Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and materials
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(4):454-465. doi:10.7150/ijbs.13379 This issue Cite
Research Paper
Alternative Roles of STAT3 and MAPK Signaling Pathways in the MMPs Activation and Progression of Lung Injury Induced by Cigarette Smoke Exposure in ACE2 Knockout Mice
Yi-Han Hung1,2,†, Wen-Yeh Hsieh 3,4,†, Jih-Sheng Hsieh1, Fon-Chang Liu1, Chin-Hung Tsai1, Li-Che Lu1,5, Chen-Yi Huang 3, Chien-Liang Wu 6, Chih-Sheng Lin 1, ![]()
1. Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan
2. Department of Medical Research, Mackay Memorial Hospital, Tamsui, Taiwan
3. Division of Chest Medicine, Department of Internal Medicine, Mackay Memorial Hospital, Hsinchu, Taiwan
4. Department of Senior Citizen Service Management , Minghsin University of Science and Technology, Hsinchu, Taiwan
5. Division of Nephrology, Department of Internal Medicine, Shin Kong Wu Ho-Su Memorial Hospital, Taiwan
6. Division of Chest Medicine, Department of Internal Medicine, Mackay Memorial Hospital, Taipei, Taiwan
† These authors contributed equally to the manuscript.
Received 2015-7-29; Accepted 2016-1-2; Published 2016-2-12
Abstract

Inflammation-mediated abnormalities in the renin-angiotensin system (RAS) and expression of matrix metalloproteinases (MMPs) are implicated in the pathogenesis of lung injury. Angiotensin converting enzyme II (ACE2), an angiotensin converting enzyme (ACE) homologue that displays antagonist effects on ACE/angiotensin II (Ang II) axis, could also play a protective role against lung diseases. However, the relationship between ACE2 and MMPs activation in lung injury is still largely unclear. The purpose of this study is to investigate whether MMPs activity could be affected by ACE2 and which ACE2 derived signaling pathways could be also involved via using a mouse model with lung injury induced by cigarette smoke (CS) exposure for 1 to 3 weeks. Wild-type (WT; C57BL/6) and ACE2 KO mice (ACE2-/-) were utilized to study CS-induced lung injury. Increases in the resting respiratory rate (RRR), pulmonary immunokines, leukocyte infiltration and bronchial hyperplasia were observed in the CS-exposed mice. Compared to WT mice, more serious physiopathological changes were found in ACE2-/- mice in the first week of CS exposure. CS exposure increased pulmonary ACE and ACE2 activities in WT mice, and significantly increased ACE in ACE2-/- mice. Furthermore, the activity of pulmonary MMPs was decreased in CS-exposed WT mice, whereas this activity was increased in ACE2-/- mice. CS exposure increased the pulmonary p-p38, p-JNK and p-ERK1/2 level in all mice. In ACE2-/- mice, a significant increase p-STAT3 signaling was detected; however, no effect was observed on the p-STAT3 level in WT mice. Our results support the hypothesis that ACE2 deficiency influences MMPs activation and STAT3 phosphorylation signaling to promote more pulmonary inflammation in the development of lung injury.
Keywords: angiotensin converting enzyme II, cigarette smoke, lung injury, matrix metalloproteinases, renin-angiotensin system
Introduction
Cigarette smoke (CS) has been used to build an animal model to study some lung diseases such as chronic obstructive pulmonary disease (COPD) [1, 2], pulmonary hypertension [3] and acute as well as chronic lung injury [4].
CS can induce inflammatory responses associated with many cytokines and chemokine, such as interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α) and tumor growth factor beta 1 (TGF-β1) [5, 6], which could participate in activations of the mitogen-activated protein kinases (MAPK) and Janus-activated kinases (JAKs) pathways [7, 8]. In addition, matrix metalloproteinases (MMPs) have also been demonstrated to be strongly associated with the lung injury [9], and could be regulated by MAKP signaling as well as signal transducer and activator of transcription 3 (STAT3) pathways [10, 11].
Angiotensin II (Ang II) combined with Ang II type I receptor (AT1R), major members in the renin-angiotensin system (RAS), can switch on several key signaling pathways, including activation of the MAPK and JAK pathways [12]. In vitro, it has been reported that STAT3 phosphorylation induced by Ang II in the renal tubular epithelial cells [13]; in vivo, angiotensin 1-7 (Ang-(1-7)) could prevent bleomycin-induced lung fibrosis and Ang II-induced lung inflammation by inhibiting the MAPK phosphorylation [14].
Abnormal RAS function has been implicated in lung injury by inducing pro-inflammatory mediators in the lung [15]. Angiotensin-converting enzyme II (ACE2), an integral component of the RAS, could degrade Ang II to a putatively protective peptide Ang-(1-7) which opposes to Ang II [16]. ACE2 is observed in the heart, kidney and lungs [17]. The actions of ACE2 appear in a variety of lung disease, including pulmonary hypertension, pulmonary fibrosis, and acute respiratory distress syndrome (ARDS) [18]. Previous studies have also demonstrated that pulmonary ACE2 could not only regulate the balance of Ang II and Ang-(1-7) [19], but also be a probable therapeutic target for pulmonary hypertension [20]. Moreover, ACE2 deficiency could exhibit more serious symptoms in a mouse model of ARDS [21]. In our previous report, we also found that ACE2 might participate in the activation of specific MMPs in fibrotic processes in vitro and in vivo [22].
In light of the above findings, we hypothesized that ACE2 might play a critical role in the pathogenesis of lung injury. Therefore, we designed a CS-induced lung injury mouse model using ACE knockout (KO) mice to investigate the molecular mechanism of ACE2 in the MMPs and the regulation of related signaling pathways. To test this hypothesis, ACE2 KO and wild-type (WT) mice were exposed to CS for 1 to 3 weeks, and the physiological parameters and activities of ACE, ACE2 as well as MMPs were measured in these mice. Furthermore, we determined the phosphorylation of p38 (p-p38), JNK (p-JNK), ERK1/2 (p-ERK1/2) and STAT3 (p-STAT3) in order to reveal the potential mechanisms underlying the protection of ACE2 against lung injury induced by CS exposure.
Methods and materials
Mouse model of lung injury induced by CS exposure
WT (C57BL/6; female) and homozygous ACE2 KO (ACE2-/- ; female) mice 6 to 8 weeks of age were used in the study. WT mice purchased from National Laboratory Animal Center (NLAC) in Taiwan. The first generation of ACE2 KO (B6; 129S5-Ace2tm1 Lex/Mmcd) mice were obtained from the Mutant Mouse Regional Resource Centers (MMRRC) and then bred in NLAC. The experimental protocol conformed to the Guide for the Care and Use of Laboratory Animals published by National Institutes of Health (NIH Publication No. 85-23, revised 1996) and was approved by the animal welfare committees of National Chiao Tung University. In each experiment, 8 mice were simultaneously exposed to CS (4 times /day and 7 days /week, for 1 to 3 weeks) using a smoke-exposure system. Each exposure lasted 30 minutes. Body weight and resting respiratory rate (RRR) were measured every week.
The brand of cigarette used in this study is “New Paradise”, a local product of Taiwan, including 0.8 mg nicotine, 10 mg tar and 12.5 mg CO per one cigarette, and around 10-20 mg total particulate matter (TPM) in the cigarette smoke per one cigarette. To simultaneously apply multiple mice of the same group in one time treatment, we set up the smoke-exposure system constructed by a transparent box, a tubular gas channel, 8 porous bottles, a shelf, a cigarette holder and one fan. This box size is 37.5 x 23.8 x 16 cm (length x width x height).
Measurement of respiratory rate
Lung respiratory rate (BPM, breaths per minute) was measured as described by a previous study with slight modifications [23]. The respiratory rate was measured using whole body unrestrained chambers (Emka, Middletown, PA, USA) on live mice exercised for 10 minutes on week 0, 1, 2 and 3 after CS exposure. Data was collected and analyzed using the BIOPAC student lab systems software package (Biopac system, Goleta, CA, USA).
Enzyme-linked immunosorbent assay (ELISA)
Tissue protein was analysed for TGF-β1, TNF-α and IL-6 using a sandwich ELISA purchased from eBioscience (San Diego, CA, USA). Tissue protein was incubated in ELISA plates in which the wells had been coated with primary antibodies. Following the addition of biotinylated antibodies, the plates were washed and reacted with horseradish peroxidase (HRP)-conjugated streptavidin. Tetramethylbenzidine (TMB) one-step substrate was used to detect the targeted protein and the samples were measured at 450 nm using a micro-plate reader.
Western blotting
Western blot analysis was performed as previously reported [24]. The lung tissue homogenates containing 20 μg of protein were electrophoresed on SDS-PAGE gels and then transferred onto polyvinylidene fluoride membranes. Primary antibodies against p-p38, p-JNK, p-STAT3, p-ERK1/2 and β-actin were purchased from Cell Signaling Technology (Beverly, MA, USA). Chemiluminescence substrates were visualized using enhanced chemiluminescence detection (Immobilon Western Chemiluminescent HRP Substrate; Millipore, Billerica, MA, USA) and exposing the membranes to Lumi-Film Chemiluminescent Detection Film (Roche, Indianapolis, IN, USA). The bands on the images were detected at the anticipated location based on size. The band intensity was quantified using Scion Image software (Scion, Frederick, MD, USA). The amounts of p-p38, p-JNK, p-ERK1/2 and p-STAT3 were expressed relative to the amount of β-actin (as the internal standard) in each sample, respectively.
ACE and ACE2 activity assay
ACE and ACE2 activities were assayed using the fluorogenic substrates Mca-YVADAPK and Mca-APK-Dnp (AnaSpec, San Jose, CA, USA), respectively, according to the report by Vickers et al. [25]. The assay was performed in a microquartz cuvette with 20 μL of lung tissue proteins and 2 μL of the fluorogenic substrates (stock concentration: 4 mM ACE substrate/1.5 mM ACE2 substrate) in ACE or ACE2 assay buffer. The reaction was followed kinetically for 1 hour using a fluorescence reader at 330 nm/390 nm. All samples were fitted and plotted using Grafit v. 4.0 (Sigma-Aldrich, St. Louis, MO, USA), and enzyme activity was expressed as RFU/hour/mL. The samples were incubated with the above mentioned reaction mixture in the presence of 1 μM captopril (Sigma-Aldrich; a specific ACE inhibitor) or 1 μM DX600 (AnaSpec; a specific ACE2 inhibitor).
Gelatin zymography assay
MMP-2 and MMP-9 activities were detected by gelatin zymography using gelatin-containing gels according to the protocol reported by Chen et al. [26]. The protein was mixed with the zymography sample buffer and incubated for 10 minutes at room temperature, and then loaded into SDS-PAGE containing 0.1% (w/v) gelatin (Sigma-Aldrich). After electrophoresis, the gel was washed twice for 30 minutes in zymography renaturing buffer to remove SDS, and then incubated in reaction buffer at 37°C for 18 hours. The gels were then stained with Coomassie blue for 30 minutes and destained with destain buffer for 12 to 16 hours. The presence of enzyme activity was evident by clear or unstained zones, indicating the action of the enzyme on the gelatin substrate. Gelatinase activities in the gel slabs were quantified by Scion Image software (NIH, Bethesda, MD, USA), which quantifies the area of bands hydrolyzed by gelatinase. A recombined MMP-2 or MMP-9 positive controls (Chemicon, Temecula, CA, USA) was contained in each gel as a standard intensity value to normalize sample intensity and express in arbitrary units.
In situ zymography
Localization of gelatinase activities in lung tissue sections was performed by in situ zymography as previously described [26, 27]. In the assay, frozen tissue sections (6 to 10 μm) were mounted onto slides with 0.05 mg/ml DQ-gelatin (for the assay of gelatinase activity) in 1% PBS containing 0.5 μg/ml propidium iodide (PI; for DNA staining) to counterstain the cell nuclei. Fresh tissues were embedded in O.C.T. (Thermo, Waltham, MA, USA), immediately frozen on dry ice, and stored at -80°C. The substrate gel solution (20 μL) was pipetted onto one slide and the zymographic reaction was carried out at 37°C overnight. Gelatinase activity resulted in the loss of quenching and was visualized by a fluorescent signal under a Leica SP5X confocal laser scanning microscope (Leica, Wetzlar, Germany).
Histological determination
The organ samples were isolated from WT and ACE2 KO mice and a part of right lung were excised and encased in 10% formaldehyde prepared for the hematoxylin-eosin (H&E) staining. The stained sections were photographed using a digital camera mounted on a microscope. Manual planimetry was performed on the microscope using PALM RoboSoftware v2.2 using H&E-stained slices. A computerized microscope equipped with a high-resolution video camera (BX 51; Olympus, Tokyo, Japan) was used for morphometric analysis. The thickness of airway epithelium was calculated by measuring the difference between the area encompassing epithelial cell basement and lumen. This expressed as the epithelial area.
Statistical analysis
All of the values are expressed as the mean±standard deviation (SD). Student's t-test was used for comparisons involving two groups and the data were compared with one-way analysis of variance (ANOVA) test to evaluate differences among multiple groups. A value of p<0.05 was considered statistically significant.
Results
CS exposure induces the features of lung injury in mice
WT and ACE2-/- mice were continuously exposed to CS for 3 weeks and the body weight and RRR were monitored each week. The average initial body weight of the WT mice and ACE2-/- mice was 18.9±0.9 g and 19.7±0.7 g, respectively. After CS exposure for 1 week, WT mice markedly lost the body weight to 17.2±0.6 g and ACE2-/- mice lost the body weight to 18.4±0.8 g. Then, the body weights increased slightly in the 2- and 3-week of CS exposure; however, the mice did not return to their initial body weight. In contrast, the air-exposed WT and ACE2-/- mice steadily gained weight during the experimental period (Figure 1A and 1B). The average RRR of WT and ACE2-/- mice was approximately 300 bpm before the experimental treatment, but the RRR of the mice exposed to CS for 1 to 2 weeks was significantly increased. The increasing RRR induced by CS exposure was more significantly in ACE2 KO mice compared to the WT mice (Figure 1C). In WT mice, RRR increased to approximate 400 BPM progressively by CS exposure for 3 weeks. By comparison, RRR of ACE2-/- mice exposed with CS for 3 weeks was significantly increased to approximate 550 BPM.
Hallmark features of lung injury induced by CS exposure in the mouse model. WT (C57BL/6, n=8 for each group) mice and ACE2 KO mice (homozygous ACE2-/-, n=8 for each group) were exposed to CS or air for 3 weeks. (A) and (B) The changes of body weight of the WT and ACE2-/- mice exposed with CS or air, respectively. † p<0.05 and ‡ p<0.01 compared with the 0 week; ** p<0.01 compared with the mice after 3 weeks of CS exposure. (C) The measurements and comparisons of the resting respiration rate (RRR) of the mice exposed to CS for 3 weeks. The animals were sacrificed to isolate the lung tissues for (D) TGF-β1, (E) TNF-α and (F) IL-6 determination using an ELISA assay. All of the values are expressed as the mean±SD from each group; † p<0.05 and ‡ p<0.01 compared with the control (i.e., 0 week); * p<0.05 and ** p<0.01 compared with the WT mice at the same CS-exposed week.
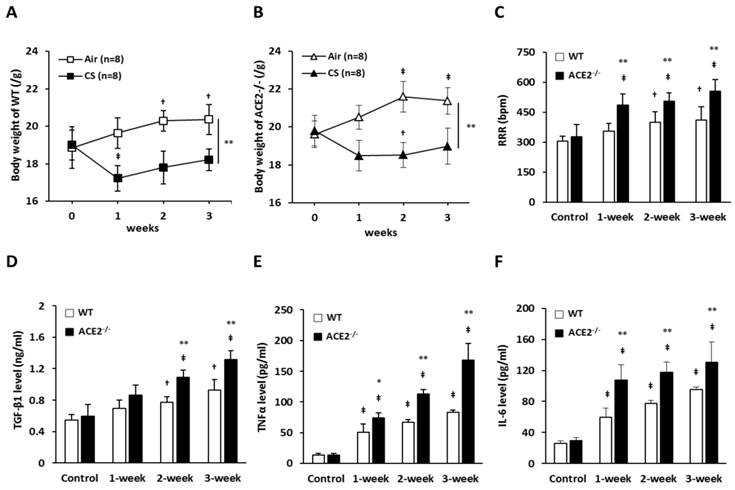
Upon analysing the expression of immunokines, we confirmed that CS exposure could induce pulmonary inflammation in the mice. The levels of pulmonary TGF-β1 (Figure 1D), TNF-α (Figure 1E) and IL-6 (Figure 1F) in the WT mice exposed to CS for 3 weeks was approximately 2, 3 and 4-folds higher than in the control mice, respectively. In the ACE2-/- mice, the increasing levels of pulmonary TGF-β1, TNF-α and IL-6 were significantly higher than in the WT mice. For example, the levels of TNF-α in the ACE2-/- mice exposed to CS for 3 weeks was 167.9±26.9 pg/ml that was almost twice as much as the level in WT mice (82.7±3.7 pg/ml).
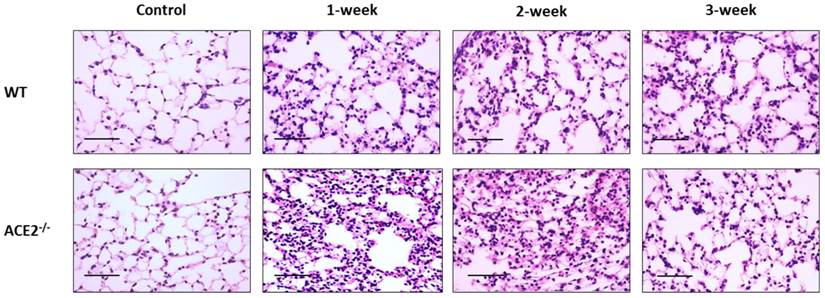
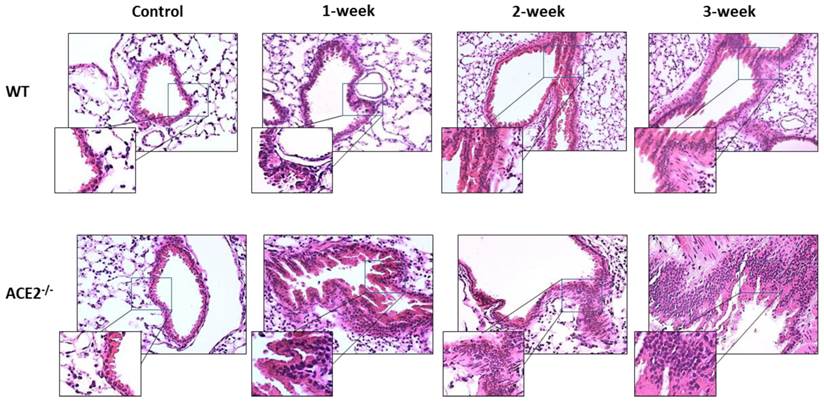
CS exposure also induced histological changes in the lungs, especially in the ACE2-/- mice. The WT and ACE2-/- mice were exposed to CS for 0 (as control), 1, 2 and 3 weeks, and then the animals were sacrificed to isolate the lung tissues for pathological examination. The lung alveolar and bronchiole sections of CS-exposed WT and ACE2-/- mice were stained by H&E and the results showed that CS exposure promotes white blood cell (WBC) infiltration around the alveolus (Figure 2) and the airway epithelial thickening (Figure 3). Moreover, the airway epithelial thickening yielded earlier and the infiltration was more severe in the CS-exposed ACE2-/- mice compared to the WT mice (Figure 2 and 3).
Alveolar infiltration of white blood cells yielded in the experimental mice with CS-induced lung injury. The lung alveolar sections of CS-exposed WT and ACE2-/- mice were stained with haematoxylin-eosin (H&E). The non-CS treated mice were defined as the control group. Relative to the control mice, there were increases in the infiltration of white blood cells around the alveolus in the CS-exposed WT and ACE2-/- mice. (Scale=50 μm)
Changes of airway epithelial tissue yielded in the experimental mice with CS-induced lung injury. The lung bronchiole sections of CS-exposed WT and ACE2-/- mice were stained with haematoxylin-eosin (H&E). The non-CS treated mice were defined as the control group. Relative to the control mice, there were increases in the infiltration of white blood cells around the airway and airway epithelial thickening in the CS-exposed WT and ACE2-/- mice.
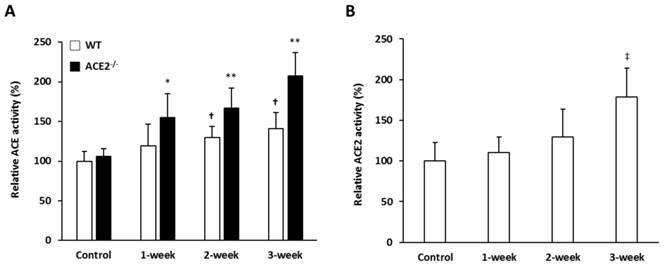
ACE and ACE2 activities in the lungs of mice are induced by CS exposure
The activities of pulmonary ACE and ACE2 in the mice exposed to CS for 0, 1, 2 and 3 weeks were investigated to test the hypothesis that RAS mediators could participate in the pathogenic induction of lung injury. The pulmonary ACE activity of WT and ACE2-/- mice increased significantly. After CS exposure for 3 weeks, the level of pulmonary ACE activity in the WT and ACE2-/- mice increased by 1.4 and 1.9-fold compared to the controls, respectively. Notably, the ACE activity induced by CS exposure in the lungs of ACE2-/- mice was significantly higher than in the WT mice (Figure 4A). Relative ACE2 activity in the lungs of WT mice was significantly upregulated to approximately 1.8-fold after 3 weeks of CS exposure (Figure 4B).
The pulmonary ACE and ACE2 activities were induced by CS exposure. WT mice and ACE2-/- mice were exposed to CS for 0 (Control), 1, 2 and 3 weeks, and then the animals were sacrificed to isolate lung tissues for ACE and ACE2 activity assays. The values of ACE and ACE2 activity in the control group were calculated as 100%. (A) The pulmonary ACE activities in WT and ACE2-/- mice were progressively increased with CS exposure. (B) The relative ACE2 activity in WT mice was increased upon CS exposure. All of the values are expressed as the mean±SD from each group (n=8 for each group); † p<0.05 and ‡ p<0.01 compared with the control group in WT mice; * p<0.05 and ** p<0.01 compared with the control group in ACE2-/- mice.
Gelatinase activities are induced in the lungs of CS-exposed ACE2 KO mice
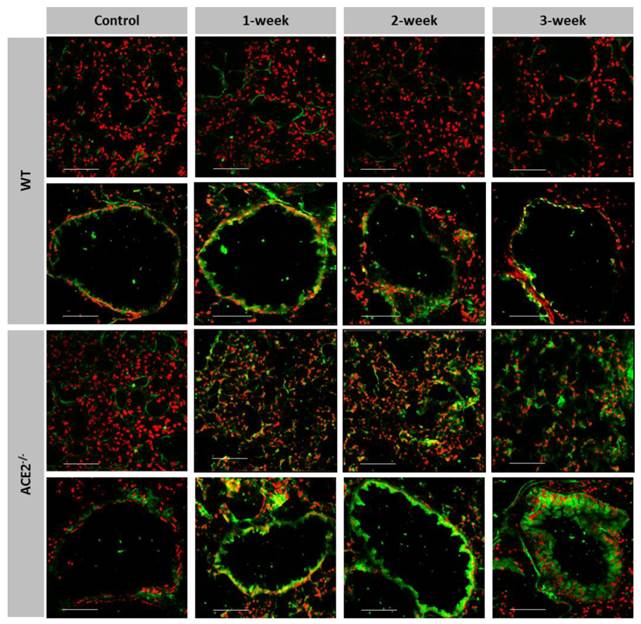
We detected the gelatinase activities, including MMP-2 and MMP-9, in the lung sections using an in situ zymography assay to locate and compare the pulmonary gelatinase activity between the WT and ACE2 KO mice. The gelatinase activity was mainly expressed near the alveolus and bronchioles (Figure 5). In the WT mice, the gelatinase activity showed little increase after espousing to CS for 1 week, but it was slightly reduced in the bronchioles after 3-week CS exposure. In contrast, the gelatinase activity near the alveolus and bronchioles was continuously and markedly increased in the ACE2-/- mice exposed to CS throughout the 3 weeks treatment period.
More gelatinase activity expressed in the pulmonary alveolus and bronchioles of CS-exposed ACE2 KO mice. The location and gelatinase activity in the lungs of WT and ACE2-/- mice exposed to CS for 1 to 3 weeks were detected using in situ zymography. The FITC signal (green) is released upon cleavage of the gelatin substrate and the nuclear signal (red) is stained. Gelatinase activity around the alveolus was significantly increased in the CS exposed ACE2-/- mice, but not in the CS-exposed WT mice. Bronchiolar gelatinase activity was significantly increased in the CS-exposed mice, especially in the ACE2-/- mice. Images were captured at low magnification (Scale=100 μm).
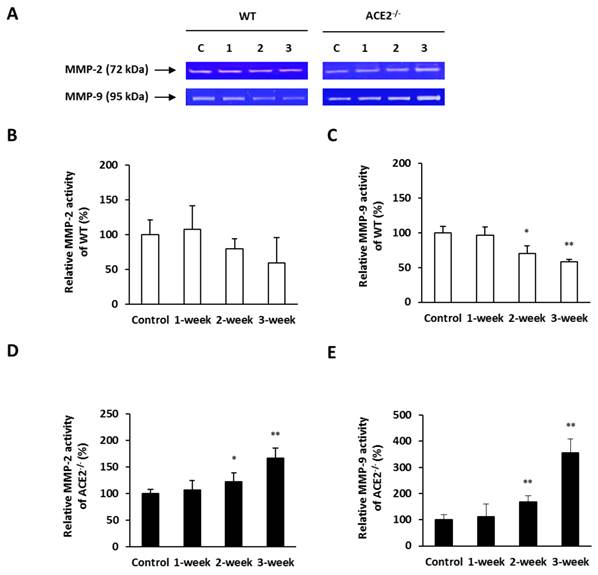
To provide further evidence that ACE2 could affect the gelatinase activity in the lungs, we assessed the MMP-2 and MMP-9 activities in the lung homogenate using a gelatin zymography assay (Figure 6A). The pulmonary MMP-2 activity in the WT mice progressively decreased with increasing CS exposure time, although the decrease did not reach statistical significance (Figure 6B). The pulmonary MMP-9 activity in the WT mice was decreased progressively with the CS exposure treatment time and the decrease was statistically significant after 2 weeks treatment (Figure 6C). On the other hand, the MMP-2 and MMP-9 activities in the lungs of CS-exposed ACE2-/- mice were proportional increase with the time of exposure. In these mice, the pulmonary MMP-2 and MMP-9 activity by the CS-exposed treatment for 3 weeks was significantly enhanced by approximately 1.5-fold and 3-fold compared to the controls, respectively (Figure 6D and 6E).
Pulmonary MMP-2 and MMP-9 activities elevated in CS-exposed ACE2 KO mice. The WT and ACE2-/- mice were exposed to CS for 1 to 3 weeks and sacrificed for the assays. (A) The activities of MMP-2 and MMP-9 in the lungs of WT and ACE2-/- mice were determined using a gelatin zymography assay. The value of MMP-2 and MMP-9 activities in the control mice was calculated as 100%. (B) CS exposure did not lead to a change in pulmonary MMP-2 activity, but (C) decreased pulmonary MMP-9 activity in the WT mice. (D) MMP-2 and (E) MMP-9 activities in the lungs of ACE2-/- mice were significantly upregulated upon CS exposure. All of the values are expressed as the mean±SD from each group (n=8); * p<0.05 and ** p<0.01 compared with the control group.
Loss of ACE2 increases pulmonary p-STAT3 expression in CS-induced lung injury
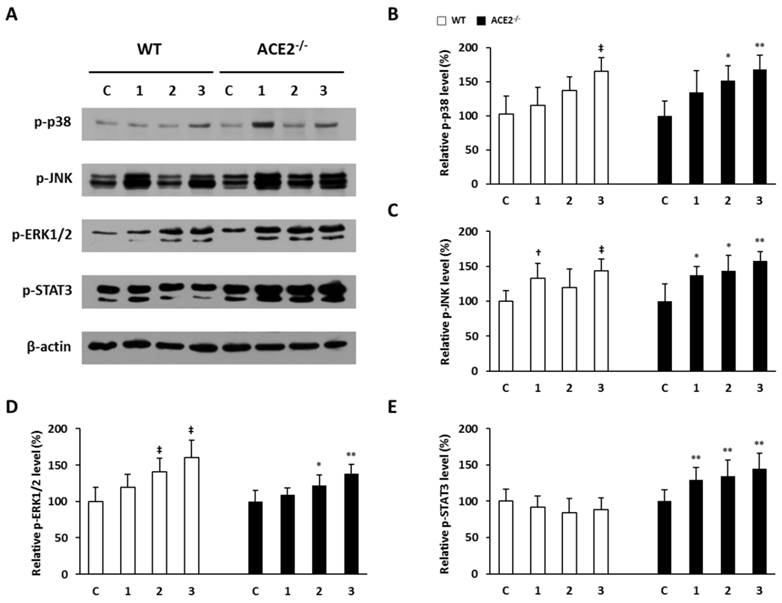
In the mice with lung injury induced by CS exposure, we found that the pulmonary immunokines, TGF-β1, TNF-α and IL-6, were increased upon CS exposure in the ACE2-/- mice. The CS-induced inflammatory response is associated with these immunokines that participate in the activation of the MAPK and JAK-STAT3 signaling pathways. To further examine whether the existence of ACE2 could also affect the mediator of MAPK and JAK-STAT3 signaling pathways in the injured lungs, we investigated the levels of p38, JNK, ERK1/2 and STAT3 phosphorylation (i.e., p-p38, p-JNK, p-ERK1/2 and p-STAT3) in the lung tissues isolated from mice via western blotting (Figure 7A). The experimental results show that CS exposure led to a significant increase in pulmonary p-p38, p-JNK and p-ERK1/2 (Figure 7B, 7C and 7D) in both of WT and ACE2 KO mice. However, the induced p-STAT3 by CS exposure was observed in the ACE2 KO mice, but not in the WT mice (Figure 7E). Taken together, the data suggest that ACE2 can affect STAT3 phosphorylation in mice upon CS-induced lung injury.
Pulmonary p-p38, p-JNK, p-ERK1/2 and p-STAT3 elevated in CS-exposed ACE2 KO mice. The WT and ACE2-/- mice were exposed to CS for 1 to 3 weeks and sacrificed for the assays. (A) The level of p-p38, p-JNK, p-ERK1/2 and p-STAT3 in the lungs was determined by immunoblotting. β-actin was used as the internal controls for p-p38, p-JNK, p-ERK1/2 and p-STAT3, respectively. The levels of p-p38, p-JNK, p-ERK1/2 and p-STAT3 expression in the non-CS exposed mice were calculated as 100% and defined as the control. The relative pulmonary (B) p-p38, (C) p-JNK, (D) p-ERK1/2 expression of the WT mice was increased upon CS exposure. (E) The relative p-STAT3 expression of WT mice showed no significant difference after CS exposure, but p-STAT3 expression was induced significantly by CS exposure in the lungs of ACE2-/- mice. All of the values are expressed as the mean±SD from each group; † p<0.05 and ‡ p<0.01 compared with the control group in WT mice; * p<0.05 and ** p<0.01 compared with the control group in ACE2-/- mice.
Discussion
In the present study, we demonstrated CS-induced lung injury in ACE2 KO mice using physiological, biochemical and pathological examinations. Our results show that induction of inflammation and lung damage (Figure 1, 2 and 3), increase in pulmonary ACE and ACE2 (Figure 4), decrease in MMP-2 and MMP-9 activities (Figure 5, 6B and 6C), and p-STAT3 level did not be enhanced (Figure 7C) in the WT mice exposed to CS for 1 to 3 weeks. In the ACE2 KO mice, more serious responses in inflammation, lung damage (Figure 1, 2 and 3) and ACE increase (Figure 4) compared to WT mice were observed. Pulmonary MMP-2 and MMP-9 activities were increased (Figure 5, 6D and 6E) and pulmonary p-p38, p-JNK, p-ERK1/2 and p-STAT3 levels were also increased (Figure 7) in the CS-exposed ACE2-/- mice. According to the results, we proposed that ACE2 deficiency could accelerate the development of lung injury via the influences on STAT3 activation and the modulation of MMPs activity.
Some studies implicated the RAS dysregulation could cause lung injury [21, 28] and RAS blockade, including ACE inhibitors (ACEIs) and angiotensin receptor blockers (ARBs), could exhibit beneficial effects in the treatments for lung diseases [29, 30]. However, these RAS blockers have been reported to induce some side effects; for example, both ACEIs and ARBs can cause cough, dizziness and headache [31, 32]. The ACE/ Ang II axis in RAS is associated with the development of several pulmonary diseases; however, much less is known about what mechanisms affected by ACE2 in the development of lung injury. ACE2, an ACE homologue, appears to be a negative regulator of the RAS and counterbalances the function of ACE. ACE2 reduces the generation of Ang II by catalyzing the conversion of angiotensin I (Ang I) to angiotensin 1-9 (Ang-(1-9)) and facilitating hydrolysis of Ang II to Ang-(1-7). Ang-(1-7) has been recognized as a potential endogenous inhibitor of the classical RAS cascade [33]. Hence, the ACE2/Ang-(1-7) axis may be an important negative modulator of Ang II bioactivity, counteracting the effects of ACE in determining Ang II levels.
Previous ARDS and SARS studies have shown that ACE2 protected murine lungs from severe acute injury [21]. Accumulating evidence has shown that ACE2 is important to lung endothelial function and pathophysiology; for instance, the level of pulmonary ACE2 decreased significantly in patients with idiopathic pulmonary fibrosis and bleomycin-induced pulmonary fibrosis was associated with ACE2 activity [34]. Additionally, some studies have reported that ACE2 was a cardiopulmonary protector because Ang II upregulated ACE and downregulated ACE2 in patients with hypertension [35] as well as higher cardiac ACE2 activity with increased Ang-(1-7) existed in patients with pulmonary hypertension [36]. These findings led us to hypothesize that ACE2 could have a protective effect against lung injury.
To study the role of ACE2 in lung injury, we utilized a mouse model of CS-induced lung injury. Previous studies have noted that TGF-β1 [37], TNF-α [38] and IL-6 [5] are associated with various pulmonary diseases, including asthma, chronic bronchitis, acute lung injury, COPD and ARDS. In our results, TGF-β1, TNF-α and IL-6 levels in the lungs of ACE2 KO mice were higher than in WT mice after CS exposure (Figure 1). Moreover, we observed that the pulmonary inflammation and pathogenic lesions in ACE2 KO mice were developed faster and were more serious than in WT mice (Figure 2 and 3). From the perspective of these pathological changes, the progression of lung injury in the ACE2 KO mice was more rapid than WT mice, demonstrating that the existence of ACE2 may play an important role to alleviate the damage from CS exposure.
Our study show that CS exposure increased pulmonary ACE and ACE2 activity and the lack of ACE2 could induce more pulmonary ACE activity in CS-exposed mice (Figure 4). In the part of CS-induced ACE is confirmed by the finding as previous study in a rat study [39]. Controversially, the ACE2 activity was decreased in the lungs of the rat treated with CS exposure for 6 months [39]. Although we did not have plasma or bronchoalveolar lavage fluid (BALF) to detect the levels of Ang II and Ang-(1-7) in mice in this study, our further study will do this part experiment to investigate whether activation of ACE2 is affected by other RAS module, e.g., Ang II, Ang-(1-7) or hypoxia during the CS exposure [40], in the mouse model of CS-induced lung injury.
MMPs, especially MMP-2 and MMP-9, have been implicated in pulmonary inflammation and diseases, including asthma, emphysema, pulmonary hypertension and lung injury [41, 42]. MMP modulation could also be a potential therapeutic strategy against lung injury [41]. Recent studies have shown that MMP-2 and MMP-9 expression can be regulated by ACE2 [20, 43]. This regulation could be responsible for an anti-inflammation effect in the lungs during CS-induced injury. We also found that CS exposure induces more gelatinase activity around the alveolus and bronchioles in the ACE2 KO mice than WT mice (Figure 5). Consistently, pulmonary MMP-2 and MMP-9 increased markedly in smoke-exposed ACE2 KO mice (Figure 6). Previous studies have reported that ACE2 could upregulate the gelatinase activation [20, 43]; nevertheless, some research has also pointed out that markedly increased MMP-2 and MMP-9 in vascular inflammation and atherosclerosis in ACE2-deficient hearts [44, 45]. The latter study is consistent with our data that absence of ACE2 could also promote the activation of MMPs in the lungs in our animal model. Inflammatory responses were induced by the abnormal activation of Ang II/AT1R axis in RAS [46] and ACE2 could degrade Ang II into Ang (1-7) to bind with Mas receptor (i.e., Ang (1-7)/Mas axis) to restrain the inflammation. Therefore, we proposed the existence of ACE2 could downregulate the gelatinase activity in a normally physiologic condition; on the contrary, we inferred that overactive Ang II/AT1R axis could consistently induce more inflammatory responses and increase activity of MMP-2 and/or MMP-9 in the ACE2 deficient mice.
Many studies have shown that the RAS takes part in the pathogenesis of many inflammatory diseases [15-20]. Increased ACE2 lessened pro-inflammatory cytokines such as TNF-α, IL-6 and TGF-β1 and elevated some anti-inflammatory cytokine [36, 47-48]. In addition, these pro-inflammatory factors were associated with MAPK pathways [49]. Previous studies have also reported that the levels of phosphorylated ERK1/2 and STAT3 were significantly increased in the hearts of Ang II-treated mice [50]. Other researchers have also proved that CS can induce p38 [51] and JNK [52] activation in experimental mice. However, the downstream signaling pathways regulated by ACE2 during lung injury are rarely reported. Therefore, it is worthy to note that the MAPK signaling p38, JNK and ERK1/2 and STAT3 phosphorylation in the lung tissue of ACE2 KO mice with CS-induced injury. Moreover, the MMPs activation participates in many biochemical responses, and many transcription factors, such as STAT3, are activated during the process [53]. MMPs could be activated indirectly via MAPK signal pathways [54]. In our study, we also provided the experimental evidence to support the CS-induced lung injury being associated with MAKP pathway. We found that increased p-ERK1/2 in the WT and ACE2 KO mice upon CS exposure. Additionally, the levels of p-p38 and p-JNK in both WT and ACE2 KO mice were also increased by CS exposure. This result is consistent with previous reports that CS exposure could enhance p38, ERK1/2 and JNK phosphorylation to regulate key inflammatory, proteolytic and apoptotic responses in the lungs [11, 55-57]. Interestingly, CS exposure induced greater p-STAT3 expression in the ACE2 KO mice, but the p-STAT3 expression had no significant changes in the CS-exposed WT mice (Figure 7). According to our results, p-p38, p-JNK and p-ERK1/2 appeared to be insignificantly affected by the presence of ACE2. By contrast, the relationship between ACE2 and STAT3 was unexpectedly close. Taken together, these evidences strongly suggest that MMPs activation could be associated with ACE2 and p-STAT3 expressions in the early stages of lung injury.
Conclusions
The present study is the first time to use CS-induced lung injury mouse model to demonstrate that ACE2 deficiency not only promoted CS-induced pulmonary inflammation and gelatinase activity but also caused STAT3 activation. However, we also found that there are no different between WT and ACE2 KO mice on activating p38, JNK, ERK1/2 from the beginning of CS-induced lung injury. We speculate that ACE2 maybe have certain capability to protect lung from CS-induced injury, but further studies are necessary to substantiate this conclusion. Moreover, the changes of circulating or pulmonary Ang II and Ang-(1-7), and the effects of both peptides reperfusion on CS-induced lung injury remain to be further study.
Abbreviations
ACE2: Angiotensin converting enzyme II; Ang II: Angiotensin II; Ang-(1-7): Angiotensin 1-7; Ang-(1-9): Angiotensin 1-9; ARDS: Acute respiratory distress syndrome; AT1R: Ang II type I receptor; ACEIs: ACE inhibitors; ARBs: Angiotensin receptor blockers; COPD: Chronic obstructive pulmonary disease; CS: Cigarette smoke;ERK1/2: Extracellular signal-regulated kinases 1 and 2; JAKs: Janus-activated kinases; KO: Knockout; IL-6: Interleukin-6; MAPKs: Mitogen-activated protein kinases; MMP-2: Matrix metalloproteinase 2; MMP-9: Matrix metalloproteinase 9; MMPs: Matrix metalloproteinases; p-ERK1/2: Phosphorylated ERK1/2; p-STAT3: Phosphorylated STAT3; RAS: Renin-angiotensin system; RRR: Resting respiratory rate; STAT3: Signal transducer and activator of transcription 3; TGF-β1: Transforming growth factor beta 1; TNF-α: Tumour necrosis factor alpha; WT: Wild-type.
Acknowledgements
This work was supported by the grants of MOST 104-2313-B-009 -001-MY3 from the Ministry of Science and Technology (MOST), Taiwan. This work is partly supported by "Aiming for the Top University Program" of the National Chiao Tung University and Ministry of Education and Mackay Memorial Hospital (MMH-CT-10503) grants. The authors thank the core facility of Multiphoton and Confocal Microscope System (MCMS) in College of Biological science and Technology, National Chiao Tung University, Hsinchu, Taiwan)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rubenfeld GD, Caldwell E, Peabody E. et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685-93
2. Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1-15
3. Wright JL, Zhou S, Churg A. Pulmonary hypertension and vascular oxidative damage in cigarette smoke exposed eNOS(-/-) mice and human smokers. Inhal Toxicol. 2012;24:732-40
4. Calfee CS, Matthay MA, Eisner MD. et al. Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am J Respir Crit Care Med. 2011;183:1660-5
5. Rincon M, Irvin CG. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci. 2012;8:1281-90
6. Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNF alpha in pulmonary pathophysiology. Respir Res. 2006;7:125
7. Schuh K, Pahl A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochem Pharmacol. 2009;77:1827-34
8. Qu P, Roberts J, Li Y. et al. Stat3 downstream genes serve as biomarkers in human lung carcinomas and chronic obstructive pulmonary disease. Lung Cancer. 2009;63:341-47
9. Davey A, McAuley DF, O'Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J. 2011;38:959-70
10. Pullamsetti S, Krick S, Yilmaz H. et al. Inhaled tolafentrine reverses pulmonary vascular remodeling via inhibition of smooth muscle cell migration. Respir Res. 2005;6:128
11. Geraghty P, Wyman AE, Garcia-Arcos I. et al. STAT3 modulates cigarette smoke-induced inflammation and protease expression. Front Physiol. 2013;4:267
12. Kemp JR, Unal H, Desnoyer R. et al. Angiotensin II-regulated microRNA 483-3p directly targets multiple components of the renin-angiotensin system. J Mol Cell Cardiol. 2014;75:25-39
13. Ni J, Shen Y, Wang Z. et al. P300-dependent STAT3 acetylation is necessary for angiotensin II-induced pro-fibrotic responses in renal tubular epithelial cells. Acta Pharmacol Sin. 2014;35:1157-66
14. Meng Y, Yu CH, Li W. et al. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway. Am J Respir Cell Mol Biol. 2014;50:723-36
15. Marshall RP. The pulmonary renin-angiotensin system. Curr Pharm Des. 2003;9:715-22
16. Wang K, Hu X, Du C. et al. Angiotensin-(1-7) suppresses the number and function of the circulating fibrocytes by upregulating endothelial nitric oxide synthase expression. Mol Cell Biochem. 2012;365:19-27
17. Tikellis C, Thomas MC. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept. 2012;2012:256294
18. Kuba K, Imai Y, Penninger JM. Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol. 2006;6:271-6
19. Brosnihan KB, Neves LA, Chappell MC. Does the angiotensin-converting enzyme (ACE)/ACE2 balance contribute to the fate of angiotensin peptides in programmed hypertension? Hypertension. 2005;46:1097-99
20. Shenoy V, Qi Y, Katovich MJ. et al. ACE2, a promising therapeutic target for pulmonary hypertension. Curr Opin Pharmacol. 2011;11:150-55
21. Imai Y, Kuba K, Rao S. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112-16
22. Pan CH, Wen CH, Lin CS. Interplay of angiotensin II and angiotensin 1-7 in the regulations of matrix metalloproteinases of human cardiocytes. Exp Physiol. 2008;93:599-612
23. Kulkarni AA, Thatcher TH, Hsiao HM. et al. The triterpenoid CDDO-Me inhibits bleomycin-induced lung inflammation and fibrosis. PLoS One. 2013;8:e63798
24. Kuan TC, Yang TH, Wen CH. et al. Identifying the regulatory element for human angiotensin-converting enzyme 2 (ACE2) expression in human cardiofibroblasts. Peptides. 2011;32:1832-39
25. Vickers C, Hales P, Kaushik V. et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838-43
26. Chen CL, Huang SK, Lin JL. et al. Upregulation of matrix metalloproteinase-9 and tissue inhibitors of metalloproteinases in rapid atrial pacing-induced atrial fibrillation. J Mol Cell Cardiol. 2008;45:742-53
27. Yan SJ, Blomme EA. In situ zymography: a molecular pathology technique to localize endogenous protease activity in tissue sections. Vet Pathol. 2003;40:227-36
28. Chen LN, Yang XH, Nissen DH. et al. Dysregulated renin-angiotensin system contributes to acute lung injury caused by hind-limb ischemia-reperfusion in mice. Shock. 2013;40:420-9
29. Ahad A, Al-Mohizea AM, Al-Jenoobi FI. et al. Transdermal delivery of angiotensin II receptor blockers (ARBs), angiotensin-converting enzyme inhibitors (ACEIs) and others for management of hypertension. Drug Deliv. 2014:1-12
30. Barreras A, Gurk-Turner C. Angiotensin II receptor blockers. Proc (Bayl Univ Med Cent). 2003;16:123-26
31. Dicpinigaitis PV. Angiotensin-converting enzyme inhibitor-induced cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129(Suppl 1):S169-73
32. Dykewicz MS. Cough and angioedema from angiotensin-converting enzyme inhibitors: new insights into mechanisms and management. Curr Opin Allergy Clin Immunol. 2004;4:267-70
33. Mercure C, Yogi A, Callera GE. et al. Angiotensin (1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ Res. 2008;103:1319-26
34. Li X, Molina-Molina M, Abdul-Hafez A. et al. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L178-85
35. Koka V, Huang XR, Chung AC. et al. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol. 2008;172:1174-83
36. Ferreira AJ, Shenoy V, Yamazato Y. et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:1048-54
37. Königshoff M, Kneidinger N, Eickelberg O. TGF-beta signaling in COPD: deciphering genetic and cellular susceptibilities for future therapeutic regimen. Swiss Med Wkly. 2009;139:554-63
38. Churg A, Wang RD, Tai H. et al. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170:492-98
