and osteoclast differentiation
For cell proliferation assays, osteoclast precursors (2 × 104 cells per well in 48-well culture plates) were incubated with M-CSF (30 ng/mL, 1.8 nM) overnight and were then treated with M-CSF (30 ng/mL) or recombinant mouse RANKL (100 ng/mL, 2 nM) for 2 days. The murine monocytic RAW264.7 cell line (5 × 103 cells per well in 48-well culture plates) was cultured overnight in α-MEM and then treated with M-CSF (30 ng/mL) or RANKL (100 ng/mL) for 2 days. Cell proliferation was then evaluated either by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay or by cell counting with a hemocytometer after staining with trypan blue. For induction of osteoclast differentiation, osteoclast precursors (2 × 104 cells per well) or RAW264.7 cells (5 × 103 cells per well) in 48-well culture plates were cultured for 4 days either with both M-CSF (30 ng/mL) and RANKL (100 ng/mL) or with RANKL (100 ng/mL) alone, respectively. TRAF6-deficient osteoclast precursors were prepared from liver-derived macrophages of TRAF6-/- mice at embryonic day 14.5 to 16.5. Flow cytometry confirmed that >95% of the cells were positive for CD11b. The TRAF6-/- and corresponding wild-type osteoclast precursors were seeded in 48-well culture plates at a density of 2 × 104 cells per well and cultured with M-CSF (30 ng/mL) and RANKL (100 ng/mL) for 4 days to induce differentiation. Fresh medium containing either M-CSF and RANKL for bone marrow-derived and TRAF6-/- osteoclast precursors or only RANKL for RAW264.7 cells was added to the cells after culture for 2 days. For assessment of osteoclast differentiation, cells were stained for TRAP (tartrate-resistant acid phosphatase) with the use of a leukocyte acid phosphatase staining kit (Sigma), and TRAP-positive multinucleated cells (TRAP+ MNCs) with more than three nuclei were counted with the use of a light microscope.
MAPK activation
Osteoclast precursors (2 × 105 cells) were seeded in 60-mm culture dishes and cultured overnight in the presence of M-CSF (30 ng/mL). The cells were then deprived of M-CSF for 6 hours before stimulation with M-CSF or RANKL at the indicated concentrations and for the indicated times.
Immunoblot analysis
Cells were washed with ice-cold phosphate-buffered saline (PBS) and suspended in a lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, 0.1% SDS, 1 mM NaF, 2 mM Na3VO4, 1 mM β-glycerophosphate, and a protease inhibitor cocktail (Roche). The lysates were centrifuged at 10,000 × g for 10 min at 4°C to remove debris, after which the protein concentration of the resulting supernatants was measured with the use of a DC protein assay (Bio-Rad). Proteins (30 µg) were fractionated by SDS-PAGE on a 10% gel, transferred to a nitrocellulose membrane, and probed with specific antibodies. Immune complexes were detected with appropriate horseradish peroxidase-conjugated secondary antibodies and ECL reagents (Abfrontier). Primary antibodies to total or phosphorylated forms of ERK, JNK, p38 or IκBα as well as those to c-Fos and flotillin-2 were obtained from Cell Signaling Technology, whereas those to RANK, c-Fms, NFATc1, p65, and ATPv0d2 were from Santa Cruz Biotechnology. Antibodies to TFIIB and β-actin were purchased from BD Transduction Laboratories and Sigma Aldrich, respectively. Band intensity was measured by densitometric analysis (Image-Pro Plus version 6.0 software).
Subcellular fractionation
A membrane fraction was prepared with the use of a Plasma Membrane Protein Extraction Kit (BioVision). In brief, detached osteoclast precursors were collected by centrifugation at 600 × g for 5 min at 4°C and washed with ice-cold PBS. The cells were resuspended in homogenizer buffer mix and disrupted with the use of a Dounce homogenizer (50 strokes) on ice. The homogenate was centrifuged at 700 × g for 10 min at 4°C, and the resulting supernatant was centrifuged again at 10,000 × g for 30 min at 4°C. The final pellet was resuspended in upper phase solution, lower phase solution was added, and the mixture was centrifuged at 1000 × g for 5 min at 4°C. The upper phase was collected and centrifuged at 10,000 × g for 10 min at 4°C, and the resulting pellet was used as the plasma membrane fraction. For nuclear fractionation, cells were lysed in an ice-cold lysis buffer containing 10 mM Hepes-KOH (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 20 mM sodium pyrophosphate, 10 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail. The lysate was centrifuged at 10,000 × g for 10 min at 4°C, the supernatant was used as the cytosolic fraction, and the resulting nuclear pellet was resuspended in nuclear extraction buffer [20 mM Hepes-KOH (pH 7.9), 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 20 mM sodium pyrophosphate, 10 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail] and centrifuged at 12,000 × g for 2 min at 4°C. The resultant supernatant was used as the nuclear fraction.
Quantitative and Semiquantitative RT-PCR Analysis
Total RNA was extracted from cells with the use of the Trizol reagent (Invitrogen), and portions (2 µg) of the RNA were subjected to reverse transcription for 1 h at 42°C with oligo(dT) and an M-MLV RT kit (Invitrogen). Semiquantitative PCR analysis was performed with the use of a Thermo Hybaid PCR Express system. Real-time (quantitative) PCR was performed with the use of SYBR Premix Ex Taq (Takara Bio, Shiga, Japan) and an Applied Biosystems 7500 Sequence Detection System. Normalized mRNA abundance was determined with the comparative delta threshold cycle method and with GAPDH mRNA as the invariant control. The sequences of PCR primers are listed in Supplementary Table S1.
Statistical analysis
Quantitative data are presented as means ± SD from at least three independent experiments and were analyzed by two-tailed Student's t test. For statistical analysis for multiple comparisons, means between groups were performed using one-way ANOVA analysis with Microsoft 2010 Excel program. A P value of <0.05 was considered statistically significant.
Results
M-CSF and RANKL mediate distinct cell fates in association with differential activation of shared MAPK signaling
M-CSF and RANKL have previously been shown to be essential for proliferation of osteoclast precursors and for the differentiation of these cells into TRAP-positive multinucleated giant osteoclasts, respectively [14-16, 20]. We first confirmed that M-CSF and RANKL induce different cell fates in two types of mouse osteoclast precursors: transformed RAW264.7 cells, which can proliferate without M-CSF treatment and differentiate into osteoclasts in response to RANKL treatment alone, and bone marrow-derived monocytes (BMMs), whose proliferation is dependent on M-CSF treatment and differentiation into osteoclasts is triggered by treatment with both M-CSF and RANKL (Fig. 1A and B). The combined results using RAW264.7 cells and BMMs showed that M-CSF and RANKL are mainly responsible for osteoclast precursor proliferation and osteoclast differentiation in cell fate decisions, respectively. All subsequent experiments were performed with BMMs as osteoclast precursors unless indicated otherwise. M-CSF and RANKL each induced activation of the MAPKs ERK, p38, and JNK in osteoclast precursors in a concentration-dependent manner (Fig. 1C). The maximal activation of each MAPK was apparent at a lower molar concentration of M-CSF (molecular mass of 16 kDa) relative to that of RANKL (molecular mass of 50 kDa), suggesting that MAPK signaling is more sensitive to M-CSF than to RANKL. We also analyzed MAPK activation using various concentrations of M-CSF or RANKL at different stages of osteoclast differentiation (Supplementary Fig. S1). Examination of the time course of MAPK phosphorylation in osteoclast precursors exposed to M-CSF or RANKL at these various concentrations revealed that (i) ERK activation induced by M-CSF was more marked and sustained than that induced by RANKL, (ii) the extent and duration of p38 activation induced by RANKL were greater than for that induced by M-CSF, and (iii) JNK activation induced by M-CSF was more prominent and persistent than that induced by RANKL, and M-CSF preferentially targeted JNK1 rather than JNK2 or JNK3 whereas RANKL showed no such preference (Fig. 1D). Similar differences between the effects of the two stimuli were also apparent in pre-osteoclasts and mature osteoclasts (Supplementary Fig. S2). In addition, M-CSF activated Akt but not NF-κB signaling in osteoclast precursors, whereas RANKL activated NF-κB signaling as evidenced by the phosphorylation and degradation of the NF-κB inhibitor IκBα but not Akt in these cells. Further, we observed that activation of MAPK upstream activators MAPK kinases (MKKs) such as MEK1/2, MKK3/6, and MKK4 and 7 (responsible for activation of ERK, p38, and JNK, respectively) by M-CSF or RANKL differed in terms of extent, duration, and isoform specificity (Supplemental Fig. S3). These data thus showed that, although M-CSF and RANKL each activate MKK and MAPK signaling in osteoclast precursors, their effects differ in terms of the levels and time course of ERK, p38, and JNK phosphorylation.
MAPK activation by M-CSF or RANKL in osteoclast precursors. (A) RAW264.7 cells and BMMs as osteoclast precursors were treated with M-CSF (30 ng/mL) or RANKL (100 ng/mL) as indicated for 2 days, after which cell proliferation was evaluated with the MTT assay or by cell counting. Data between groups were analyzed by one-way ANOVA comparison from a representative experiment run in triplicate. *P < 0.01. (B) RAW264.7 cells and BMMs as osteoclast precursors were cultured in the presence of M-CSF (30 ng/mL) or RANKL (100 ng/mL) as indicated for 4 days, after which the cells were stained for TRAP and the number of TRAP-positive multinucleated cells (TRAP+ MNCs) with more than three nuclei was counted. Data are means ± SD (n = 3) for three independent experiments. ND, none detected. Representative images of the stained cells are also shown. Scale bar, 100 µm. (C) Osteoclast precursors (BMMs) were treated with various concentrations of M-CSF or RANKL for 10 min, after which cell lysates were prepared and subjected to immunoblot analysis with antibodies to total or phosphorylated (p-) forms of the MAPKs ERK, p38, and JNK. The blots were also subjected to densitometric analysis for determination of each p-MAPK/MAPK ratio. The quantitative data are means ± SD (n = 3) of three independent experiments with similar results. Overall P value for fold induction by M-CSF or RANKL stimuli compared to unstimulated control is < 0.05 (Student t test). (D) Osteoclast precursors (BMMs) were treated with M-CSF (25 ng/mL) or RANKL (500 ng/mL) for the indicated times, after which cell lysates were subjected to immunoblot analysis with antibodies to total or phosphorylated forms of MAPKs, Akt, and IκBα as well as to β-actin (loading control). The gel images are representative of three independent experiments.
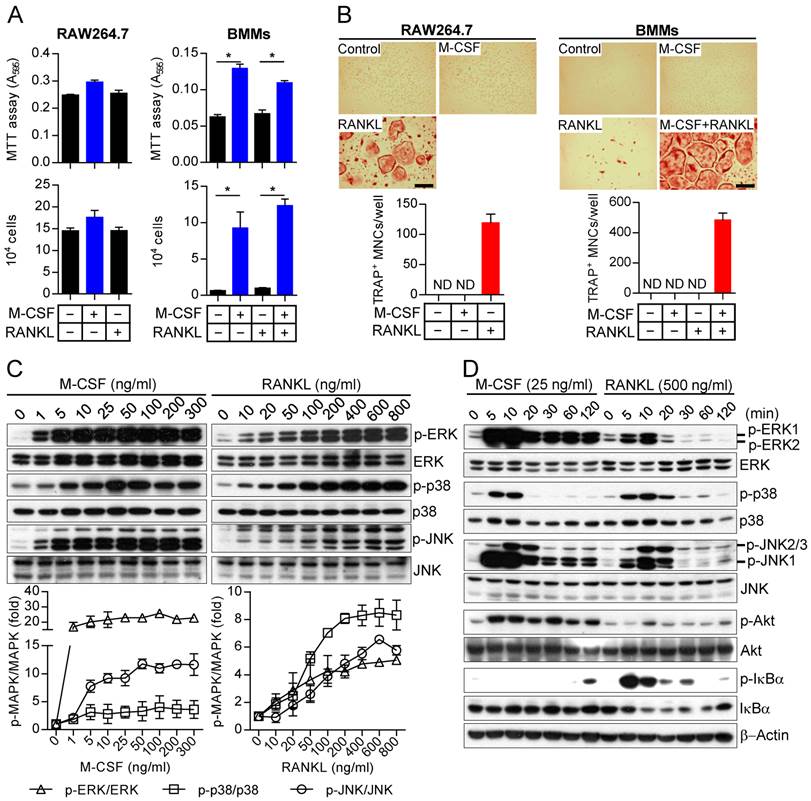
Differences in the long-term effects of M-CSF and RANKL on MAPK activation as well as in the effects of restimulation
We next examined in more detail the kinetics of MAPK activation by M-CSF or RANKL in relation to those of cell fate determination in osteoclast precursors. In general, M-CSF had an immediate effect (5 to 20 min) on MAPK phosphorylation, whereas RANKL induced both an immediate activation (5 to 20 min) and a delayed activation (8 to 24 hours) (Fig. 2). The timing of this secondary response to RANKL coincided with the initiation of differentiation induced by the combination of M-CSF and RANKL, as evaluated by detection of TRAP+ mononuclear cells and gene expression for osteoclastogenic markers, including TRAP, NFATc1, NF-κB, and AP-1, ATPv0d2, cathepsin K (CathK), carbonic anhydrase II (CAII), and matrix metalloproteinase-9 (MMP9) (Supplementary Fig. S4). We next explored the effects of restimulation with M-CSF or RANKL on MAPK activation at 2 h after the initial stimulation, when MAPK phosphorylation had returned to basal levels. Whereas the effects of restimulation with M-CSF on MAPK activation were similar to those of the initial stimulation, the effects of restimulation with RANKL were less pronounced than those of the initial stimulation (Fig. 3). These results suggested that osteoclast precursors remain refractory to a second stimulation with RANKL for a longer period than they do to a second stimulation with M-CSF.
Kinetics of MAPK activation during long-term treatment of osteoclast precursors with M-CSF or RANKL. (A and B) Osteoclast precursors were stimulated with M-CSF at 1 ng/mL (A) or RANKL at 100 ng/mL (B) for the indicated times, after which cell lysates were subjected to immunoblot analysis of MAPK activation. The blots were also subjected to densitometric analysis for determination of each p-MAPK/MAPK ratio. The quantitative data are means ± SD (n = 3) for three independent experiments with similar results. *P < 0.01 and †P < 0.05 versus unstimulated control (Student t test).

Effects of restimulation of osteoclast precursors with M-CSF or RANKL on MAPK activation. (A and B) Osteoclast precursors were treated with M-CSF (M) at 25 ng/mL (A) or RANKL (R) at 500 ng/mL (B) for 2 hours and then restimulated with M-CSF (25 ng/mL) or RANKL (500 ng/mL) for 5 or 10 min, as indicated. Cell lysates were then subjected to immunoblot analysis of MAPK activation. The blots were also subjected to densitometric analysis for determination of each p-MAPK/MAPK ratio. The quantitative data between groups were analyzed by one-way ANOVA comparison. *P < 0.01.
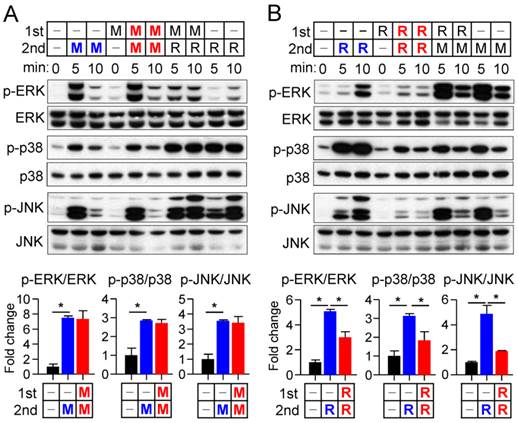
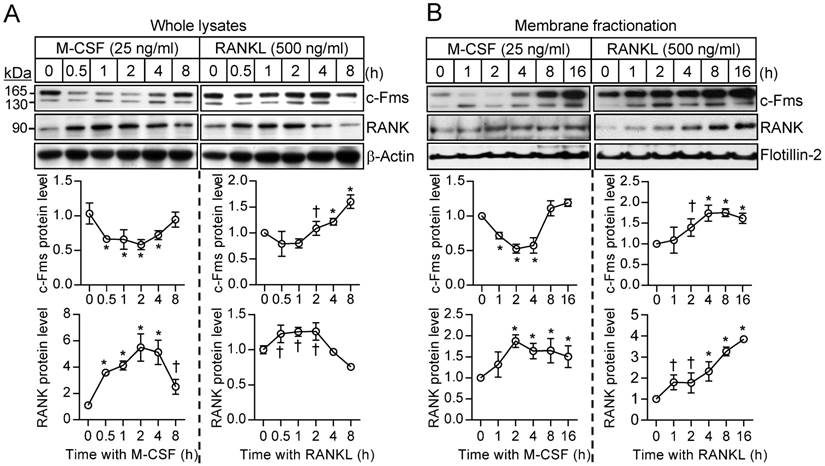
Cross talk between M-CSF and RANKL signaling at the receptor level
To investigate further the difference in the sensitivity of osteoclast precursors to restimulation with M-CSF or RANKL, we examined the effects of these ligands on the corresponding receptors. The abundance of the M-CSF receptor (c-Fms), a membrane-targeted receptor tyrosine kinase, underwent a transient decrease in both whole cell lysates and a membrane fraction in response to M-CSF exposure, having returned to basal levels by 8 hours (Fig. 4A and B). M-CSF had no effect on the abundance of c-Fms mRNA during this period (Supplementary Fig. S5). In contrast, M-CSF increased expression at both mRNA and protein levels of the RANKL receptor (RANK) (Fig. 4A and B), which is localized to the plasma membrane of both osteoclast precursors and mature osteoclasts [21]. Exposure of osteoclast precursors to RANKL increased the abundance of both c-Fms and RANK in both whole cell lysates and a membrane fraction (Fig. 4A and B) as well as increased the amounts of the corresponding mRNAs (Supplementary Fig. S5). We also examined the stability of c-Fms and RANK in osteoclast precursors by blocking de novo protein synthesis with cycloheximide. c-Fms was rapidly degraded on exposure of the cells to cycloheximide in the absence or presence of M-CSF (Supplementary Fig. S6A), whereas the abundance of RANK was little affected by cycloheximide in the absence or presence of RANKL (Supplementary Fig. S6B), suggesting that RANK is more stable than c-Fms.
Effects of M-CSF and RANKL on the expression of c-Fms and RANK. (A and B) Osteoclast precursors were treated with M-CSF (25 ng/mL) or RANKL (500 ng/mL) for the indicated times, after which whole cell lysates (A) or a membrane fraction derived therefrom (B) were subjected to immunoblot analysis with antibodies to c-Fms and to RANK as well as with those to β-actin and to flotillin-2 as loading controls. The blots were also subjected to densitometric analysis for determination of c-Fms or RANK abundance. The quantitative data are means ± SD (n = 3) of three independent experiments with similar results. *P < 0.01 and †P < 0.05 versus unstimulated control (Student t test).
TRAF6 deficiency prevents osteoclast differentiation by inhibiting RANKL-induced activation of p38 and down-regulating osteoclastogenic transcription factors
TRAF6 (tumor necrosis factor receptor-associated factor 6) functions as an adapter molecule for the intracellular region of RANK and mediates osteoclastogenic signaling downstream of the RANKL-RANK axis [22-24]. To certify the functional role of TRAF6 in osteoclastogenesis, we analyzed osteoclast formation, MAPK activation, and osteoclastogenic signaling pathways in TRAF6-deficient osteoclast precursors. TRAF6-deficient (TRAF6-/-) osteoclast precursors thus failed to form osteoclasts in response to stimulation with RANKL and M-CSF (Fig. 5A), whereas they still showed a proliferative response to M-CSF alone (Fig. 5B).
TRAF6-dependent activation of p38 by RANKL. (A) Osteoclast precursors from wild-type (WT) or TRAF6-/- mice were cultured in the presence of M-CSF (30 ng/mL) either alone or together with RANKL (100 ng/mL) for 4 days, after which the proportion of TRAP+ MNCs was determined. Data are means ± SD for three independent experiments. ND, none detected. Representative images of the stained cells are also shown. Scale bar, 100 µm. (B) Osteoclast precursors from WT or TRAF6-/- mice were incubated with or without M-CSF (30 ng/mL) for 2 days, after which the number of cells was counted. Data are expressed as a percentage of the corresponding control value and are means ± SD (n = 3) for three independent experiments. *P < 0.01 (Student t test). (C) Osteoclast precursors from WT or TRAF6-/- mice were incubated with M-CSF (25 ng/mL) or RANKL (500 ng/mL) for the indicated times, after which cell lysates were subjected to immunoblot analysis of MAPK activation. Data are representative of three independent experiments. (D) Osteoclast precursors from WT or TRAF6-/- mice were incubated with M-CSF (30 ng/mL) alone or together with RANKL (100 ng/mL) for the indicated times, after which cell lysates were subjected to immunoblot analysis of NFATc1, p65, and c-Fos. Data are representative of three independent experiments. (E) Osteoclast precursors from WT or TRAF6-/- mice were incubated with M-CSF (30 ng/mL) and RANKL (100 ng/mL) for 2 days (to allow the formation of TRAP+ mononuclear cells), deprived of M-CSF and RANKL for 6 h, and then stimulated with M-CSF (25 ng/mL) or RANKL (500 ng/mL) for the indicated times. Cytosolic and nuclear fractions were then prepared from the cells and subjected to immunoblot analysis of NFATc1, p65, and c-Fos. β-Actin and TFIIB were examined as loading controls. The gel images for C, D, and E are representative of three independent experiments.

Of note, RANKL-induced p38 activation was not apparent in TRAF6-/- cells, whereas RANKL-induced activation of ERK and JNK as well as M-CSF-induced activation of ERK, p38, and JNK appeared to be unaffected by the loss of TRAF6 (Fig. 5C). Examination of signaling downstream of MAPK activation revealed that expression of NFATc1 and c-Fos (a component of AP-1) was down-regulated in the TRAF6-/- cells (Fig. 5D), and that the nuclear import of NFATc1, c-Fos, and the p65 subunit of NF-κB induced by RANKL was blocked in the absence of TRAF6 (Fig. 5E). M-CSF did not induce the nuclear translocation of these osteoclastogenic transcription factors in wild-type or TRAF6-/- cells. The nuclear import of activated MAPKs did not appear to differ between wild-type osteoclast precursors stimulated with RANKL and those stimulated with M-CSF (Supplementary Fig. S7). These results thus indicate that RANKL activates p38 signaling via TRAF6 in osteoclast precursors, and that such signaling maintains the expression and triggers the nuclear translocation of osteoclastogenic transcription factors, resulting in a shift in cell fate from M-CSF-dependent proliferation toward RANKL-dependent osteoclast differentiation.
