Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(4):397-408. doi:10.7150/ijbs.13475 This issue Cite
Research Paper
MiR-487a Promotes TGF-β1-induced EMT, the Migration and Invasion of Breast Cancer Cells by Directly Targeting MAGI2
Mengtao Ma1*, Miao He1*, Qian Jiang1, Yuanyuan Yan1, Shu Guan2, Jing Zhang1, Zhaojin Yu1, Qiuchen Chen1, Mingli Sun1, Weifan Yao1, Haishan Zhao1, Feng Jin2, Minjie Wei1 ![]()
1. Department of Pharmacology, School of Pharmacy, China Medical University, Shenyang, Liaoning Province, China;
2. Department of Surgical Oncology, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, China.
*These authors contributed equally to this work.
Received 2015-8-6; Accepted 2015-11-29; Published 2016-2-5
Abstract

Tumor metastasis is a complex and multistep process and its exact molecular mechanisms remain unclear. We attempted to find novel microRNAs (miRNAs) contributing to the migration and invasion of breast cancer cells. In this study, we found that the expression of miR-487a was higher in MDA-MB-231breast cancer cells with high metastasis ability than MCF-7 breast cancer cells with low metastasis ability and the treatment with transforming growth factor β1 (TGF-β1) significantly increased the expression of miR-487a in MCF-7 and MDA-MB-231 breast cancer cells. Subsequently, we found that the transfection of miR-487a inhibitor significantly decreased the expression of vimentin, a mesenchymal marker, while increased the expression of E-cadherin, an epithelial marker, in both MCF-7 cells and MDA-MB-231 cells. Also, the inactivation of miR-487a inhibited the migration and invasion of breast cancer cells. Furthermore, our findings demonstrated that miR-487a directly targeted the MAGI2 involved in the stability of PTEN. The down-regulation of miR-487a increased the expression of p-PTEN and PTEN, and reduced the expression of p-AKT in both cell lines. In addition, the results showed that NF-kappaB (p65) significantly increased the miR-487a promoter activity and expression, and TGF-β1 induced the increased miR-487a promoter activity via p65 in MCF-7 cells and MDA-MB-231 cells. Moreover, we further confirmed the expression of miR-487a was positively correlated with the lymph nodes metastasis and negatively correlated with the expression of MAGI2 in human breast cancer tissues. Overall, our results suggested that miR-487a could promote the TGF-β1-induced EMT, the migration and invasion of breast cancer cells by directly targeting MAGI2.
Keywords: miR-487a, MAGI2, EMT, TGF-β1, p65, Breast cancer.
Introduction
At present, metastasis is the leading cause of breast cancer deaths among women [1]. Tumor metastasis is modulated by various factors, such as connected angiogenesis, signal transduction, cell adhesion, metastasis related factors [2]. Recent studies found that epithelial mesenchymal transition (EMT) played an important role in the process of tumor metastasis.
During the process of EMT, epithelial markers, such as tight junction protein zonula occluden-1 (ZO-l) and E-cadherin are down-regulated, while mesenchymal markers, such as N-cadherin and vimentin, or signal transduction proteins, such as Snail and Twist, are up-regulated. Eventually, epithelial tumor cells lost cell polarity [3], the connection between cells become loose [4] and tumor cells are more prone to invasion and metastasis [5, 6]. The occurrence of EMT is related to complex signal transduction pathways and molecular mechanisms, such as transforming growth factor-beta (TGF-β), Wnt and Notch signaling pathways. TGF-β, a cytokine with multiple biological activities, can induce and maintain EMT via autocrine and paracrine effects on a variety of tumor cells [7]. It was reported that not only the smad pathway but also the non-smad pathways, such as the MAPK pathway, Rho pathway, phosphatidylinositol-3-kinase (PI3K) signaling pathway, were involved in TGF-β-induced EMT. At present, several findings supported that TGF-β could promote tumor cell migration and invasion through PI3K/AKT pathway [8, 9]. However, the molecular mechanisms underlying TGF-β inducing EMT by PI3K/AKT pathway remain unclear.
MicroRNAs (miRNAs) regulate gene expression through mRNA degradation or translational repression. Many miRNAs were contributed to regulating the process of EMT in cancer cells. The cluster of miR-200 was reported to inhibit the migration of breast cancer cells by directly targeting E-cadherin and increasing the ZEB1/ZEB2 expression [10]. It had also been found that miR-10b [11], miR-23a [12] and miR-181a [13] played an important role in TGF-β-induced EMT of breast cancer cells. In previous studies, we found that miR-487a increased the sensitivity of resistant breast cancer cells MCF-7/MX to MX by targeting breast cancer resistance protein (BCRP) [14]. Recently we found that the expression of miR-487a was significantly higher in MDA-MB-231breast cancer cells with high metastasis ability than MCF-7 breast cancer cells with low metastasis ability, and TGF-β1 could promote the expression of miR-487a in breast cancer cells. So we hypothesized that miR-487a might be involved in the process of EMT of breast cancer cells.
In this study, MCF-7 and MDA-MB-231 breast cancer cells were transfected with miR-487a inhibitor and treated with TGF-β1, to investigate the role of miR-487a in TGF-β1 induced EMT, migration and invasion of breast cancer cells. Bioinformatics analysis and luciferase reporter assay demonstrated MAGI2 was a direct target of miR-487a. Furthermore, we measured the effects of the down-regulation of miR-487a on the activation of PI3K/AKT pathway in MCF-7 and MDA-MB-231 cells, to investigate the mechanisms of miR-487a regulating the migration and invasion of breast cancer cells. In general, our findings showed that miR-487a promoted the TGF-β1-induced EMT, the migration and invasion of breast cancer cells by directly targeting MAGI2.
Materials and methods
Cell culture
The human embryonic kidney 293T cells (HEK 293T cells), the human breast cancer MCF-7, T47D, MDA-MB-435s, BT549 and MDA-MB-231 cell lines were all purchased from the American Type Culture Collection (ATCC). MCF-7, T47D, and 293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Hyclone, USA) high glucose medium with 10% fetal bovine serum (FBS, Hyclone, USA). BT549 cells were cultured in RPMI-1640 medium (Hyclone, USA) with 10% FBS. MDA-MB-435s and MDA-MB-231 cells were grown in L15 culture medium (GIBCO, USA) with 10% FBS.
Cell transient transfection and treatment with TGF-β1
MCF-7 and MDA-MB-231 cells were transiently transfected with miR-487a inhibitor or negative control inhibitor (NC inhibitor) from RiboBio (Guangzhou, China) using Lipofectamine 2000 (Life Technologies, USA). The detailed procedure was described as our previous reports [14]. After the normal culture for 24 h, MCF-7 and MDA-MB-231 cells were cultured in medium without serum for 24 h, then treated with TGF-β1 (PeproTech, USA) at a concentration of 10 ng/ml in serum-free medium for indicated time periods.
Western blot analysis
Western blot analysis was performed as described previously [14]. The primary antibody included E-cadherin (1:1000; 3195, CST, USA), vimentin (1:1000; 5741, CST, USA), MAGI2 (1:1000; YT2627, Immunoway, USA), p-AKT (1:1000; 4060, CST, USA), AKT (1:1000; 9272, CST, USA), p-PTEN (1:1000; 9554, CST, USA), PTEN (1:1000; 9559, CST, USA), p65 (1:1000; ab19870, Abcam, USA) and β-actin (1:1000; 4967, CST, USA). β-actin was used as a normalization control. The protein expression was quantitatively analyzed using FluorChem V2.0 software (Alpha Innotech Corp, USA).
QRT-PCR analysis
The reaction conditions and procedure of qRT-PCR were previously described [14]. The PCR primers were listed in table 1. The β-actin gene was used as an internal control for MAGI2 mRNA expression analysis. U6 snRNA was used as an internal control for the expression analysis of miR-487a. The 2-∆∆Ct method was used to calculate the fold change for miR-487a and MAGI2 mRNA relative to the control.
Primer sequences used for the qRT-PCR analysis.
| Application | Oligonudeotides | Sequences (5'-3') |
|---|---|---|
| miR-487a | RT | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAACTGG |
| Forward | CGCTGGCAATCATACAGGGACAT | |
| Reverse | GTGCAGGGTCCGAGGT | |
| U6 | RT | AACGCTTCACGAATTTGCGT |
| Forward | CTCGCTTCGGCAGCACA | |
| Reverse | AACGCTTCACGAATTTGCGT | |
| MAGI2 | Forward | ACCCTTCGCATCATTCCTC |
| Reverse | CTTGCCTTGCTTTCACTTCC | |
| β-actin | Forward | CTGGCCGGGACCTGACT |
| Reverse | TCCTTAATGTCACGCACGATTT |
Scratch migration analysis
The scratch migration analysis was performed as described previously [15]. Briefly, MCF-7 and MDA-MB-231 cells were allowed to grow until confluent. After making a scratch in the middle of the cells in straight lines, the cells were treated with or without 10 ng/ml TGF-β1. The microscopic images of the cells were collected before and after the treatment for 48 h. Wound closure was quantitatively analyzed by Image J software.
Invasion analysis
Invasion assays were performed as previously described [16]. Briefly, cells were serum-starved overnight. Cells (1×105) were plated in serum-free medium on the top Transwell precoated with Matrigel, and then were stimulated with or without 10 ng/ml TGF-β1 for 48 h. The cells invading to the lower chamber were stained with crystal violet solution and counted using a microscope.
Dual-luciferase reporter assay
The GV354-basic vector containing upstream promoter regions of pri-miR-487a was constructed by Shanghai Genechem Co., Ltd of China. The pGL3-MAGI2-3'UTR plasmids were constructed by Ribo Life Science Co., Ltd of China. For the effects of miR-487a on the MAGI2-3'UTR activity, 293T cells were transfected with pGL3-MAGI2-3'UTR plasmids (wild type or mutant) and miR-487a mimic. For the effects of p65 on the miR-487a promoter activity, the GV354-miR-487a promoter plasmids or NC promoter plasmids and p65 cDNA were transfected into MCF-7 and MDA-MB-231 cells treated with or without p65 inhibitor. For the effects of TGF-β1 on the miR-487a promoter activity, the GV354-miR-487a promoter plasmids or NC promoter plasmids were transfected into MCF-7 and MDA-MB-231 cells treated with or without p65 inhibitor and TGF-β1. Luciferase and renilla signals were detected after the transfection for 48 h according to the protocol of the Dual Luciferase Reporter Assay Kit (Promega, USA).
Patients and tissue samples
Breast cancer tissues were obtained from 119 breast cancer patients, who operated at First Affiliated Hospital of China Medical University from 2004 to 2008. The study has attained the approval of the Institutional Review Board of China Medical University, and the informed consent from all subjects in the study.
Immunohistochemistry
The procedure for immunohistochemistry was performed as previously described [17]. The immunoreactivity was evaluated according to the percentage and intensity of the stained cells. The intensity scores: 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). The percentage of stained cells was scored from 0 to 100% of stained cells. The final score was attained by multiplying the intensity score and the percentage of stained cells. The minimum score was 0 and the maximum score was 300%.
In situ hybridization
In situ hybridization was performed according to the protocol of Enhanced Sensitive ISH Detection KitⅡ (Boster, China). The sequence of the synthetic oligonucleotide probe labeled by 3' and 5'-tailing with digoxin was 5'-aactttatgtccctgtatgatt-3'. The staining scores for in situ hybridization were the same as those for immunohistochemistry.
Statistical analysis
Data analyses were carried out using SPSS17.0 software. Student's t-test was used to analyze two groups of data in vitro experiments. One-way analysis of variance (ANOVA) was used to evaluate the differences among three or more groups. Pearson chi-square test was used for categorical data. The Pearson rank correlation analysis was used to evaluate the association between the expression of miR-487a and MAGI2. Probability values <0.05 was regard as statistically different significance.
Results
TGF-β1 induces miR-487a over-expression in MCF-7 and MDA-MB-231 breast cancer cells
In the previous studies we found that miR-487a expression was significantly lower in MCF-7/MX breast resistant cells than the parental MCF-7 cells. Ectopic miR-487a expression inhibited the expression of BCRP in MCF-7/MX cells and enhanced the sensitivity of MCF-7/MX cells to mitoxantrone (MX) [14]. In the present study, we detected the expression of miR-487a in breast cancer cells with different metastasis ability, including MCF-7, T47D, MDA-MB-435s, BT549 and MDA-MB-231 cells by qRT-PCR. Surprisingly, we found that the expression of miR-487a was significantly higher in MDA-MB-231 breast cancer cells with high metastasis ability than MCF-7 breast cancer cells with low metastasis ability (Fig. 1A). The data suggested that miR-487a may be associated with the metastasis phenotype of breast cancer cells.
TGF-β1 induces the over-expression of miR-487a in breast cancer cells. (A) The expression of miR-487a was measured in MCF-7, T47D, MDA-MB-435s, BT549 and MDA-MB-231 cells breast cancer cells by qRT-PCR. The mesenchymal marker vimentin and epithelial marker E-cadherin were detected by western blot in MCF-7 (B) and MDA-MB-231 cells (C) treated with 10 ng/ml TGF-β1 for 24 h or 48 h. β-actin was used as an internal control. (D) The expression of miR-487a was detected by qRT-PCR in MCF-7 and MDA-MB-231 cells treated with 10 ng/ml TGF-β1 for 24 h or 48 h. *P<0.05, **P<0.01.
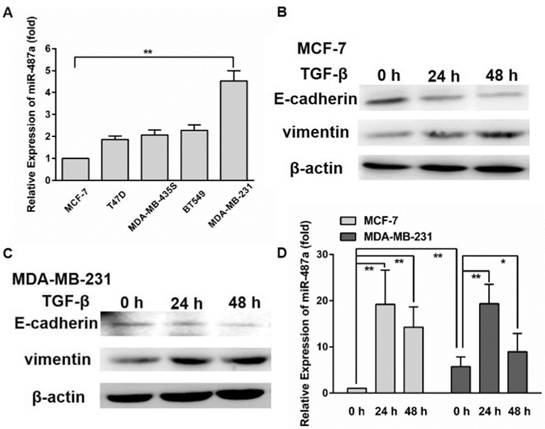
To investigate whether miR-487a was involved in the formation of EMT, we first detected the proteins expression of mesenchymal marker vimentin and epithelial marker E-cadherin in MCF-7 and MDA-MB-231 cells treated with 10 ng/ml TGF-β1 for 24 h or 48 h. The results showed that the treatment of TGF-β1 for 24 h or 48 h significantly increased the expression of vimentin and decreased the expression of E-cadherin in two cell lines (Fig. 1B and 1C). The results demonstrated that TGF-β1 induced the occurrence of EMT in two cell lines.
Next, we detected the expression of miR-487a in MCF-7 and MDA-MB-231 cells treated with TGF-β1 by qRT-PCR and found that the miR-487a expression was increased by approximately 19-fold and 17-fold in MCF-7 cells, 3.5-fold and 2-fold in MDA-MB-231 cells after the treatment with TGF-β1 for 24 h and 48 h, respectively (Fig. 1D). The findings showed that the over-expression of miR-487a was associated with the occurrence of EMT induced by TGF-β1 in breast cancer cells.
Inhibition of miR-487a expression suppresses EMT induced by TGF-β1 in breast cancer cells
To further analyze the role of miR-487a in TGF-β1-induced EMT, we first transfected MCF-7 and MDA-MB-231 cells with miR-487a inhibitor. After 24 h, the cells were starved with serum-free medium for 24 h and then treated with 10 ng/ml TGF-β1 for 24 h. The results showed that the transfection of miR-487a inhibitor significantly decreased the expression of miR-487a in both MCF-7 cells and MDA-MB-231 cells with or without TGF-β1 treatment, compared with the NC group. The inhibitory extent of miR-487a was more obvious in the MCF-7 cells and MDA-MB-231 cells treated with TGF-β1 (40%, 50%) than those treated without TGF-β1 (30%, 40%; Fig. 2A and 2B).
Inhibition of miR-487a expression suppresses TGF-β1-induced EMT in MCF-7 and MDA-MB-231 cells. The expression of miR-487a was detected by qRT-PCR in MCF-7 (A) and MDA-MB-231 cells (B) transfected with miR-487a inhibitor for 24 h, starved with serum-free medium for 24 h, then treated with 10 ng/ml TGF-β1 for 24 h. The expression of miR-487a in the cells transfected with NC and treated without TGF-β1 was set as 1. The expression of E-cadherin and vimentin was measured by western blot in MCF-7 (C) and MDA-MB-231 cells (D) transfected with miR-487a inhibitor for 24 h, starved with serum-free medium for 24 h, then treated with 10 ng/ml TGF-β1 for 24 h. β-actin was used as an internal control. The protein expression of E-cadherin or vimentin normalized to β-actin in the cells transfected with NC was set as 1. 1 for blank group, 2 for NC group, 3 for miR-487a inhibitor group. *P<0.05, **P<0.01 compared with NC group.
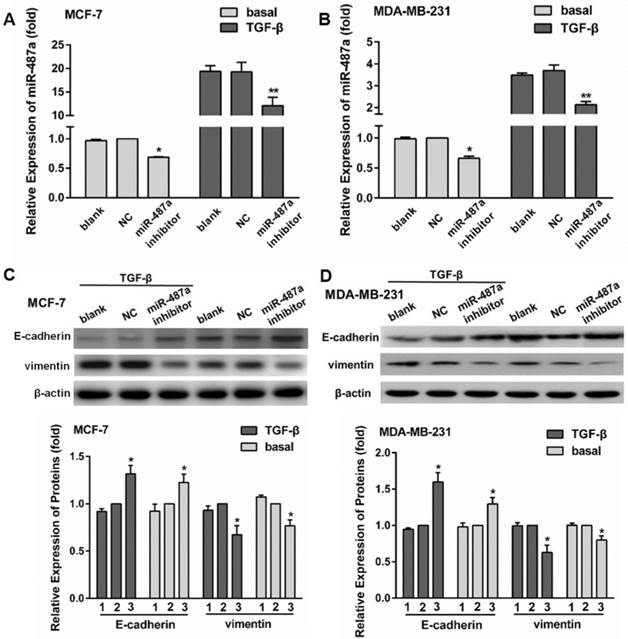
In addition, we found that the transfection of miR-487a inhibitor significantly decreased the expression of vimentin, while increased the expression of E-cadherin in both MCF-7 cells and MDA-MB-231 cells (Fig. 2C and 2D). Especially the changes were more obvious in the cells treated with TGF-β1 than those treated without TGF-β1.
The down-regulation of miR-487a inhibits breast cancer cells migration and invasion induced by TGF-β1
To further clarify the role of miRNA-487a in the process of TGF-β1 induced EMT in MCF-7 and MDA-MB-231 cells, we detected the changes of the migration and invasion abilities of the cells treated with or without TGF-β1 after transfected with miR-487a inhibitor. We found that the treatment of TGF-β1 for 48 h significantly increased the cells migration abilities in MCF-7 cells by 40% and MDA-MB-231 cells by 20% by wound healing analysis (Fig. 3A and 3B). In addition, the increased migration abilities of MCF-7 and MDA-MB-231 cells were both significantly inhibited by the transfection with miR-487a inhibitor.
The down-regulation of miR-487a inhibits the migration and invasion induced by TGF-β1 in breast cancer cells. The migration abilities were measured by wound healing analysis in MCF-7 cells (A) and MDA-MB-231 cells (B) transfected with miR-487a inhibitor or NC, and treated with or without TGF-β1. The wound closure in the cells transfected with NC and treated without TGF-β1 was set 1. The invasion abilities were measured by transwell invasion assay in MCF-7 cells (C) and MDA-MB-231 cells (D) transfected with miR-487a inhibitor or NC, and treated with or without TGF-β1. The invasion ability of the cells transfected with NC and treated without TGF-β1 was set 1. *P<0.05.
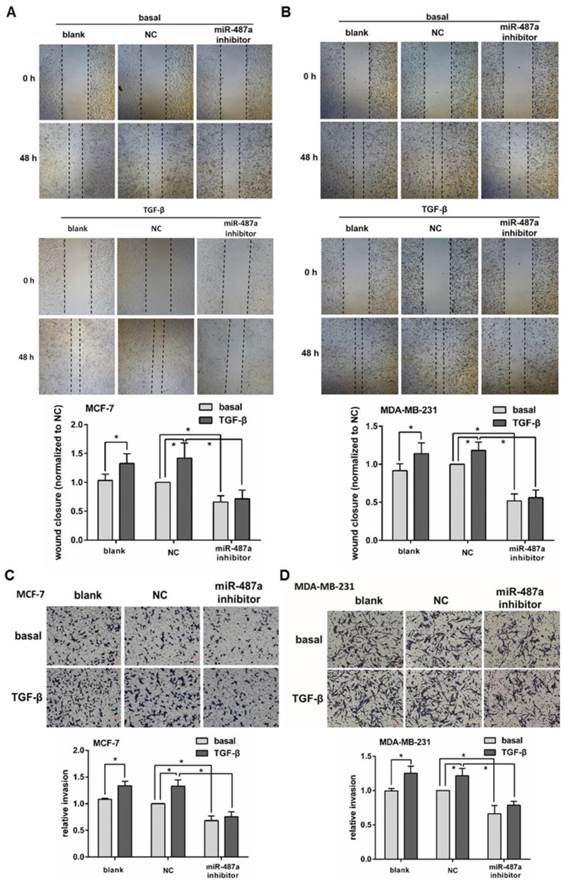
We also observed that the treatment of TGF-β1 for 48 h significantly increased the cells invasion abilities in MCF-7 cells by 40% and MDA-MB-231 cells by 25% by transwell invasion assay (Fig. 3C and 3D). The miR-487a inhibitor obviously suppressed the increased invasion abilities of MCF-7 and MDA-MB-231 cells.
MAGI2 is a direct target of miR-487a
To investigate the mechanisms of miR-487a regulating the migration and invasion of breast cancer cells, we first applied targetscan to predict the possible targets of miR-487a. We found that MAGI2 mRNA-3'-untranslated region (3'-UTR) had a binding site of miR-487a (Fig. 4A). MAGI2 (Membrane-associated guanylate kinase inverted 2) was a tumor suppressor and could be combined with the PTEN in HCC cells. MAGI2 could inhibit the invasion and migration of tumor cells by maintaining the stability of PTEN to inhibit the activity of PI3K/AKT pathway [18, 19]. To determine whether MAGI2 was a direct target of miR-487a, we constructed a luciferase reporter plasmid containing MAGI2-3'UTR harboring a conserved miR-487a binding site (pGL3-MAGI2-3'UTR) and a plasmid containing MAGI2-3'UTR with miR-487a target sequences mutated (pGL3-MAGI2-3'UTR mu). The binding ability of miR-487a to MAGI2-3'UTR was detected by double luciferase reporter assay.
MAGI2 is a direct target of miR-487a. (A) Bioinformatics predicted the binding site of miR-487a with MAGI2 mRNA-3'UTR. (B) Double luciferase reporter assay in 293T cells co-transfected with miR-487a mimic or miR-NC and PGL3-MAGI2-3'UTR or pGL3-MAGI2-3'UTR mu. Relative luciferase activity was measured at 48h after transfection. *P<0.05 compared with NC group.
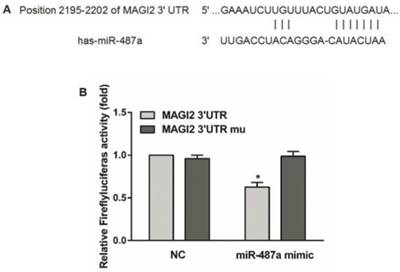
The results showed that the luciferase activity was significantly decreased by approximately 40% in 293T cells co-transfected with miR-487a mimic and the pGL3-MAGI2-3'UTR compared with NC. However, the luciferase activity was not significantly reduced in the cells co-transfected with miR-487a mimic the MAGI2-3'UTR mu (Fig. 4B). The data suggested that MAGI2 was a direct target of miR-487a.
The down-regulation of miR-487a inhibits TGF-β1-induced EMT by increasing the expression of MAGI2 in breast cancer cells
To further confirm whether miR-487a was involved in TGF-β1-induced EMT through targeting MAGI2, we performed the following experiments. We first found that the treatment of TGF-β1 significantly reduced the expression of MAGI2 mRNA by qRT-PCR analysis in both MCF-7 cells and MDA-MB-231 cells (Fig. 5A and 5B). MiR-487a inhibitor significantly increased the expression of MAGI2 mRNA by 40% in MCF-7 cells treated with TGF-β1 and by 20% in MCF-7 cells treated without TGF-β1. Similarly, miR-487a inhibitor significantly increased the expression of MAGI2 mRNA by 50% in MDA-MB-231cells treated with TGF-β1 and by 20% in MDA-MB-231 cells treated without TGF-β1 (Fig. 5A and 5B). The changes of protein expression levels of MAGI2 were consistent with the expression levels of MAGI2 mRNA (Fig. 5C and 5D).
The down-regulation of miR-487a inhibits TGF-β1-induced EMT by increasing the expression of MAGI2 in breast cancer cells. The expression of MAGI2 mRNA was detected by qRT-PCR in MCF-7cells (A) and MDA-MB-231cells (B) transfected with miR-487a inhibitor or NC, and treated with or without TGF-β1. The expression of MAGI2 mRNA in the cells transfected with NC and treated without TGF-β1 was set 1. The protein expression of MAGI2 was detected by western blot in MCF-7 cells (C) and MDA-MB-231cells (D) transfected with miR-487a inhibitor or NC, and treated with or without TGF-β1. The proteins expression of p-PTEN, PTEN, p-AKT and AKT was detected by western blot in MCF-7 cells (E) and MDA-MB-231cells (F) transfected with miR-487a inhibitor or NC, and treated with or without TGF-β1. β-actin was used as an internal control. The proteins expression of p-PTEN, PTEN, or p-AKT normalized to β-actin in the cells transfected with NC was set as 1. 1 for blank group, 2 for NC group, 3 for miR-487a inhibitor group. *P<0.05 compared with NC group.
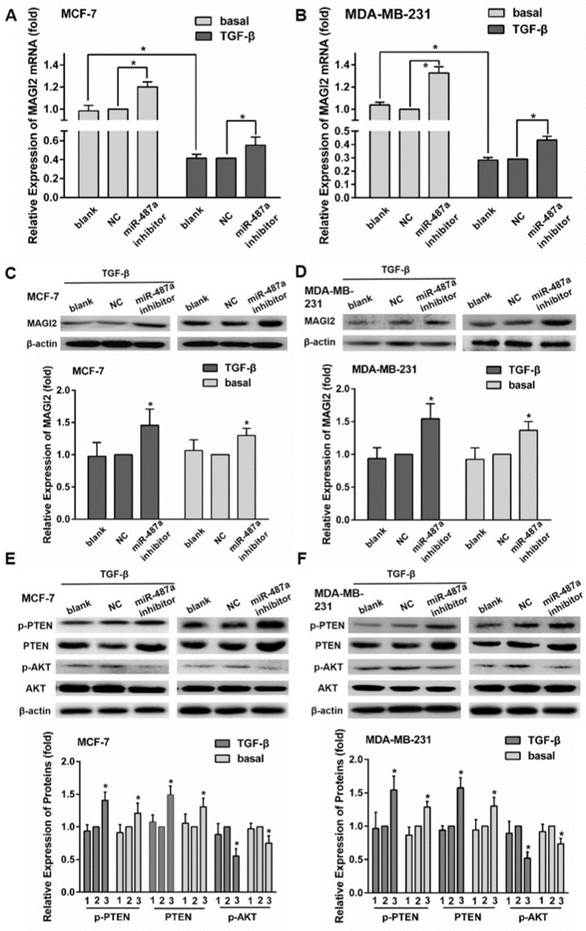
Next, we detected the expression of MAGI2 downstream proteins, p-PTEN, p-AKT, PTEN, and AKT. The results showed that miR-487a inhibitor significantly increased the expression levels of PTEN and p-PTEN and reduced the expression of p-AKT compared with NC inhibitor (Fig. 5E and 5F). These results suggested that miR-487a could be involved in the process of EMT mediated by TGF-β1 by targeting MAGI2 and regulating MAGI2/PETN/AKT pathway.
TGF-β1-induced the increased NF-kappaB (p65) expression transcribes miR-487a in breast cancer cells
To further explore how TGF-β1 regulates the expression of miR-487a, we next sought to identify transcription factor-binding motifs in the region 2000 bp upstream of the hsa-miR-487a promoter using Jaspar and Promo (ALGGEN) software. The analysis revealed that NF-kappaB (p65) had three binding sites in the upstream of the hsa-miR-487a (Fig 6A). Luciferase reporter assay showed that p65 cDNA transfection significantly increased the miR-487a promoter activity and the p65 inhibitor decreased the miR-487a promoter activity in MCF-7 cells and MDA-MB-231 cells (Fig 6B). In addition, we found that the over-expression of p65 by transfection of p65 cDNA elevated the expression of miR-487a in MCF-7 cells and MDA-MB-231 cells (Fig 6C). The data suggested that transcription factor p65 contributed to the miR-487a transcriptional activation.
TGF-β1-induced the increased NF-kappaB (p65) expression transcribes miR-487a in breast cancer cells. (A) The transcription factor-binding motifs in the region 2kp upstream of the hsa-miR-487a promoter with NF-kappaB (p65) using Jaspar and Promo (ALGGEN) software. (B) The miR-487a promoter activity was measured in MCF-7 cells and MDA-MB-231 cells transfected with p65 cDNA and miR-487a promoter plasmids or NC promoter plasmids. (C) The expression of miR-487a was measured in MCF-7 cells and MDA-MB-231 cells transfected with p65 cDNA or control cDNA by qRT-PCR. (D) The protein expression of p65 was detected by western blot in MCF-7 cells and MDA-MB-231 cells treated with TGF-β1. (E) The miR-487a promoter activity was measured in MCF-7 cells and MDA-MB-231 cells treated with TGF-β1 or p65 inhibitor. Set the luciferase activity in the cells transfected with NC promoter plasmids as 1. **P<0.01 compared with NC group, ##P<0.01 compared with miR-487a promoter plasmids and no p65 inhibitor group.
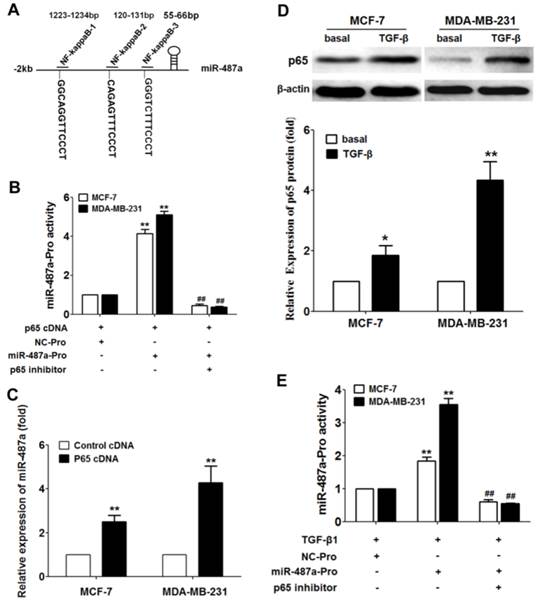
To further evaluate how TGF-β1 affects the expression of miR-487a, we measured the effects of TGF-β1 on the expression of p65 protein in breast cancer cells by Western blot. The results showed that TGF-β1 significantly induced the increased expression of p65 in MCF-7 cells and MDA-MB-231 cells (Fig 6D). Furthermore, we surprisingly found that TGF-β1 significantly increased the miR-487a promoter activity in MCF-7 cells and MDA-MB-231 cells, and the p65 inhibitor eliminated TGF-β1-induced the upregulated miR-487a promoter activity by luciferase reporter assay (Fig 6E). These results suggested that TGF-β1 increased the miR-487a promoter activity and expression by inducing the p65 upregulation in breast cancer cells.
MiR-487a expression is negatively correlated with MAGI2 expression in human breast cancer tissues
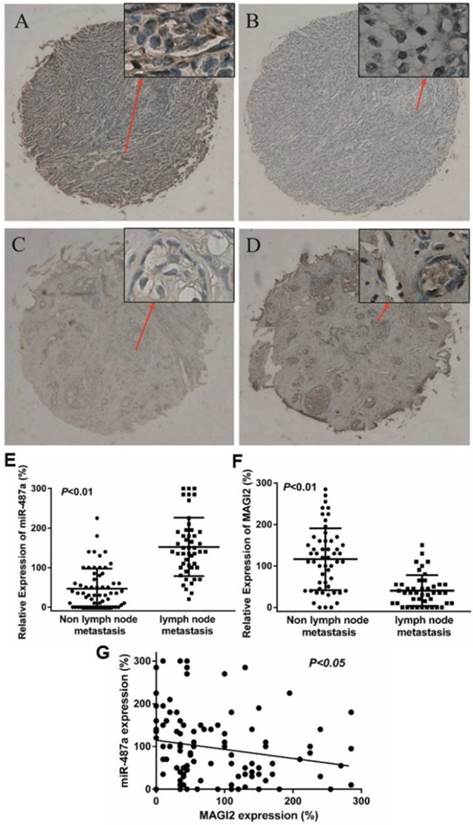
We further detected the miR-487 expression by in situ hybridization and the MAGI2 expression by immunohistochemistry in 119 cases of breast cancer samples with tissue microarray (TMA) to assess the correlation of endogenous expression of miR-487a with MAGI2. The positive expression of miR-487a and MAGI2 were mainly in cytoplasm (Fig. 7A-7D). According to the optimal sensitivity and specificity of the receiver operating characteristic (ROC) curve analysis with respect to lymph nodes metastasis, 65% and 67.5% were defined as the optimal cut point for miR-487a and MAGI2, respectively. MAGI2 staining of 13 cases of sample sections occurred off chip and the information for Lymph node metastasis of 11 cases of breast cancer patients was not got. As shown in Table 2, the positive expression rate for miR-487a and MAGI2 was 52.9% (63/119) and 45.3% (48/106), respectively. Pearson chi square (χ2) test showed that the expression of miR-487a was positively correlated with the metastasis of lymph nodes and tumor size (P<0.01, P<0.05, Table 2 and Fig. 7E), the expression of MAGI2 was negatively correlated with the metastasis of lymph nodes (P<0.01, Fig. 7F), and miR-487a expression was negatively correlated with the expression of MAGI2 in human breast cancer tissues (Fig. 7G).
The expression of miR-487a is negatively correlated with MAGI2 expression in human breast cancer tissues. (A) The positive and (B) negative expression of miR-487a was measured by in situ hybridizationin in 119 cases of breast cancer samples with tissue microarray (TMA). (C) The negative and (D) positive expression of MAGI2 was measured by immunohistochemistry in 119 cases of breast cancer samples with TMA. (E) The expression of miR-487a was compared between the breast cancer tissues with lymph node metastases and non-lymph node metastases. (F) The expression of MAGI2 was compared between the breast cancer tissues with lymph node metastases and non-lymph node metastases. (G) The correlation analysis of the expression miR-487a and the expression of MAGI2 in 119 cases of breast cancer samples.
The association of the expression of miR-487a with the characteristics and the MAGI2 expression in 119 cases of breast cancer patients.
| Characteristics | No. of cases (%) | miR-487a | |||
|---|---|---|---|---|---|
| Positive (%) | Negative (%) | P-value | |||
| MAGI2 | |||||
| Negative | 58 (54.7) | 38 (65.5) | 20 (34.5) | 0.042 | |
| Positive | 48 (45.3) | 22 (45.8) | 26 (54.2) | ||
| lymphatic metastasis | |||||
| Negative | 61 (56.5) | 16 (26.2) | 45 (73.8) | <0.001 | |
| Positive | 47 (43.5) | 42 (89.4) | 5 (10.6) | ||
| Age at diagnosis | |||||
| <50 | 45 (42.9) | 22 (48.9) | 23 (51.1) | 0.535 | |
| ≥50 | 60 (57.1) | 33 (55.0) | 27 (45.0) | ||
| TNM stage | |||||
| I | 22 (24.7) | 11 (50.0) | 11 (50.0) | ||
| II | 45 (50.6) | 20 (44.4) | 25 (55.6) | 0.727 | |
| III | 22 (24.7) | 12 (54.5) | 10 (45.5) | ||
| Tumor size, cm | |||||
| <2 | 35 (36.8) | 13 (37.1) | 22 (62.9) | 0.046 | |
| ≥2 | 60 (63.2) | 35 (58.3) | 25 (41.7) | ||
| ER | |||||
| Negative | 38 (39.2) | 21 (55.3) | 17 (44.7) | 0.793 | |
| Positive | 59 (60.8) | 31 (52.5) | 28 (47.5) | ||
| PR | |||||
| Negative | 52 (52.5) | 24 (46.2) | 28 (53.8) | 0.121 | |
| Positive | 47 (47.5) | 29 (61.7) | 18 (38.3) | ||
| C-erbB-2 | |||||
| Negative | 29 (29.9) | 15 (51.7) | 14 (48.3) | 0.808 | |
| Positive | 68 (70.1) | 37 (54.4) | 31 (45.6) | ||
| Ki67 | |||||
| Negative | 20 (23.3) | 12 (60.0) | 8 (40.0) | 0.367 | |
| Positive | 66 (76.7) | 32 (48.5) | 34 (51.5) | ||
Discussion
Considering the metastasis as the main cause of death for breast cancer patients, it is necessary to define the molecular mechanisms of metastasis to cure metastatic breast cancers. The EMT process in which epithelial cells get mesenchymal phenotypes is considered as an important step in tumor metastasis. TGF-β has been shown to play an important role in EMT process [20]. A variety of miRNAs were found to contribute to the regulation of EMT induced by TGF-β. It was reported that TGF-β could regulate the expression of miR-181 family and promote the migration and invasion in breast cancer cells [15]. MiR-181a was also found to participate in mediating the induction of EMT stimulated by TGF-β in breast cancer cells by targeting the pro-apoptotic protein Bim [21]. Gregory et al. reported that TGF-β signaling could maintain the mesenchymal state through up-regulating ZEB1 and ZEB2 and repressing miR-200 in breast cancer [22]. In the present study, we found that miR-487a acted as one of the "metastamirs". Inactivation of miR-487a could inhibit the EMT mediated by TGF-β1 by regulating the expression of MAGI2.