39. Han SX, He GM, Wang T. et al. Losartan attenuates chronic cigarette smoke exposure-induced pulmonary arterial hypertension in rats: possible involvement of angiotensin-converting enzyme-2. Toxicol Appl Pharmacol. 2010;245:100-7
40. Zhang R, Wu Y, Zhao M. et al. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L631-40
41. Davey A, McAuley DF, O'Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J. 2011;38:959-70
42. Vandenbroucke RE, Dejonckheere E, Libert C. A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur Respir J. 2011;38:1200-14
43. Kuan TC, Chen MY, Liao YC. et al. Angiotensin II downregulates ACE2-mediated enhancement of MMP-2 activity in human cardiofibroblasts. Biochem Cell Biol. 2013;91:1-8
44. Kassiri Z, Zhong J, Guo D. et al. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2:446-55
45. Zhang Z, Chen L, Zhong J. et al. ACE2/Ang-(1-7) signaling and vascular remodeling. Sci China Life Sci. 2014;57:802-8
46. George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer. 2010;10:745-59
47. Shenoy V, Ferreira AJ, Qi Y. et al. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2010;182:1065-72
48. Sukumaran V, Veeraveedu PT, Gurusamy N. et al. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1-7/mas receptor axis. Int J Biol Sci. 2011;7:1077-92
49. Simões e Silva AC, Silveira KD, Ferreira AJ. et al. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013;169:477-92
50. Jiang HM, Wang HX, Yang H. et al. Role for granulocyte colony stimulating factor in angiotensin II-induced neutrophil recruitment and cardiac fibrosis in mice. Am J Hypertens. 2013;26:1224-33
51. Marumo S, Hoshino Y, Kiyokawa H. et al. p38 mitogen-activated protein kinase determines the susceptibility to cigarette smoke-induced emphysema in mice. BMC Pulm Med. 2014;14:79
52. Barnes PJ. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat Rev Drug Discov. 2013;12(7):543-59
53. Sun J. Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J Signal Transduct. 2010;2010:985132
54. Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;87:69-98
55. Lemire BB, Debigaré R, Dubé A. et al. MAPK signaling in the quadriceps of patients with chronic obstructive pulmonary disease. J Appl Physiol. 2012;113(1):159-66
56. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396-405
57. Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838-49
Author biography
 Chih-Sheng Lin received the Ph.D. degree from the Department of Animal Science, National Chung Hsing University, Taiwan, in 1998. He is currently a Professor of the Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan. He has authored more than 100 scientific publications and 12 patents. His major research spans two fields, including (i) biological medicine to investigate the molecular pathogenesis of inflammatory fibrotic diseases in heart and lung using mouse models including transgenic and gene knock out mice, and (2) biosensing technology combined with gold nanoparticles (AuNPs) for the detection of pathogens and biomarkers in blood.
Chih-Sheng Lin received the Ph.D. degree from the Department of Animal Science, National Chung Hsing University, Taiwan, in 1998. He is currently a Professor of the Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan. He has authored more than 100 scientific publications and 12 patents. His major research spans two fields, including (i) biological medicine to investigate the molecular pathogenesis of inflammatory fibrotic diseases in heart and lung using mouse models including transgenic and gene knock out mice, and (2) biosensing technology combined with gold nanoparticles (AuNPs) for the detection of pathogens and biomarkers in blood.
 Yi-Han Hung received the B.S. degrees in Molecular Biology and Human Genetics from the Department of Molecular Biology and Human Genetics, Tzu Chi University, Hualien,Taiwan and M.S. degrees in Molecular Medicine and Bioengineering from the Department of Biological Science and Technology, National Chiao-Tung University, Hsinchu, Taiwan, in 2012 and 2014, respectively. She is currently working at Department of Medical Research in the Mackay Memorial Hospital, Taiwan, focusing on the association with cell biology and cancer research. Her research interests include molecular biology, bioengineering and regenerative medicine.
Yi-Han Hung received the B.S. degrees in Molecular Biology and Human Genetics from the Department of Molecular Biology and Human Genetics, Tzu Chi University, Hualien,Taiwan and M.S. degrees in Molecular Medicine and Bioengineering from the Department of Biological Science and Technology, National Chiao-Tung University, Hsinchu, Taiwan, in 2012 and 2014, respectively. She is currently working at Department of Medical Research in the Mackay Memorial Hospital, Taiwan, focusing on the association with cell biology and cancer research. Her research interests include molecular biology, bioengineering and regenerative medicine.
 Wen-Yeh Hsieh is qualified from the School of Chinese Medicine, China Medical College in 1996. He is a board certified internal medicine physician. In 1998, Dr. Hsieh entered the Mackay Memorial Hospital for specialist registrar training in respiratory and general internal medicine and went on to study for a PhD at the Institute of Biological Science and Technology within National Chiao Tung University in 2009. He now serves at the head for the chest medicine department at Mackay Memorial Hospital, Hsinchu, Taiwan. In addition to his clinical work, which involves the medical management of a wide spectrum of pulmonary diseases, Dr. Hsieh has strong research interests in pleural diseases and inflammations related to lung injury.
Wen-Yeh Hsieh is qualified from the School of Chinese Medicine, China Medical College in 1996. He is a board certified internal medicine physician. In 1998, Dr. Hsieh entered the Mackay Memorial Hospital for specialist registrar training in respiratory and general internal medicine and went on to study for a PhD at the Institute of Biological Science and Technology within National Chiao Tung University in 2009. He now serves at the head for the chest medicine department at Mackay Memorial Hospital, Hsinchu, Taiwan. In addition to his clinical work, which involves the medical management of a wide spectrum of pulmonary diseases, Dr. Hsieh has strong research interests in pleural diseases and inflammations related to lung injury.
![]() Corresponding author: Chih-Sheng Lin, Ph.D. Department of Biological Science and Technology, National Chiao Tung University, No.75 Po-Ai Street, Hsinchu 30068, Taiwan. Tel.: +886-3-5131338; Fax: +886-3-5729288; E-mail: lincsnctu.edu.tw
Corresponding author: Chih-Sheng Lin, Ph.D. Department of Biological Science and Technology, National Chiao Tung University, No.75 Po-Ai Street, Hsinchu 30068, Taiwan. Tel.: +886-3-5131338; Fax: +886-3-5729288; E-mail: lincsnctu.edu.tw