Discussion
MAPKs play an essential role in the transduction of extracellular stimuli into intracellular responses and thereby contribute to the regulation of diverse cellular activities including gene expression, cell cycle progression, metabolism, motility, survival, apoptosis, and differentiation [1, 25]. We showed that osteoclast precursors display differential patterns of MAPK activation by M-CSF or RANKL and thereby may be involved in cell fate decisions in terms of proliferation or differentiation. Activation of MAPKs by M-CSF or RANKL thus differed in terms of the extent and duration of ERK, p38, and JNK phosphorylation as well as the selective phosphorylation of JNK isoforms; the activation of MAPKs via the M-CSF-c-Fms axis occurred at lower molar concentrations of ligand compared with that induced by RANKL-RANK; RANKL induced a second phase of MAPK activation coincident with the onset of osteoclast differentiation, whereas M-CSF exhibited only an initial phase of such activation; activation of p38 by RANKL was dependent on TRAF6 recruitment to the cytoplasmic tail of RANK; and osteoclast fate was determined by differential gene expression and nuclear import of osteoclastogenic factors that function downstream of MAPK activation. Our results thus suggest that the switch from proliferation to differentiation in osteoclast precursors is dependent on sustained activation of p38 via the RANKL-RANK-TRAF6 axis. Interestingly, TGF-β has been reported to induce p38 signaling in human and mouse monocytes, playing a role in promoting the osteoclastogenesis [26-28]. It is possible that TGF-β may contribute to osteoclast differentiation through p38 activation, as was the case in the RANKL.
Cells manifest numerous signaling responses to external stimuli, and they adopt different fates depending on the signaling networks and genetic programs activated [29-31]. Our results regarding M-CSF-induced osteoclast precursor proliferation and RANKL-dependent osteoclast differentiation are consistent with this notion. The M-CSF receptor (c-Fms) is synthesized as a 130-kDa precursor in the endoplasmic reticulum, processed to yield a 165-kDa mature form by posttranslational modification, and then targeted to the plasma membrane [32]. Mature c-Fms comprises an extracellular domain that binds M-CSF, a hydrophobic transmembrane domain, and an intracellular domain that mediates activation of downstream signaling. On binding of M-CSF, c-Fms forms a homodimer and undergoes transphosphorylation at a specific tyrosine residue in its intracellular domain, resulting in the recruitment of Src homology 2 (SH2) domain-containing signaling proteins such as Src family kinases, Grb2, phosphatidylinositol 3-kinase, Mona, phospholipase C (PLC), c-Cbl, and STAT1 [15, 33, 34] as well as activation of downstream signals such as MAPKs, Akt, and STAT3 [35, 36]. The M-CSF-c-Fms axis thereby induces differentiation of monocytes into macrophages and regulates their survival and proliferation [13, 15, 20]. Macrophage osteoclast precursors undergo further differentiation into mature osteoclasts in response to RANKL stimulation. Interaction of RANKL with the extracellular domain of RANK induces the assembly of a RANK homotrimer and the recruitment of TRAF adapter proteins (in particular, TRAF6), c-Src, and Gab2 to the cytoplasmic tail of the receptor. Such recruitment triggers the activation of numerous downstream signaling molecules including PTK2, c-Cbl, protein kinase C, phosphatidylinositol 3-kinase, Akt, Rho/Rac GTPases, and MAPKs as well as that of the Syk-PLC-Ca2+-calcineurin-NFATc1 and IKK-IκB-NF-κB pathways [14, 16]. Such signaling leads to changes in the expression of RANKL-targeted osteoclastogenic genes including those for TRAP, αvβ3 integrin, osteopontin, OSCAR, and NFATc1, which in turn support pre-osteoclast migration, cell-cell contact, and cell fusion to form multinucleated mature osteoclasts [37]. MAPK activation by M-CSF or RANKL has previously been found to play essential roles in osteoclast precursor survival and proliferation and in osteoclast differentiation, respectively [14-16]. It is also reported that three isoforms of JNK, JNK1 and JNK2/3 (corresponding to phospho-p46 band and phospho-p54 band, respectively), can be specifically activated in response to extracellular stimuli [38, 39]. Our findings support that differential preference of JNK isoforms by M-CSF or RANKL stimulation may affect their downstream signal molecules during osteoclast proliferation and differentiation. Collectively, differences in the pattern of MAPK activation and the differential regulation of signaling molecules that function upstream or downstream of MAPKs have been thought to determine the transition between osteoclast precursor proliferation and osteoclast differentiation.
