Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(7):812-823. doi:10.7150/ijbs.15229 This issue Cite
Research Paper
Transient Receptor Potential Ankyrin 1 Channel Involved in Atherosclerosis and Macrophage-Foam Cell Formation
Jin-Feng Zhao1, Song-Kun Shyue2, Yu Ru Kou1, Tse-Min Lu3,4 ![]() , Tzong-Shyuan Lee1,5
, Tzong-Shyuan Lee1,5 ![]()
1. Department of Physiology, School of Medicine, National Yang-Ming University, Taipei, Taiwan
2. Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
3. Faculty of Medicine, School of Medicine, National Yang-Ming University, Taipei, Taiwan
4. Division of Cardiology, Department of Internal Medicine, Taipei Veterans General Hospital, Taipei, Taiwan
5. Genome Research Center, National Yang-Ming University, Taipei, Taiwan
Received 2016-2-8; Accepted 2016-4-8; Published 2016-5-25
Abstract

Transient receptor potential ankyrin 1 channel (TRPA1) plays an important role in the pathogenesis of inflammatory diseases, yet its role and the underlying mechanism in atherosclerosis remain unclear. We aimed to investigate the role of TRPA1 in atherosclerosis and foam-cell formation in vivo in mice and in vitro in mouse macrophages. Histopathology was examined by hematoxylin and eosin staining, levels of cytokines and lipid profile were evaluated by assay kits, and protein expression was determined by western blot analysis. TRPA1 expression was increased in macrophage foam cells in atherosclerotic aortas of apolipoprotein E-deficient (apoE-/-) mice. Atherosclerotic lesions, hyperlipidemia and systemic inflammation were worsened with chronic administration of the TRPA1 channel antagonist HC030031 or genetic ablation of TRPA1 (TRPA1-/-) in apoE-/- mice. Treatment with allyl isothiocyanate (AITC, a TRPA1 agonist) retarded the progression of atherosclerosis in apoE-/- mice but not apoE-/-TRPA1-/- mice. Mouse macrophages showed oxidized low-density lipoprotein (oxLDL) activated TRPA1 channels. OxLDL-induced lipid accumulation of macrophages was exacerbated by HC030031 or loss of function of TRPA1. Inhibition of TRPA1 activity did not alter oxLDL internalization but impaired cholesterol efflux by downregulating the ATP-binding cassette transporters. Furthermore, tumor necrosis factor-α-induced inflammatory response was attenuated in AITC-activated macrophages. TRPA1 may be a pivotal regulator in the pathogenesis of atherosclerosis and cholesterol metabolism of macrophage foam cells.
Keywords: TRPA1, atherosclerosis, foam cell, cholesterol efflux, inflammation
Introduction
Transient receptor potential ankyrin 1 (TRPA1) is a type of non-selective cation channel. This transmembrane channel protein is characterized by a large number of N-terminal ankyrin repeats and is mainly permeable to Ca2+, which suggests its importance in Ca2+ signaling [1, 2]. Although TRPA1 cDNA was initially cloned from cultured human lung fibroblasts [3], the protein is thought to be predominately expressed in sensory neurons. TRPA1 channels play a crucial role in activating sensory neurons and are implicated in acute inflammatory pain and nociception induced by a wide variety of endogenous or exogenous stimuli such as reactive oxygen species (ROS) or allyl isothiocyanate (AITC) [4-6]. Growing evidence suggests that TRPA1 is also expressed in certain types of non-neuronal cells such as endothelial cells and helps regulate the function of cardiovascular system [7-10]. Activation of endothelial TRPA1 channels elicits vasodilation of cerebral arteries via Ca2+-activated K+ channels [7, 10]. The possible involvement of TRPA1 channels in the development of atherosclerosis and the cellular and molecular mechanism are unclear. Further investigation delineating the role and molecular mechanisms of TRPA1 in atherogenesis is warranted.
Complications of atherosclerosis are the most common causes of death from coronary artery diseases and cerebrovascular diseases in developed countries. The most important event of early-stage atherosclerosis is the accumulation of lipid-laden macrophages in the subendothelial space [11, 12]. Both macrophage-mediated cholesterol metabolism and pro-inflammatory cytokine secretion are central steps in the initiation and progression of atherosclerosis [13, 14]. Cholesterol levels of macrophage foam cells are tightly regulated by a complex mechanism that involves internalization of oxidized low-density lipoprotein (oxLDL) and efflux of excess cholesterol [11-14]. Scavenger receptors (SRs) such as SR-A and CD36 are responsible for internalizing oxLDL [15, 16]. In contrast, the efflux of intracellular cholesterol to apolipoprotein AI (apoAI) or high-density lipoprotein (HDL) is mediated by reverse cholesterol transporters such as ATP-binding cassette transporter A1 (ABCA1) or ABCG1 [17-19]. Modulating the functions of SRs and reverse cholesterol transporters has been suggested as a therapeutic strategy for treating or preventing atherosclerosis [11, 19]. However, less is known about the interlocking biology of TRPA1 channels and cholesterol metabolism of macrophage foam cells as well as the development of atherosclerosis.
In this study, we aimed to characterize the role and the potential mechanisms of TRPA1 channels in the development of atherosclerosis in apolipoprotein E-deficient (apoE-/-) mice and the formation of macrophage foam cells. We first investigated the effect of modulating TRPA1 activation on the development of atherosclerosis in HC030031 (TRPA1 antagonist)-treated apoE-/- mice, apoE-/-TRPA1-/- mice or AITC (TRPA1 agonist)-treated apoE-/- mice. We then determined whether ablation of TRPA1 activity by pharmacological inhibition or genetic manipulation affected oxLDL-induced foam-cell formation. