Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Maintenance of cholesterol...
LDLR pathway
The structure and physiological...
Transcriptional regulation of...
The post-transcriptional...
Dysregulation of the LDLR pathway
Inflammation mediates the LDLR...
Activation of RAS induces the...
High glucose disrupts the...
Activation of mTORC1 is involved...
Dysregulation of the LDLR...
Dysregulation of the LDLR...
Dysregulation of the LDLR...
Dysregulation of the LDLR...
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(5):569-579. doi:10.7150/ijbs.14027 This issue Cite
Research Paper
Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury
Yang Zhang1, Kun Ling Ma1 ![]() , Xiong Zhong Ruan2, Bi Cheng Liu1
, Xiong Zhong Ruan2, Bi Cheng Liu1
1. Institute of Nephrology, Zhong Da Hospital, School of Medicine, Southeast University, Nanjing City, Jiangsu Province, China.
2. Centre for Nephrology, University College London (UCL) Medical School, Royal Free Campus, UK.
Received 2015-10-2; Accepted 2016-2-26; Published 2016-3-21
Abstract
The low-density lipoprotein receptor (LDLR) pathway is a negative feedback system that plays important roles in the regulation of plasma and intracellular cholesterol homeostasis. To maintain a cholesterol homeostasis, LDLR expression is tightly regulated by sterol regulatory element-binding protein-2 (SREBP-2) and SREBP cleavage-activating protein (SCAP) in transcriptional level and by proprotein convertase subtilisin/kexin type 9 (PCSK9) in posttranscriptional level. The dysregulation of LDLR expression results in abnormal lipid accumulation in cells and tissues, such as vascular smooth muscle cells, hepatic cells, renal mesangial cells, renal tubular cells and podocytes. It has been demonstrated that inflammation, renin-angiotensin system (RAS) activation, and hyperglycemia induce the disruption of LDLR pathway, which might contribute to lipid disorder-mediated organ injury (atherosclerosis, non-alcoholic fatty liver disease, kidney fibrosis, etc). The mammalian target of rapamycin (mTOR) pathway is a critical mediator in the disruption of LDLR pathway caused by pathogenic factors. The mTOR complex1 activation upregulates LDLR expression at the transcriptional and posttranscriptional levels, consequently resulting in lipid deposition. This paper mainly reviews the mechanisms for the dysregulation of LDLR pathway and its roles in lipid disorder-mediated organ injury under various pathogenic conditions. Understanding these mechanisms leading to the abnormality of LDLR expression contributes to find potential new drug targets in lipid disorder-mediated diseases.
Keywords: dysregulation, low-density lipoprotein receptor, cholesterol homeostasis, lipid disorder, organ injury.
Introduction
The disorder of lipid metabolism implies that the lipids and their metabolites in the blood and tissues become abnormal at quality and quantity due to either a congenital condition or an acquired change. Lipid disorders are major risk factors of metabolic diseases, such as atherosclerosis, fatty liver disease, and diabetes. Recent reports have suggested that abnormal lipid homeostasis may contribute to the pathogenesis of renal fibrosis and vascular calcification [1-3].
Traditionally, lipid metabolism disorders have been associated with an improper diet, a lack of exercise, gene inheritance, and metabolic syndrome. Recently, clinical and animal studies have demonstrated that many factors, including chronic inflammation, immune disorder[4], renin-angiotensin system (RAS) activation[5], and hormone dysregulation[6], may affect lipid metabolism. A high-fat diet induces adipose to produce inflammatory cytokines[7]. In turn, inflammation modifies lipid metabolism. The studies reported that inflammation alters the HDL structure[8], reduces the levels of apolipoprotein A-I[9] and apolipoprotein B-100[9], increases cholesterol de novo synthesis and uptake, and increases the serum levels of triglycerides[10]. Similarly, the lipid-RAS interaction and lipid-hormone crosstalk in various diseases might modify lipid homeostasis and subsequently cause tissue lipid accumulation, thus exacerbating the progression of the diseases. As a key cell surface receptor for cholesterol uptake and traffic, low-density lipoprotein receptor (LDLR) is susceptible to be attacked by detrimental factors. The LDLR regulates the homeostasis of plasma and intracellular cholesterol through intaking plasma-derived LDL cholesterol and mediating its internalization and metabolism. The LDLR gene transcription is determined by cholesterol availability via a negative feedback mechanism that is tightly controlled by two proteins and their interaction, sterol regulatory element-binding protein-2 (SREBP-2) and SREBP cleavage-activating protein (SCAP)[11]. The posttranscriptional regulation of LDLR expression is modulated by proprotein convertase subtilisin/kexin type 9 (PCSK9)[12]. The disruption of this tightly regulated LDLR pathway may induce cholesterol accumulation and foam cell formation in organs. Many factors can disrupt the negative feedback regulation of LDLR pathway, which results in target organs injury. Here, we focus on the physiological regulation of LDLR pathway, the disruption of LDLR pathway in various conditions, and the clinical significance of this pathway in various diseases.
Maintenance of cholesterol homeostasis
Cholesterol is a necessary component of mammalian cell membranes and it is also a precursor of many bioactive substances, such as steroid hormones, bile acids, and vitamin D. These varied and essential functions require that the cellular cholesterol levels be maintained within a stable range. The sources of cholesterol include synthesis by the cells themselves and absorption in the small intestine mediated by the Niemann Pick Disease Type C1 Like-1(NPC1L1) protein[13]. Cholesterol is transported between the intracellular and extracellular compartments by various receptors and channels.
For most cells in mammals, intracellular cholesterol homeostasis is primarily governed by the tight regulation of cholesterol biosynthesis and transfer inside and outside cells involving influx and effux pathway. The cellular requirements for cholesterol are mainly satisfied through two mechanisms: 1) exogenously by cholesterol influx pathways involving cholesterol uptake mediated by various lipoprotein receptors; 2) endogenously by cholesterol synthesis regulated by the rate-limiting enzyme of this process, 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR)[14]. The cholesterol efflux pathways are crucial in the prevention of excessive cholesterol accumulation in the cells. The cholesterol and phospholipid efflux pathways are mediated by transport proteins, such as adenosine triphosphate (ATP)-binding cassette transporter A1 (ABCA1) and ABCG1.
Thus, at both the cell and the intact organism levels, the overall cholesterol homeostasis depends on the contributions of cholesterol synthesis, absorption, elimination, and storage.
LDLR pathway
Under physiological conditions, there is a intricate lipoprotein transport system distributing on various of cells and tissues responsible for the transport of cholesterol and fatty acids, from centre to the peripheral and from extracellular to intracellular. The key lipoprotein receptors responsible for lipid uptake include very-low density lipoprotein receptor (VLDLR), LDLR, and scavenger receptor.
The LDLR and its feedback regulation system is crucial to maintain cholesterol homeostasis[15]. As a transmembrane protein and important cholesterol transfer tools for LDL cholesterol, it is itself in the control of regulation at the cellular level. The intracellular feedback control system sense the content of LDL cholesterol and its oxidized derivatives and keep the level of cholesterol uptake by LDLR, thereby protecting the cell from excessive accumulation of cholesterol. Therefore, the LDLR-mediated regulatory system of cholesterol contributes to rational utilization of intra-and extra celluar cholesterol, which is important to keep a balance of cholesterol at cell and systemic level.
The structure and physiological function of LDLR
The LDLR is a transmembrane glycoprotein widely expressed in mammals that function in uptaking and internalizing of lipoproteins specially LDL-cholesterol, which is one of the important members in the LDLR family[16]. The LDLR is firstly synthesized as a 120 kDa precursor protein in the rough endoplasmic reticulum (ER), and then is transported to the Golgi apparatus for a series of processing and modification to be matured [17]. The mature LDLR protein is 160 kDa and consists of five functional domains. The first domain is ligand-binding repeats R1-R7 riching in cysteine, the second is a epidermal growth factor (EGF) precursor homology domain, including 3 EGF-like repetitive sequences (EGF-A, EGF-B and EGF-C) and a β-propeller structure which lies between EGF-B and EGF-C, followed by an O-linked polysaccharide domain, a transmembrane domain, and an intracellular domain[18]. The mature LDLR then presents on the cell surface and mediates the internalization and degradation of plasma LDL-cholesterol and other apolipoprotein-containing lipoproteins. The LDL combines with the ligand binding domain of LDLR and internalizes into cells by clathrin alveolus. Subsequently, the LDLR-lipid complexes enter into lysosomes, where the ligand uncouples from the receptor since the lower PH. After dissociation, the lipid part is degraded and the receptor recycling to the cell surface for binding lipid again [19].
Transcriptional regulation of the LDLR
As eukaryotic genes mediating lipid metabolism, the expression of LDLR is also under intricate regulation at both the transcriptional and posttranscriptional levels, which are mediated through precise signalling pathways (Fig.1). Cholesterol and its derivatives, as well as nonsterol factors, including inflammation mediator, growth factors, certain hormones, glucose and its metabolites are able to regulate LDLR expression[20, 21].
The transcriptional and posttranscriptional regulation of LDLR expression. The expression of LDLR is tightly regulated at transcriptional and posttranscriptional levels. When the intracellular concentration of cholesterol is high, the sterol sensor protein SCAP undergoes a conformational change that permits it to bind to Insig-1, which then forms SREBP-2/SCAP/Insig-1 ternary complex. The binding of the Insig proteins traps SREBP-2/SCAP in the ER membrane so that the SREBP-2 is not able to reach the Golgi apparatus for cleavage, and the LDLR gene expression decreases accordingly. As a result, cholesterol uptake is inhibited, and the cells establish cholesterol homeostasis. In contrast, when the cells are low on cholesterol, SCAP does not interact with the Insig proteins. The SREBP-2/SCAP complex is therefore free to leave the ER. After reaching the Golgi apparatus, the transcriptionally active domain of the SREBP-2 precursor will be proteolytically cleaved to release the mature form of nSREBP-2 in the Golgi membrane, while SCAP returns to the ER for recycling. The nSREBP-2 enters into the nucleus and binds with SRE to activate LDLR gene transcription. At the posttranscriptional level, the secreted form of PCSK9 specifically binds to LDLR to form the PCSK9/LDLR complex. In the presence of ARH, the LDLR/PCSK9 complex is internalized into the cells. After entering the endosome, the binding of PCSK9 and LDLR is further strengthened, preventing recycling of the LDLR from the endosome back to the cell surface and/or directing the LDLR to the lysosome where it is degraded. Alternatively, in an intracellular pathway, the nascent PCSK9 combines directly with LDLR in the Golgi apparatus, which mediates degradation in the lysosomes. LDLR, low density lipoprotein receptor; SREBP-2, sterol regulatory element-binding protein-2; nSREBP-2, nuclear SREBP-2; SCAP, SREBP cleavage activating protein; SRE, sterol regulatory element; ARH, autosomal recessive hypercholesterolaemia; PCSK9, proprotein convertase subtilisin kexin 9; ER, endoplasmic reticulum.
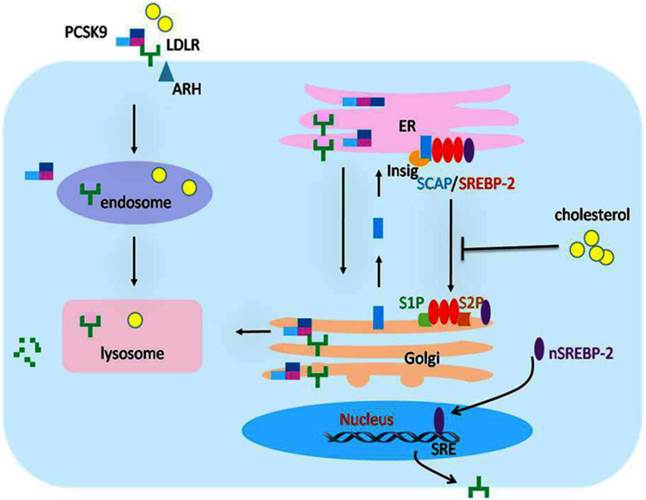
The SREBPs are the crucial elements for the modulation of LDLR gene transcription [22]. These proteins were identified by their ability to interact with the sterol regulatory element(SRE), which presents in the promoter of gene code for LDLR and other genes of proteins involving in lipid biosynthesis and uptake, such as the LDLR, the HMGR, and the acetyl coenzyme A synthetase [22-25]. The SREBPs have three members in mammalian cells, including SREBP-1a, SREBP-1c, and SREBP-2. The SREBP-2 is the major promoter of the LDLR gene. [26]. The SREBPs are generated as forms of inactive precursors with a molecular weight of approximately 125kDa, which are embedded in the ER membrane. To be matured and functional, the amino terminal domain of SREBP precursors is cleaved by site-1 protease (S1P) and site-2 protease (S2P). Since the two proteases are located in Golgi membrane, SREBP precursors need to be translocated from the ER to the Golgi for maturity [27], which requires escorting by another ER membrane protein termed SCAP. The amino terminal domain of SCAP has a sterol-sensing domain(SSD) serve as sterol sensors, which is sensitive to cholesterol in cells [28]. And the carboxy terminal domain of SCAP is crucial for the form of SREBP/SCAP complex through combined with the SREBP precursor [28, 29]. When cells are rich in cholesterol or its derivatives, the SSD of SCAP sense and bind to the sterol, resulting in a conformational change in SCAP and binding of the SREBP/SCAP with proteins termed as insulin induced genes (Insig 1 or 2) on ER membrane [26-30]. The formation of SREBP/SCAP/Insig ternary complex in the ER membrane impedes the SREBPs reaching the Golgi apparatus for cleavage, thereby suppressing the transcription of LDLR gene or other genes necessary to lipid synthesis. As a result, the cells are not able to generate and uptake cholesterol, and then establish cholesterol homeostasis [30-34]. In contrast, when intracellular sterols are exhausted, the SCAP dissociates with the protein of Insigs, permitting the SREBP/SCAP complex transfer to the Golgi apparatus [35]. In Golgi apparatus, the SREBP precursors are cleaved by the S1P and S2P proteases sequentially, producing a fragment weighing approximately 68kDa, which is designated nuclear SREBP (nSREBP), while SCAP returns to the ER for recycling [11, 36]. As a matured and functional form of SREBP, the nSREBP subsequently binds to its target genes and activates the transcription in nucleus.[15, 36-38]. Then, the intracellular cholesterol level is increased and returns to normal steady status. In the condition of diseases, the homeostasis of cholesterol and transcription of LDLR gene were broken by damage factors [20, 26, 40], which may be the cause of organ damage mediated by lipids.
The post-transcriptional regulation of LDLR
Posttranscriptional regulation of LDLR expression is also crucial for keeping itself in a stable level in intracellular, which was accompanied by remove of cholesterol.
The PCSK9 was firstly described as the ninth member of proprotein convertase subtilisin/kexin family in 2003[41]. Studies have shown that PCSK-9 exerts its role on cholesterol metabolism through posttranslational downregulation of the LDLR.
The immature precursor of PCSK9, synthesized as a72 kDa in the ER, contains an N-terminal signal peptide sequence, followed by a prodomain, a catalytic domain, and a c-terminal domain rich in cysteine[41]. The precursor form of PCSK9 undergoes autocatalytic cleavage at its 152th residue between the prodomain and catalytic domain, forming a 14 KDa prodomain fragment and a 63 KDa mature fragment that contains catalytic structure domain and the c-terminal domain [41]. The prodomain fragment remains bound to the mature protein through a covalent bond after cleavage, forming a complex with the mature segment. The bound fragment serves as a molecular chaperone that accompanies the mature protein during the translocation from the ER to the Golgi[42, 43]. The mature PCSK9 and the associated prodomain both undergo tyrosine sulfation, acetylation, and a series of modification in the Golgi and are finally secreted into the blood circulation [43].
The secreted form of PCSK9 specifically binds to the N-terminal region of the first EGF-A domain of LDLR at the cell membrane in a calcium-dependent process, thus forming the PCSK9/LDLR complex. The internalization of the complex also required for an endocytic adaptor protein named autosomal recessive hypercholesterolaemia (ARH). In the presence of this adaptor, the LDLR/PCSK9 complex is internalized as a unit into the cell [44]. After entering the endosome, the interaction of PCSK9 and LDLR is further strengthened due to the reduced pH. This stable complex inhibits the conformational change of the LDLR, preventing the recycling of the LDLR from the endosome to the cell surface and/or directing the LDLR enter into the lysosome where it is degraded[45]. Alternatively, an intracellular pathway directs the nascent PCSK9 combined with LDLR in Golgi apparatus and mediates degradation in the lysosome.
PCSK9 is associated with atherosclerosis, inflammation, infection, diabetes and other metabolic diseases. Inflammation can stimulate the expression of PCSK9, causing the degradation of LDLR, which increases plasma LDL levels. The mRNA level of PCSK9 was found to be increased in the livers of cholesterol-fed mice treated with lipopolysaccharide (LPS)[46].
As a key lipoprotein receptor in hepatic cells, the LDLR is crucial for liver clearing cholesterol. It not only clears LDL-cholesterol in circulation, but also maintains peripheral cellular cholesterol homeostasis. The LDLR gene mutation causes familial hypercholesterolemia (FH), an autosomal dominant disease. Forty years ago, Watanabe firstly found out a kind of rabbits with genetic defect of functional LDLR resulted from an in-frame deletion of part LDLR structural gene, which showed FH and cholesterol metabolism disorder similar to humans FH[47]. Ishibashi et al [48] produced mice lacking functional LDL receptor genes (LDLR-/- mice), accompanied by two folds higher plasma cholesterol than those of wild-type littermates, which are subsequently accepted as classical atherosclerosis models. Plasma lipoproteins revealed a remarkable increase in LDL-cholesterol levels in LDLR -/- mice, while unregulated hepatic LDLR expression significantly decreased the hepatic lipid content[49]. Bombo et al[50] found that LDLR defect led to increased lipid content and macrophage infiltration in the artery, displayed by obvious atherosclerotic lesion. In addition, abnormal overexpression of LDLR in peripheral organs or some cells which originally do not express LDLR also causes serious metabolic disorder. These studies in LDLR knockout mice indirectly demonstrated that the negative-feedback regulation of LDLR provides a protective effect on cholesterol homeostasis. Thus, the disruption of LDLR pathway may be involved in the mechanism for atherosclerosis.
Dysregulation of the LDLR pathway
Studies of LDLR-mediated endocytosis have provided much of our understanding of lipoprotein clearance. In addition to geneogenous abnormal expression, the stimulator-mediated dysregulation of the LDLR pathway disrupts the intracellular cholesterol homeostasis, which results in excessive uptake of cholesterol, lipid accumulation, foam cell formation, and organ injury.
Inflammation mediates the LDLR pathway disruption
Chronic low-grade inflammation manifests a persistent inflammatory reaction, which can be induced by a various of metabolic disorders and involved into the pathogenesis of insulin resistance, atherosclerosis, non-alcoholic fatty liver disease (NAFLD) and diabetes, which are commonly regarded as chronic inflammatory diseases[51]. Recently, it has been confirmed that chronic inflammation deteriorates the progression of diseases mediated by lipid disorder, such as NAFLD and atherosclerosis, as well as involved in the lipotoxicity in diabetes and chronic kidney disease[52-54].
Ruan et al [55, 56] firstly focuses on clarifying the relationship between lipid-mediated injury and inflammation in human mesangial cells (HMCs). They found that both tumor necrotic factor-α (TNF-α) and interleukin-1β (IL-1β) overrode the LDL loading-mediated LDLR suppression to upregulate the LDLR pathway, thereby increasing native LDL uptake by HMCs. As a result, LDLR gene expression was increased permitting the intracellular accumulation of unmodified LDL in vascular smooth muscle cells (VSMCs)[57]. These findings were further confirmed in hepatic cells by Ma et al [58]. To verify the results of the in vitro studies, Ma et al induced an inflamed model of NAFLD in apolipoprotein E knockout (ApoE KO) mice treated by Western diet and subcutaneous casein injection for 8 weeks, which results in a chronic, systemic inflammation. This inflammatory response upregulates cholesterol uptake mediated by LDLR [58]. In the livers of normal chow diet (NCD)-fed C57BL/6J mice and HepG2 cells in absence of lipid loading, IL-1β disrupts the LDLR feedback regulation and then causes statin resistance[59, 60]. It is accepted that macrophage scavenger receptors are crucial transporters in internalization of chemically modified lipoproteins which induces macrophages transform into foam cells. However, excess accumulation of unmodified LDL uptaked by LDLR under inflammatory conditions also mediates the transition of THP-1 originated macrophages to foam cells. This increased uptake results from significant upregulation of the LDLR pathway[61]. Furthermore, in end-stage renal disease (ESRD) patients accompanied with increased plasma level of C-reactive protein (CRP), the expression of LDLR is increased, which contributes to vascular calcification[2]. Recently, Ma et al[62] found that chronic inflammation redistributed lipid from the plasma to the kidneys and other organs in db/db mice, which was caused by the disruption of LDLR pathway. In addition, these inflammatory mediators inhibited the expression of the PCSK9 protein, which attenuated its efficacy in degrading LDLR[63]. However, LPS treatment increased the PCSK9 mRNA levels in the livers and kidneys of cholesterol-fed mice, which led to increased LDLR degradation [46]. Thus, the role of inflammation in regulating PCSK9 remains uncertain.
Hence, inflammatory stress modifies cholesterol homeostasis by disrupting LDLR at the transcriptional and posttranscriptional levels and causes unrestrained LDL-cholesterol uptake, subsequently resulting in redistribution of cholesterol from the blood to the tissues. This migration results in cholesterol accumulation in the peripheral tissues including kidneys, blood vessels, and liver, finally leading to excessive lipid deposition and damage to the tissues.
Activation of RAS induces the dysregulation of LDLR pathway
RAS activation is crucial in the development of atherosclerotic diseases. Increasing evidence shows that there is crosstalk between lipid disorder and RAS activation in atherogenesis[64, 65]. The expression of RAS components in human endothelial cells is increased under the stimulation of native or oxidized LDL[66]. Interestingly, in patients with hypercholesterolaemia, lipid-lowering agent statins inhibited circulatory RAS activity, and decreased circulatory RAS activity is associated with lowering serum cholesterol by statins [67]. Meanwhile, the administration of Ang II induces lipid accumulation in rat heart and liver, and Ang II-infusion also causes lipid deposition in kidneys through increasing the expression of SREBP-1 and fatty acid synthase [3, 68, 69]. Ang II stimulation causes accumulation of lipids in the renal vascular cells and tubular epithelial cells [69]. Ang II also promotes the LDL oxidation and its uptake in VSMCs and macrophages[70]. Ma et al.[71] investigated the association between RAS and LDLR pathway in HMCs. They demonstrated that lipid accumulation in HMCs induced by Ang II was strongly associated with the upregulaiton of LDLR pathway (Fig.2). However, these effects were ameliorated by telmisartan. In contrast, cholesterol loading enhanced the expression of RAS components in HMCs. These evidence shows that dysregulation of the LDLR pathway plays a crucial role in lipid disorder mediated by RAS activation.
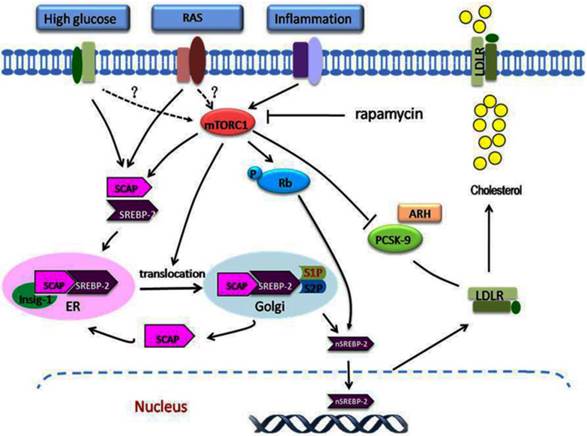
The role of mTORC1 in dysregulation of the LDLR pathway. A variety of stimulating factors, including inflammation, hyperglycaemia, and RAS activation, may lead to disruption of the regulation of LDLR. Under these stimuli, mTORC1 is activated, which modulates the dysregulation of LDLR at the transcriptional and posttranscriptional levels. At the transcriptional level, inflammation or other stimuli activate the mTORC1, which phosphorylates Rb. This increases the expression of SCAP and SREBP-2, increases the translocation of SCAP/SREBP-2 complex from the ER to the Golgi, and then enhances the nuclear translocation of SREBP-2, which upregulates LDLR gene expression. At the posttranscriptional level, the increased mTORC1 activity increases the LDLR protein level by inhibiting the PCSK9 protein expression. The overexpression of LDLR in the cells results in excess cholesterol accumulation. mTORC1, mammalian target of rapamycin complex1; Rb, retinoblastoma tumour suppressor protein.
In the past two decades, local or intracellular RAS has received increasing attention and acquaintance especially considering its important role in the pathogenesis and the clinical. Local RAS can be produced by some specific tissues and organs instead of circulation, such as kidney, heart, liver, vascular, adipose tissue, and nervous system [72]. It is detected that Ang II level in kidney is three to five times higher that in the circulation. The components of local RAS directly affect on the tissue itself or the nearby tissues similar to some endocrine hormone, which may carry diverse physiological effects at the cellular and tissue levels by interacting with other signalling pathways. Besides the effects on the cardiovascular/body haemodynamic systems, local RAS also involves multiple conditions of disease status, such as inflammation, oxidative stress, fibrosis, and lipid metabolism disorder[73]. Therefore, these local or intracellular components of RAS facilitate the interaction between RAS activation and the lipid disorders directly mediated by various receptors.
High glucose disrupts the negative feedback regulation of LDLR
Dyslipidaemia is one of the main risk factors for cardiovascular complications in patients with diabetes. Dyslipidaemia is more atherogenic in diabetic patients than in non-diabetic patients[74]. High glucose promotes macrophage transfer to foam cell by inducing expression of scavenger receptors[75]. Xue et al[76] demonstrated that the lipid accumulation in VSMCs was increased significantly under high glucose stimulation. They found that this consequence may mediated by increasing expression of CD36 and decreasing expression of ABCG1 in VSMCs. In renal diseases, high glucose promotes mesangial cell transform into foam cell through destroying the balance of cholesterol influx and efflux pathways [77].
Yuan et al[78] investigated the effect of Nε-(carboxymethyl) lysine (CML, a member of the advanced glycation end products (AGEs) family) on lipid deposition in HMCs. CML stimulation increased lipid deposition in HMCs wich showed similar effects with the IL-1β. In addition, CML prevented the degradation of SCAP by increasing the Golgi mannosidase activity and enhanced the availability of SCAP in Golgi. In diabetic rats induced by a high fat/sucrose diet combined with low-dose intraperitoneal injection of streptozocin (STZ), Sun et al[79] observed lipid droplet accumulation in kidneys accompanied with increased the expression of HMGR and LDLR, whereas lipid-lowering agent, atorvastatin, alleviated the lipid disorder and renal injury. Using type 2 diabetic db/db mice and cell culture model of podocytes, Ma et al.[80] found that lipid accumulation in kidneys and in podocytes was increased under the high glucose stimulation, which is mediated through the disuption of LDLR pathway. Interestingly, using siRNA to silence the LDLR gene, the lipid deposition in podocytes was inhibited [80]. These studies reveal that high glucose-induced lipid accumulation and foam cell formation might be correlated with the imbalance of intracellular cholesterol homeostasis. The LDLR, as a major lipoprotein receptor, is involved in governing excess cholesterol uptake in podocytes (Fig.2).
Activation of mTORC1 is involved in the dysregulation of LDLR pathway
The mammalian target of rapamycin(mTOR), as an crucial protein kinase in mammals, has multiple functions, including regulating cell growth, initiating mRNA translation, protein synthesis, cell cycle progression, and DNA synthesis. The mTOR consists of two distinct multi-protein complexes according to its constitutive protein: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2)[81]. The mTORC1 is activated by insulin, environmental nutrients, and other growth factors. After activation, mTORC1 phosphorylates p70 S6 kinase 1 and eukaryotic initiation factor 4E-binding protein 1 to induce messenger RNA translation and protein synthesis, ultimately promote cell growth, cell proliferation, energy balance and ribosome biosynthesis[82-84]. The mTORC2 primarily modulates embryonic development, cell polarity, cell cytoskeleton reorganization, cell mifration, and protein synthesis[85]. The multiple signals can activate the mTOR. When activated, it regulates cell growth and proliferation by targeting its downstean factors, thus maintain cell growth in metabolic homeostasis. Rapamycin, a potent immunosuppressive agent, is an inhibitor of the mTORC1, which inactivates the mTOR through its binding to FK506 binding protein (FKBP12).
Studies have reported that the anti-atherosclerotic effects of rapamycin are mediated through its ability to suppress the proliferation and migration of VSMCs [86-88]. Clinical trials have demonstrated that treatment with rapamycin releasing stent significant reduces the secondary stenosis, late lumen loss and repeat revascularization rates in patient with coronary disease compared with traditional standard coronary stents. [89, 90]. Moreover, rapamycin reduces the atherosclerotic lesion size in ApoE KO mice when administered either intraperitoneally [87, 91] or orally [92]. Therefore, the activation of mTOR pathway involves the dysregulation of lipid homoeostasis.
Recently, some studies suggest that mTORC1 modulates insulin-mediated fatty acid synthesis [93, 94]. Akt-induced nuclear localization of SREBP-1c can be blocked by rapamycin, resulting in upregulation of lipogenic genes and lipid synthesis. In addition, the mTORC1 upregulates expression of cholesterol biosynthetic gene in hepatic cells by inducing SREBP-2 processing, an effect that leads to the accumulation of intracellular cholesterol.
Ma et al.[95] showed that rapamycin inhibited the LDLR-mediated cholesterol accumulation in hepatic cells induced by IL-1β. This inhibitory effect was mediated by the down-regulation of LDLR pathway. Recently, Ma et al.[96] further examined how the mTOR pathway function in the progression of atherosclerosis in both ApoE KO mice and VSMCs in present of inflammatory stress. They demonstrated for the first time that mTORC1 is the key downstream signal of inflammation in modulating LDLR negative feedback pathway. Reponses to IL-1β stimulation, the mTOR was activated and subsequently phosphorylated the retinoblastoma tumour suppressor protein which exerts crucial roles in cell cycle control process governing entry into S phase. These effects eventually resulted in increased nuclear SREBP-2 expression and subsequent LDLR-mediated cholesterol uptake. Ma et al.[63] further investigated the role of mTORC1 in NAFLD under the condition of inflammation. They found that the effect of IL-1β on the LDLR pathway was meditaed by upregulating mTORC1 activity. Interestingly, Ma et al. also observed that inhibition of mTORC1 reversed the down-regulation of PCSK9 in the livers of casein-injected mice or in IL-1β-stimulated HepG2 cells, which partly mediated the LDLR overexpression. Similarly, Ai et al [97] also reported that the activation of mTORC1 increased LDLR protein expression via a posttranscriptional mechanism involving the inhibition of PCSK9 protein expression, resulting in a decreased level of circulating LDL cholesterol. These studies highlight the crucial role of mTORC1 in disruption of the LDLR pathway at both transcriptional and posttranscriptional mechanisms (Fig.2).
Dysregulation of the LDLR pathway exacerbates lipid disorder-mediated organ injury
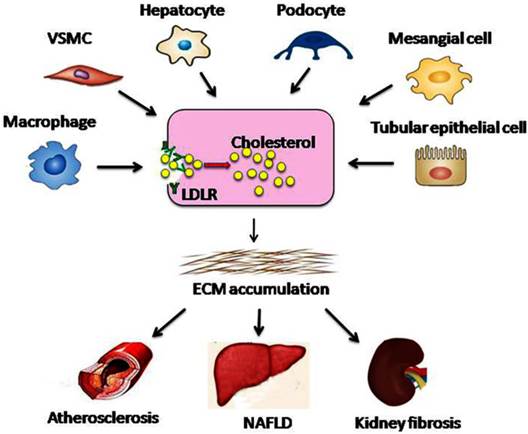
Studies have shown that the dysregulation of LDLR pathway occurs in various types of cells, ranging from pluripotent precursors to foam cells (Fig.3). Therefore, dysregulation of the LDLR pathway might be a critical process in accelerating the progression of lipid disorder-mediated target organ injuries (atherosclerosis, glomerulosclerosis, and NAFLD, etc.).
Dysregulation of the LDLR pathway exacerbates lipid disorder-mediated organ injury. Under conditions of hyperglycaemia, RAS activation, or chronic inflammation, feedback regulation of the LDLR pathway is disrupted at the transcriptional or post-transcriptional level. Therefore, native LDL-cholesterol is taken up by macrophages, VSMCs, hepatocytes, podocytes, mesangial cells, and tubular epithelial cells without limitation, which may result in the conversion of these cells into foam cells. Aggregation of the foam cells in the arteries, liver, or kidney leads to ECM deposition and tissue sclerosis, which ultimately accelerates the progression of atherosclerosis, NAFLD, and kidney fibrosis. VSMC, vascular smooth muscle cell; ECM, extracellular matrix.
Dysregulation of the LDLR pathway in cardiovascular diseases
The cardiovascular system is the primary system that is vulnerable to lipid-mediated damage, as indicated by the increasing prevalence of atherosclerosis and atherosclerotic cardiovascular disease. It is accepted that atherosclerosis is a multifactorial disease that involves the disorder of endothelial function, inflammatory cell recruitment, local cytokine production, and lipid accumulation inside the intima of the vessel wall[98]. Ma et al[96] found that the inflammation-mediated disruption of LDLR feedback regulation contributes to atherosclerosis in ApoE KO mice and in VSMCs. When the expression of LDLR was upregulated, the foam cells and lipid plaques were significantly increased in the ApoE knockout mice. Rapamycin alleviated atherosclerosis by inhibiting the expression of LDLR and the intracellular lipid accumulation [44].
In addition to directly mediating atherosclerotic plaque formation, Ma et al[2] found inflammation-mediated LDLR disruption contributed to vascular calcification in ESRD patients. Thus, dysregulation of the LDLR pathway is critical for atherosclerosis, especially under conditions of chronic inflammation.
Dysregulation of the LDLR pathway in NAFLD
It is well accepted that triglyceride accumulation in the liver is the major hallmark of NAFLD. However, increasing evidence has recently linked disrupted hepatic cholesterol homeostasis and free cholesterol accumulation to the pathogenesis of NAFLD. Under inflammatory stress, LDLR-mediated cholesterol influx promotes a number of hepatic cells convert to foam cells, resulting in lipid over-accumulation in ApoE KO mice and C57BL/6 mice [60, 63]. Moreover, inflammatory cytokines also cause statin resistance [59]. Therefore, a higher concentration of statins may be required to achieve the same lipid-lowering effects when accompanied with inflammation. Although statin therapy lowers LDL cholesterol in subjects with NAFLD, LDLR expression remains at the level seen in those not on statins. This LDLR overexpression mediated by cholesterol trafficking disturbance may be a contributing factor for the progression of NAFLD. However, rapamycin prevents the inflammation-induced cholesterol accumulation in HepG2 cells that results from the disruption of LDLR pathway[95]. Taken together, these studies showed that dysregulation of LDLR pathway and increased accumulation of lipid exacerbated the progression of NAFLD.
Dysregulation of the LDLR pathway in kidney diseases
The concept of the 'lipid nephrotoxicity hypothesis' is now accepted by increasing numbers of researchers[99]. In patients with CKD or ESRD, a number of metabolic disorders involve the proression of glomerulosclerosis and tubulointerstitial fibrosis, including atherosclerosis, dyslipidaemia, oxidative stress, and inflammatory stress[99]. In previous studies, Ruan et al confirmed that LDLR dysregulation mediated the HMCs foam cell transition, which contributes to lipid-mediated renal damage [3, 10]. Ang II also induced HMCs to produce excessive extracellular matrix and accelerated the progression of glomerulosclerosis by disturbing LDLR pathway. Furthermore, lipid accumulation induced by IL-1β and TNF-α leads to increased reactive oxygen species (ROS) and ER stress in the HMCs and in the kidneys of C57BL/6J mice[100]. In the kidneys of diabetic rats induced by STZ injection, we found significant lipid accumulation and increased expressions of LDLR, SCAP, and SREBP-2[79]. Ma et al [62, 100] demonstrated that excessive lipid accumulation induces epithelial mesenchymal transition of podocytes. These effects were closely correlated with the inflammation- or high glucose-mediated upregulation of the LDLR pathway. Finally, these change results in effacement of the foot processes and phenotype change of podocyte, mesangial matrix expansion, basement membrane thickening, glomerulosclerosis, increased serum creatinine, and proteinuria.
In summary, the negative feedback regulation of LDLR pathway in the liver and other peripheral organs provides a mechanism to regulate the plasma cholesterol at aconstant level and maintain intracellular cholesterol homeostasis. Under the pathological conditions, disruption of the LDLR pathway may result in cholesterol accumulation in the cells, which results in the acceleration of foam cell formation and exacerbation of the progression of target organ injury.
Conclusion
Dysregulation of the LDLR pathway contributes to lipid disorder-mediated organ injury. The activation of mTORC1 is involved in the dysregulation of the LDLR pathway. Increased understanding of the mechanisms of LDLR dysregulation will encourage researchers to identify reliable strategies to prevent lipid disorder-mediated organ injury. In addition, in some clinical patients, effective control of hyperglycaemia, RAS activation, and chronic inflammation, which are independent risk factors for dysregulation of the LDLR pathway, are essential.
Acknowledgements This work was supported by the Jiangsu Province Ordinary University Graduate Research Innovation Project (KYZZ15-0061), the National Natural Science Foundation of China (grants 81170792 and 81470957), the Natural Science Foundation of Jiangsu Province (BK20141343), and the Clinical Medical Science Technology Special Project of Jiangsu Province (BL2014080).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37-46
2. Liu J, Ma KL, Gao M, Wang CX, Ni J, Zhang Y. et al. Inflammation disrupts the LDL receptor pathway and accelerates the progression of vascular calcification in ESRD patients. PloS one. 2012;7:e47217
3. Ni J, Ma KL, Wang CX, Liu J, Zhang Y, Lv LL. et al. Activation of renin-angiotensin system is involved in dyslipidemia-mediated renal injuries in apolipoprotein E knockout mice and HK-2 cells. Lipids Health Dis. 2013;12:49
4. Campbell LA, Rosenfeld ME. Infection and Atherosclerosis Development. Arch Med Res. 2015;46(5):339-50
5. Koh KK, Han SH, Oh PC, Shin EK, Quon MJ. Combination therapy for treatment or prevention of atherosclerosis: focus on the lipid-RAAS interaction. Atherosclerosis. 2010;209:307-13
6. Traish AM, Abdou R, Kypreos KE. Androgen deficiency and atherosclerosis: The lipid link. Vascul Pharmacol. 2009;51:303-13
7. Gao M, Ma Y, Liu D. High-fat diet-induced adiposity, adipose inflammation, hepatic steatosis and hyperinsulinemia in outbred CD-1 mice. PloS one. 2015;10:e0119784
8. Kaysen GA, Eiserich JP. The role of oxidative stress-altered lipoprotein structure and function and microinflammation on cardiovascular risk in patients with minor renal dysfunction. J Am Soc Nephrol. 2004;15:538-48
9. Ettinger WH, Varma VK, Sorci-Thomas M, Parks JS, Sigmon RC, Smith TK. et al. Cytokines decrease apolipoprotein accumulation in medium from Hep G2 cells. Arterioscler Thromb. 1994;14:8-13
10. Chen Y, Chen Y, Zhao L, Chen Y, Mei M, Li Q. et al. Inflammatory stress exacerbates hepatic cholesterol accumulation via disrupting cellular cholesterol export. J Gastroenterol Hepatol. 2012;27:974-84
11. Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc Natl Acad Sci U S A. 1999;96:11235-40
12. Lambert G, Charlton F, Rye KA, Piper DE. Molecular basis of PCSK9 function. Atherosclerosis. 2009;203:1-7
13. Wang DQ. Regulation of intestinal cholesterol absorption. Annu Rev Physiol. 2007;69:221-48
14. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425-30
15. Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241-60
16. Sudhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985;228:815-22
17. Tolleshaug H, Goldstein JL, Schneider WJ, Brown MS. Posttranslational processing of the LDL receptor and its genetic disruption in familial hypercholesterolemia. Cell. 1982;30:715-24
18. Defesche JC. Low-density lipoprotein receptor-its structure, function, and mutations. Semin Vasc Med. 2004;4:5-11
19. Soutar AK. Intracellular transport of the low-density lipoprotein receptor. Biochem Soc Trans. 1996;24:547-52
20. Catapano AL. The low density lipoprotein receptor: structure, function and pharmacological modulation. Pharmacol Ther. 1989;43:187-219
21. Soutar AK, Knight BL. Structure and regulation of the LDL-receptor and its gene. Br Med Bull. 1990;46:891-916
22. Rawson RB. The SREBP pathway-insights from Insigs and insects. Nat Rev Mol Cell Biol. 2003;4:631-40
23. Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X. et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75:187-97
24. Briggs MR, Yokoyama C, Wang X, Brown MS, Goldstein JL. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. I. Identification of the protein and delineation of its target nucleotide sequence. J Biol Chem. 1993;268:14490-6
25. Wang X, Briggs MR, Hua X, Yokoyama C, Goldstein JL, Brown MS. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. II. Purification and characterization. J Biol Chem. 1993;268:14497-504
26. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125-31
27. Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53-62
28. Nohturfft A, Brown MS, Goldstein JL. Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. J Biol Chem. 1998;273:17243-50
29. [29] Hua X, Nohturfft A, Goldstein JL, Brown MS. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell. 1996;87:415-26
30. [30] Adams CM, Goldstein JL, Brown MS. Cholesterol-induced conformational change in SCAP enhanced by Insig proteins and mimicked by cationic amphiphiles. Proc Natl Acad Sci U S A. 2003;100:10647-52
31. [31] Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell. 2002;10:237-45
32. [32] Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol Cell. 2004;15:259-68
33. [33] Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A. 2002;99:12753-8
34. [34] Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R. et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489-500
35. [35] Espenshade PJ, Li WP, Yabe D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc Natl Acad Sci U S A. 2002;99:11694-9
36. [36] Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A. 1999;96:11041-8
37. [37] Wilson GM, Vasa MZ, Deeley RG. Stabilization and cytoskeletal-association of LDL receptor mRNA are mediated by distinct domains in its 3' untranslated region. J Lipid Res. 1998;39:1025-32
38. [38] Armiger LC, Headrick JP, Jordan LR, Willis RJ. Bound inorganic phosphate and early contractile failure in global ischaemia. Basic Res Cardiol. 1995;90:482-8
39. [39] Hirano Y, Yoshida M, Shimizu M, Sato R. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem. 2001;276:36431-7
40. Auwerx JH, Chait A, Wolfbauer G, Deeb SS. Involvement of second messengers in regulation of the low-density lipoprotein receptor gene. Mol Cell Biol. 1989;9:2298-302
41. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S. et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100:928-33
42. Naureckiene S, Ma L, Sreekumar K, Purandare U, Lo CF, Huang Y. et al. Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch Biochem Biophys. 2003;420:55-67
43. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W. et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865-75
44. Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB. et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995-3005
45. Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71-7
46. Feingold KR, Moser AH, Shigenaga JK, Patzek SM, Grunfeld C. Inflammation stimulates the expression of PCSK9. Biochem Biophys Res Commun. 2008;374:341-4
47. Watanabe Y. Serial inbreeding of rabbits with hereditary hyperlipidemia (WHHL-rabbit). Atherosclerosis. 1980;36:261-8
48. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883-93
49. Goedeke L, Rotllan N, Canfran-Duque A, Aranda JF, Ramirez CM, Araldi E. et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat Med. 2015;21:1280-9
50. Bombo RP, Afonso MS, Machado RM, Lavrador MS, Nunes VS, Quintao ER. et al. Dietary phytosterol does not accumulate in the arterial wall and prevents atherosclerosis of LDLr-KO mice. Atherosclerosis. 2013;231:442-7
51. Andel M, Polak J, Kraml P, Dlouhy P, Stich V. [Chronic mild inflammation links obesity, metabolic syndrome, atherosclerosis and diabetes]. Vnitr Lek. 2009;55:659-65
52. Fernando RL, Varghese Z, Moorhead JF. Oxidation of low-density lipoproteins by rat mesangial cells and the interaction of oxidized low-density lipoproteins with rat mesangial cells in vitro. Nephrol Dial Transplant. 1993;8:512-8
53. Moorhead JF, Chan MK, El-Nahas M, Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet. 1982;2:1309-11
54. Diamond JR. Analogous pathobiologic mechanisms in glomerulosclerosis and atherosclerosis. Kidney Int Suppl. 1991;31:S29-34
55. Ruan XZ, Varghese Z, Powis SH, Moorhead JF. Dysregulation of LDL receptor under the influence of inflammatory cytokines: a new pathway for foam cell formation. Kidney Int. 2001;60:1716-25
56. Ruan XZ, Varghese Z, Fernando R, Moorhead JF. Cytokine regulation of low-density lipoprotein receptor gene transcription in human mesangial cells. Nephrol Dial Transplant. 1998;13:1391-7
57. Ruan XZ, Moorhead JF, Tao JL, Ma KL, Wheeler DC, Powis SH. et al. Mechanisms of dysregulation of low-density lipoprotein receptor expression in vascular smooth muscle cells by inflammatory cytokines. Arterioscler Thromb Vasc Biol. 2006;26:1150-5
58. Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology. 2008;48:770-81
59. Chen Y, Ruan XZ, Li Q, Huang A, Moorhead JF, Powis SH. et al. Inflammatory cytokines disrupt LDL-receptor feedback regulation and cause statin resistance: a comparative study in human hepatic cells and mesangial cells. Am J Physiol Renal Physiol. 2007;293:F680-7
60. Zhao L, Chen Y, Tang R, Li Q, Gong J, Huang A. et al. Inflammatory stress exacerbates hepatic cholesterol accumulation via increasing cholesterol uptake and de novo synthesis. J Gastroenterol Hepatol. 2011;26:875-83
61. Ye Q, Chen Y, Lei H, Liu Q, Moorhead JF, Varghese Z. et al. Inflammatory stress increases unmodified LDL uptake via LDL receptor: an alternative pathway for macrophage foam-cell formation. Inflamm Res. 2009;58:809-18
62. Zhang Y, Ma KL, Liu J, Wu Y, Hu ZB, Liu L. et al. Inflammatory stress exacerbates lipid accumulation and podocyte injuries in diabetic nephropathy. Acta Diabetol. 2015