So far, the reports about miR-487a are very rare except that it was found up-regulated in clear cell renal cell carcinoma (ccRCC) tissues [23], and increased in atypical coronary artery disease (ACAD) patients [24]. In our studies, we found that the expression of miR-487a was significantly higher in MDA-MB-231 breast cancer cells with high metastasis ability than MCF-7 breast cancer cells with low metastasis ability. We further demonstrated that the inhibition of miR-487a suppressed the migration, invasion and EMT induced by TGF-β1 in MCF-7 and MDA-MB-231 cells, suggesting that miR-487a might play an important role in the metastasis of breast cancer cells. Furthermore, we also found that the expression of miR-487a was positively correlated with the metastasis of lymph nodes in human breast cancer patients. Previously, we demonstrated that miR-487a could sensitize MCF-7/MX breast cancer cells to MX by targeting BCRP in vitro and in vivo [14]. Evidently, the roles of miR-487a are bidirectional. As we all know, individual miRNAs can regulate different mRNAs and individual mRNAs can also be targeted by different miRNAs. Similarly, miR-181a was found to have contrary effects in different cells. It was reported that miR-181a improved the sensitivity of radiotherapy by suppressing Bcl-2 in malignant glioma as a tumor suppressor [25], while promoted tumor occurrence in breast cancer, oral cancer, liver cancer and leukemia as an oncogene [26-30].
MAGI2, also called synaptic scaffolding molecule (S-SCAM), belongs to membrane-associated guanylate kinases (MAGUKa) protein superfamily member, with multiple protein-protein interaction domains [31]. Several studies showed that MAGI2 could inhibit the EMT process by maintaining the stability of PTEN in different cells [18, 31]. To elucidate the mechanisms of miR-487a on regulating the EMT in breast cancer, we screened the targets of miR-487a by bioinformatic analysis and found that MAGI2 was one of the targets of miR-487a. Further studies demonstrated that MAGI2 was a direct target of miR-487a by luciferase reporter assay. In addition, the transfection of miR-487a inhibitor increased the expression of MAGI2, PTEN, p-PTEN, and decreased the expression of p-AKT in MCF-7 and MDA-MB-231 cells, suggesting the MAGI2 could be a key factor in miR-487a regulating the EMT process in breast cancer cells. Furthermore, we also found that miR-487a expression was negatively correlated with the expression of MAGI2 in human breast cancer tissues. Other studies showed that miR-134/487b/655 contributed to the TGF-β1-induced EMT process by targeting MAGI2 in lung cancer cells [32].
Currently, we found that the effects of the transfection of miR-487a inhibitor on decreasing the expression of vimentin and increasing the expression of E-cadherin were more obvious in MDA-MB-231 cells than in MCF-7 cells after TGF-β1 treatment. The possible reason is that the expression of miR-487a is higher in MDA-MB-231 cells with higher metastasis ability and suppressing the expression of miR-487a by the transfection of miR-487a inhibitor can induce more obvious biological effects.
Furthermore, we found that transcription factor p65 could significantly increase the miR-487a promoter activity and expression in MCF-7 cells and MDA-MB-231 cells. In addition, TGF-β1 significantly induced the increased p65 expression and miR-487a promoter activity in MCF-7 cells and MDA-MB-231 cells, while the effects could be eliminated by p65 inhibitor. We demonstrated that TGF-β1 upregulated the miR-487a expression via p65 in breast cancer cells. Similarly, Gong et al. reported that TGF-β1 enhanced the transcription of miR-106b by activating c-jun [33].
In summary, our findings suggest that miR-487a can promote the TGF-β1-induced EMT, the migration and invasion of breast cancer cells by directly targeting MAGI2. It will provide a great help to explore new molecular mechanisms on the breast cancer metastasis. However, further studies must be performed to explore the utility of miR-487a as a predictive biomarker for breast cancer metastasis.
Abbreviations
TGF-β1, transforming growth factor β1; miRNA, microRNA; EMT, Epithelial-Mesenchymal Transition; DAB, 3,3-diaminobenzidine; PTEN, phosphatase and tensin homolog deleted on chromosome ten; AKT, protein kinase B; PI3K, phosphatidylinositol 3 kinase; MAGI2, Membrane-associated guanylate kinase inverted 2; wt, wild type; mu, mutant; UTR, untranslated region.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81173092 and 81572898), Program for Liaoning Innovative Research Team in University, LNIRT, China (Grant No. LT2014016), the Liaoning Provincial Science and Technology Program, China (Grant No. 2013225079) and the S&T Projects in Shenyang, China (Grant No. F14-232-6-05).
Authors' contributions
Minjie Wei, Mengtao Ma and Miao He designed the experiments. Mengtao Ma, Qian Jiang, Yuanyuan Yan, Jing Zhang, Qiuchen Chen, Weifan Yao and Haishan Zhao performed the experiments. Mengtao Ma, Zhaojin Yu, Mingli Sun analyzed the data. Shu Guan and Feng Jin gave technical and material support. Mengtao Ma, Miao He and Minjie Wei wrote and reviewed the manuscript.