The specificity of MAPK activation is thought to be determined at several levels including the continuity and intensity of the activating signal, the direct interaction of MAPKs with scaffold proteins, the differential subcellular localization of MAPK isoforms, cross talk between MAPKs, interplay between MAPKs and other signal transducers, and posttranslational modification [1, 40]. Our results not only support such previous findings but also emphasize a role for resensitization of MAPKs and the subcellular localization of MAPK-modulating factors. The binding of M-CSF to the extracellular region of c-Fms eventually leads to degradation of the receptor mediated both by lysosomal digestion of internalized vesicles and by the ubiquitin-proteasome system [41-44]. Indeed, we observed that M-CSF stimulation induced rapid degradation of c-Fms, with the abundance of the receptor having been restored by 8 hours after M-CSF binding. This down-regulation of c-Fms is thought to attenuate M-CSF signaling. Of note, the ability of M-CSF restimulation to activate MAPKs was fully restored at 2 hours after the initial stimulation (that is, at the time of maximal c-Fms down-regulation) whereas that of RANKL restimulation was not, even though the abundance of RANK was unchanged at this time. The c-Fms levels may be sufficient to support normal levels of signaling even when the receptor levels are reduced by 50%. Alternatively, TRAF6 may be involved in RANKL-restimulated MAPK desensitization. When analyzing whether or not binding of RANKL to its receptor RANK differentially recruits TRAF6 during the first and second stimulation of RANKL, treatment with M-CSF or RANKL did not affect expression of TRAF6 (Supplementary Fig. S8A). Initial binding of RANKL to RANK transiently recruited TRAF6 in oscillatory mode, whereas TRAF6 during the second RANKL stimulation persistently interacted with RANK with no oscillation (Supplementary Fig. S8B), suggesting that oscillatory recruitment of TRAF6 by RANK may mediate RANKL-induced MAPK restimulation.
The EGF-induced proliferation of PC12 cells can be tilted toward neuronal differentiation by switching from transient to sustained activation of ERK [9]. This PC12 cell fate decision is determined by changes to sequential events including ERK interactions, nuclear localization of ERK, and phosphorylation of transcription factors. Furthermore, activation of p38 as a result of ERK inhibition was previously shown to accelerate osteoclast formation [19]. We have now found that long-lasting activation of p38 by RANKL-RANK-TRAF6 signaling gives rise to up-regulation of osteoclastogenic transcription factors and promotes osteoclast differentiation. Further studies are now warranted to provide insight into dynamic changes in the interactions of p38 with other proteins during osteoclast differentiation. Together, these observations suggest that changes in the duration and extent of MAPK activity are able to determine cell fate.
Our findings suggest that osteoclast precursor proliferation is achieved by M-CSF-induced activation of MAPKs and that osteoclast differentiation is regulated by a more persistent biphasic activation of MAPKs by RANKL, with that of p38 playing a prominent role and being mediated by a RANKL-RANK-TRAF6 axis. This pathway for p38 activation is thus a potential therapeutic target for osteoporotic bone defects due to increased osteoclast formation and activation. Regulatory factors linked to p38 signaling have previously been proposed as therapeutic targets for rheumatoid arthritis [45]. The distinct patterns of MAPK signaling induced by M-CSF and by RANKL in osteoclast precursors support a role for MAPKs in cell fate determination.
Supplementary Material
Supplementary Figures and Table.
Abbreviations
M-CSF: macrophage colony-stimulating factor; RANKL: receptor activator of NF-κB ligand; JNK: c-Jun NH2-terminal kinase; NF-κB: nuclear factor-κB; NFATc1: nuclear factor of activated T cells c1; α-MEM: α-minimum essential medium; TRAP: tartrate-resistant acid phosphatase; CathK: cathepsin K; CAII: carbonic anhydrase II; MMP9: matrix metalloproteinase-9.