63. Liu J, Ma KL, Zhang Y, Wu Y, Hu ZB, Lv LL. et al. Activation of mTORC1 disrupted LDL receptor pathway: a potential new mechanism for the progression of non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2015;61:8-19
64. Singh BK, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in atherogenesis and therapy of coronary heart disease. Indian Heart J. 2001;53:511-8
65. Singh BM, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in the therapy of hypertension and coronary heart disease. Arch Intern Med. 2003;163:1296-304
66. Catar RA, Muller G, Heidler J, Schmitz G, Bornstein SR, Morawietz H. Low-density lipoproteins induce the renin-angiotensin system and their receptors in human endothelial cells. Horm Metab Res. 2007;39:801-5
67. Long H, Wang L, Su H, Xu J, Li J, Peng Q. et al. Increased circulatory RAS activity can be inhibited by statins in patients with hypercholesterolemia. J Renin Angiotensin Aldosterone Syst. 2015;16:126-30
68. Hongo M, Ishizaka N, Furuta K, Yahagi N, Saito K, Sakurai R. et al. Administration of angiotensin II, but not catecholamines, induces accumulation of lipids in the rat heart. Eur J Pharmacol. 2009;604:87-92
69. Ishizaka N, Hongo M, Sakamoto A, Saito K, Furuta K, Koike K. Liver lipid content is reduced in rat given 7-day administration of angiotensin II. J Renin Angiotensin Aldosterone Syst. 2011;12:462-8
70. Wang X, Phillips MI, Mehta JL. LOX-1 and angiotensin receptors, and their interplay. Cardiovasc Drugs Ther. 2011;25:401-17
71. Ma KL, Ni J, Wang CX, Liu J, Zhang Y, Wu Y. et al. Interaction of RAS activation and lipid disorders accelerates the progression of glomerulosclerosis. Int J Med Sci. 2013;10:1615-24
72. Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747-803
73. Leung PS. The peptide hormone angiotensin II: its new functions in tissues and organs. Curr Protein Pept Sci. 2004;5:267-73
74. Krentz AJ. Lipoprotein abnormalities and their consequences for patients with type 2 diabetes. Diabetes Obes Metab. 2003;5(Suppl 1):S19-27
75. Moheimani F, Tan JT, Brown BE, Heather AK, van Reyk DM, Davies MJ. Effect of exposure of human monocyte-derived macrophages to high, versus normal, glucose on subsequent lipid accumulation from glycated and acetylated low-density lipoproteins. Exp Diabetes Res. 2011;2011:851280
76. Xue JH, Yuan Z, Wu Y, Liu Y, Zhao Y, Zhang WP. et al. High glucose promotes intracellular lipid accumulation in vascular smooth muscle cells by impairing cholesterol influx and efflux balance. Cardiovasc Res. 2010;86:141-50
77. Song KH, Park J, Ha H. High glucose increases mesangial lipid accumulation via impaired cholesterol transporters. Transplant Proc. 2012;44:1021-5
78. Yuan Y, Zhao L, Chen Y, Moorhead JF, Varghese Z, Powis SH. et al. Advanced glycation end products (AGEs) increase human mesangial foam cell formation by increasing Golgi SCAP glycosylation in vitro. Am J Physiol Renal Physiol. 2011;301:F236-43
79. Sun H, Yuan Y, Sun ZL. Cholesterol Contributes to Diabetic Nephropathy through SCAP-SREBP-2 Pathway. Int J Endocrinol. 2013;2013:592576
80. Zhang Y, Ma KL, Liu J, Wu Y, Hu ZB, Liu L. et al. Dysregulation of low-density lipoprotein receptor contributes to podocyte injuries in diabetic nephropathy. Am J Physiol Endocrinol Metab. 2015;308:E1140-8
81. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471-84
82. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926-45
83. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163-75
84. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296-302
85. Lieberthal W, Levine JS. Mammalian target of rapamycin and the kidney. I. The signaling pathway. Am J Physiol Renal Physiol. 2012;303:F1-10
86. Degertekin M, Regar E, Tanabe K, Lee CH, Serruys PW. Sirolimus eluting stent in the treatment of atherosclerosis coronary artery disease. Minerva Cardioangiol. 2002;50:405-18
87. Elloso MM, Azrolan N, Sehgal SN, Hsu PL, Phiel KL, Kopec CA. et al. Protective effect of the immunosuppressant sirolimus against aortic atherosclerosis in apo E-deficient mice. Am J Transplant. 2003;3:562-9
88. Zhao L, Ding T, Cyrus T, Cheng Y, Tian H, Ma M. et al. Low-dose oral sirolimus reduces atherogenesis, vascular inflammation and modulates plaque composition in mice lacking the LDL receptor. Br J Pharmacol. 2009;156:774-85
89. Marx SO, Marks AR. Bench to bedside: the development of rapamycin and its application to stent restenosis. Circulation. 2001;104:852-5
90. Morice MC, Serruys PW, Sousa JE, Fajadet J, Ban Hayashi E, Perin M. et al. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N Engl J Med. 2002;346:1773-80
91. Castro C, Campistol JM, Sancho D, Sanchez-Madrid F, Casals E, Andres V. Rapamycin attenuates atherosclerosis induced by dietary cholesterol in apolipoprotein-deficient mice through a p27 Kip1 -independent pathway. Atherosclerosis. 2004;172:31-8
92. Pakala R, Stabile E, Jang GJ, Clavijo L, Waksman R. Rapamycin attenuates atherosclerotic plaque progression in apolipoprotein E knockout mice: inhibitory effect on monocyte chemotaxis. J Cardiovasc Pharmacol. 2005;46:481-6
93. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107:3441-6
94. Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE. et al. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci U S A. 2012;109:16184-9
95. Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Sirolimus modifies cholesterol homeostasis in hepatic cells: a potential molecular mechanism for sirolimus-associated dyslipidemia. Transplantation. 2007;84:1029-36
96. Ma KL, Liu J, Wang CX, Ni J, Zhang Y, Wu Y. et al. Activation of mTOR modulates SREBP-2 to induce foam cell formation through increased retinoblastoma protein phosphorylation. Cardiovasc Res. 2013;100:450-60
97. Ai D, Chen C, Han S, Ganda A, Murphy AJ, Haeusler R. et al. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest. 2012;122:1262-70
98. Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012;18:4266-88
99. Ruan XZ, Varghese Z, Moorhead JF. An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol. 2009;5:713-21
100. Zhong S, Zhao L, Li Q, Yang P, Varghese Z, Moorhead JF. et al. Inflammatory Stress Exacerbated Mesangial Foam Cell Formation and Renal Injury via Disrupting Cellular Cholesterol Homeostasis. Inflammation. 2015;38:959-71
Author contact
![]() Corresponding author: Kun Ling Ma. Institute of Nephrology, Zhong Da Hospital, School of Medicine, Southeast University, NO. 87, Ding Jia Qiao Road, Nan Jing City, Jiangsu Province, China, 210009. Tel: 0086 25 83262442 Fax: 0086 25 83262442 E-mail: Kun Ling Ma: klma05com.
Corresponding author: Kun Ling Ma. Institute of Nephrology, Zhong Da Hospital, School of Medicine, Southeast University, NO. 87, Ding Jia Qiao Road, Nan Jing City, Jiangsu Province, China, 210009. Tel: 0086 25 83262442 Fax: 0086 25 83262442 E-mail: Kun Ling Ma: klma05com.