Competing Interests
The authors have no conflicts of interests to declare.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: A Cancer Journal for Clinicians. 2011;61(2):69-90
2. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009;139(5):871-890
3. Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307(5715):1603-1609
4. Ikenouchi J. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. Journal of Cell Science. 2003;116(10):1959-1967
5. Guarino M. Epithelial-mesenchymal transition and tumour invasion. The International Journal of Biochemistry & Cell Biology. 2007;39(12):2153-2160
6. Yang J, Weinberg RA. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Developmental Cell. 2008;14(6):818-829
7. Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Research. 2009;19(2):156-172
8. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-Kinase Function Is Required for Transforming Growth Factor -mediated Epithelial to Mesenchymal Transition and Cell Migration. Journal of Biological Chemistry. 2000;275(47):36803-36810
9. Chen X-F, Zhang H-J, Wang H-B, Zhu J, Zhou W-Y, Zhang H, Zhao M-C, Su J-M, Gao W, Zhang L. et al. Transforming growth factor-β1 induces epithelial-to-mesenchymal transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling pathways. Molecular Biology Reports. 2011;39(4):3549-3556
10. Korpal M, Lee ES, Hu G, Kang Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2. Journal of Biological Chemistry. 2008;283(22):14910-14914
11. Han X, Yan S, Weijie Z, Feng W, Liuxing W, Mengquan L, Qingxia F. Critical role of miR-10b in transforming growth factor-β1-induced epithelial-mesenchymal transition in breast cancer. Cancer Gene Therapy. 2014;21(2):60-67
12. Cao M, Seike M, Soeno C, Mizutani H, KITAMURA K, MINEGISHI Y, NORO R, YOSHIMURA A, CAI L, GEMMA A. MiR-23a regulates TGF-β-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. International Journal of Oncology. 2012
13. Brockhausen J, Tay SS, Grzelak CA, Bertolino P, Bowen DG, d'Avigdor WM, Teoh N, Pok S, Shackel N, Gamble JR. et al. miR-181a mediates TGF-β-induced hepatocyte EMT and is dysregulated in cirrhosis and hepatocellular cancer. Liver International. 2015;35(1):240-253
14. Ma M-T, He M, Wang Y, Jiao X-Y, Zhao L, Bai X-F, Yu Z-J, Wu H-Z, Sun M-L, Song Z-G. et al. MiR-487a resensitizes mitoxantrone (MX)-resistant breast cancer cells (MCF-7/MX) to MX by targeting breast cancer resistance protein (BCRP/ABCG2). Cancer Letters. 2013;339(1):107-115
15. Neel J-C, Lebrun J-J. Activin and TGFβ regulate expression of the microRNA-181 family to promote cell migration and invasion in breast cancer cells. Cellular Signalling. 2013;25(7):1556-1566
16. Li Y, Li W, Ying Z, Tian H, Zhu X, Li J, Li M. Metastatic heterogeneity of breast cancer cells is associated with expression of a heterogeneous TGFbeta-activating miR424-503 gene cluster. Cancer research. 2014;74(21):6107-6118
17. Yu Z, Xiao Q, Zhao L, Ren J, Bai X, Sun M, Wu H, Liu X, Song Z, Yan Y. et al. DNA methyltransferase 1/3a overexpression in sporadic breast cancer is associated with reduced expression of estrogen receptor-alpha/breast cancer susceptibility gene 1 and poor prognosis. Molecular Carcinogenesis. 2014
18. Valiente M. Binding of PTEN to Specific PDZ Domains Contributes to PTEN Protein Stability and Phosphorylation by Microtubule-associated Serine/Threonine Kinases. Journal of Biological Chemistry. 2005;280(32):28936-28943
19. Hu Y, Li Z, Guo L, Wang L, Zhang L, Cai X, Zhao H, Zha X. MAGI-2 Inhibits cell migration and proliferation via PTEN in human hepatocarcinoma cells. Archives of Biochemistry and Biophysics. 2007;467(1):1-9
20. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell research. 2009;19(2):156-172
21. Molly A. Taylor, Khalid Sossey-Alaoui, Cheryl L. Thompson, David Danielpour, Schiemann WP: TGF-β upregulates miR-181a expressionto promote breast cancer metastasis. The Journal of Clinical Investigation. 2013
22. Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY. et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Molecular biology of the cell. 2011;22(10):1686-1698
23. He H, Wang L, Zhou W, Zhang Z, Xu S, Wang D, Dong J, Tang C, Tang H, Yi X. et al. MicroRNA Expression Profiling in Clear Cell Renal Cell Carcinoma: Identification and Functional Validation of Key miRNAs. PloS one. 2015;10(5):e0125672
24. Wang J, Pei YH, Zhong Y, Jiang SS, Shao JQ, Gong JB. Altered Serum MicroRNAs as Novel Diagnostic Biomarkers for Atypical Coronary Artery Disease. PloS one. 2014:9 (9)
25. Chen G, Zhu W, Shi D, Lv L, Zhang C, Liu P, Hu W. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncology Reports. 2010:23 (4)
26. Wang Y. et al. Transforming growth factor β regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene. 2011
27. Yang C-C, Hung P-S, Wang P-W, Liu C-J, Chu T-H, Cheng H-W, Lin S-C. miR-181 as a putative biomarker for lymph-node metastasis of oral squamous cell carcinoma. J Oral Pathol Med. 2011;40:397-404
28. Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, Ghoshal K. TGFβ-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. 2009;29(12):1787-1797
29. Pons A ea. Hematopoiesis-related microRNA expression in myelodysplastic syndromes. Leuk Lymphoma. 2009;50(11):1854-1859
30. Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP. et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proceedings of the National Academy of Sciences. 2008;105(35):12885-12890
31. Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y. A Novel Multiple PDZ Domain-containing Molecule Interacting with N-Methyl-D-aspartate Receptors and Neuronal Cell Adhesion Proteins. The Journal of Biological Chemistry. 1998;273:21105-21110
32. Kitamura K, Seike M, Okano T, Matsuda K, Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K, Gemma A. MiR-134/487b/655 Cluster Regulates TGF- -Induced Epithelial-Mesenchymal Transition and Drug Resistance to Gefitinib by Targeting MAGI2 in Lung Adenocarcinoma Cells. Molecular Cancer Therapeutics. 2013;13(2):444-453
33. Gong C, Qu S, Liu B, Pan S, Jiao Y, Nie Y, Su F, Liu Q, Song E. MiR-106b expression determines the proliferation paradox of TGF-beta in breast cancer cells. Oncogene. 2015;34(1):84-93
Author contact
![]() Corresponding author: Minjie Wei; Department of Pharmacology, School of Pharmacy, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, Liaoning Province, P.R. China. Tel: +86/24/23256666-5318; Fax: +86/24/23255471. E-mail address: weiminjiecmucom
Corresponding author: Minjie Wei; Department of Pharmacology, School of Pharmacy, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, Liaoning Province, P.R. China. Tel: +86/24/23256666-5318; Fax: +86/24/23255471. E-mail address: weiminjiecmucom