Acknowledgements
We thank Y. Choi (University of Pennsylvania, Philadelphia, PA) for kindly providing M-CSF and RANKL. This study was supported by a grant (no. A120596) from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare, and Family Affairs, Republic of Korea, as well as by a grant (no. 2013R1A1A2A10006677) from the National Research Foundation of Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci. 2008;65:3525-44
2. Katz M, Amit I, Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta. 2007;1773:1161-76
3. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911-2
4. Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320-44
5. Chen JY, Lin JR, Cimprich KA, Meyer T. A two-dimensional ERK-AKT signaling code for an NGF-triggered cell-fate decision. Mol Cell. 2012;45:196-209
6. Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648-9
7. Qui MS, Green SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705-17
8. York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ. Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol. 2000;20:8069-83
9. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179-85
10. Kunath T, Saba-El-Leil MK, Almousailleakh M, Wray J, Meloche S, Smith A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134:2895-902
11. Jaiswal RK, Jaiswal N, Bruder SP, Mbalaviele G, Marshak DR, Pittenger MF. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. The Journal of biological chemistry. 2000;275:9645-52
12. Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791-801
13. Blair HC, Robinson LJ, Zaidi M. Osteoclast signalling pathways. Biochem Biophys Res Commun. 2005;328:728-38
14. Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231:241-56
15. Ross FP. M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann N Y Acad Sci. 2006;1068:110-6
16. Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12:17-25
17. Sankar U, Patel K, Rosol TJ, Ostrowski MC. RANKL coordinates cell cycle withdrawal and differentiation in osteoclasts through the cyclin-dependent kinase inhibitors p27KIP1 and p21CIP1. J Bone Miner Res. 2004;19:1339-48
18. Mizoguchi T, Muto A, Udagawa N, Arai A, Yamashita T, Hosoya A. et al. Identification of cell cycle-arrested quiescent osteoclast precursors in vivo. J Cell Biol. 2009;184:541-54
19. Hotokezaka H, Sakai E, Kanaoka K, Saito K, Matsuo K, Kitaura H. et al. U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast-like cells. The Journal of biological chemistry. 2002;277:47366-72
20. Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457-84
21. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
22. Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. The Journal of biological chemistry. 2002;277:44347-56
23. Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S. et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001;20:1271-80
24. Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H. et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4:1041-9
25. Aouadi M, Binetruy B, Caron L, Le Marchand-Brustel Y, Bost F. Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie. 2006;88:1091-8
26. Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F. et al. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. The Journal of experimental medicine. 2005;202:589-95
27. Karsdal MA, Hjorth P, Henriksen K, Kirkegaard T, Nielsen KL, Lou H. et al. Transforming growth factor-beta controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. The Journal of biological chemistry. 2003;278:44975-87
28. Huang H, Ryu J, Ha J, Chang EJ, Kim HJ, Kim HM. et al. Osteoclast differentiation requires TAK1 and MKK6 for NFATc1 induction and NF-kappaB transactivation by RANKL. Cell death and differentiation. 2006;13:1879-91
29. Friedman A, Perrimon N. Genetic screening for signal transduction in the era of network biology. Cell. 2007;128:225-31
30. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241-5
31. von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N. et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol. 2009;11:1458-64
32. Sherr CJ, Rettenmier CW, Sacca R, Roussel MF, Look AT, Stanley ER. The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell. 1985;41:665-76
33. Jacquel A, Benikhlef N, Paggetti J, Lalaoui N, Guery L, Dufour EK. et al. Colony-stimulating factor-1-induced oscillations in phosphatidylinositol-3 kinase/AKT are required for caspase activation in monocytes undergoing differentiation into macrophages. Blood. 2009;114:3633-41
34. Huynh J, Kwa MQ, Cook AD, Hamilton JA, Scholz GM. CSF-1 receptor signalling from endosomes mediates the sustained activation of Erk1/2 and Akt in macrophages. Cell Signal. 2012;24:1753-61
35. Hamilton JA. CSF-1 signal transduction. J Leukoc Biol. 1997;62:145-55
36. Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14:628-38
37. Nakashima T, Takayanagi H. Osteoclasts and the immune system. J Bone Miner Metab. 2009;27:519-29
38. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239-52
39. Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell research. 2005;15:36-42
40. Agell N, Bachs O, Rocamora N, Villalonga P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal. 2002;14:649-54
41. Lee PS, Wang Y, Dominguez MG, Yeung YG, Murphy MA, Bowtell DD. et al. The Cbl protooncoprotein stimulates CSF-1 receptor multiubiquitination and endocytosis, and attenuates macrophage proliferation. EMBO J. 1999;18:3616-28
42. Wang Y, Yeung YG, Stanley ER. CSF-1 stimulated multiubiquitination of the CSF-1 receptor and of Cbl follows their tyrosine phosphorylation and association with other signaling proteins. J Cell Biochem. 1999;72:119-34
43. Manger R, Najita L, Nichols EJ, Hakomori S, Rohrschneider L. Cell surface expression of the McDonough strain of feline sarcoma virus fms gene product (gp 140fms). Cell. 1984;39:327-37
44. Guilbert LJ, Stanley ER. The interaction of 125I-colony-stimulating factor-1 with bone marrow-derived macrophages. The Journal of biological chemistry. 1986;261:4024-32
45. Yoshizawa T, Hammaker D, Boyle DL, Corr M, Flavell R, Davis R. et al. Role of MAPK kinase 6 in arthritis: distinct mechanism of action in inflammation and cytokine expression. J Immunol. 2009;183:1360-7
Author contact
![]() Corresponding author: Daewon Jeong, Department of Microbiology, Yeungnam University College of Medicine, 170 Hyunchung-Ro, Nam-Gu, Daegu 705-717, Korea. E-mail: dwjeongac.kr
Corresponding author: Daewon Jeong, Department of Microbiology, Yeungnam University College of Medicine, 170 Hyunchung-Ro, Nam-Gu, Daegu 705-717, Korea. E-mail: dwjeongac.kr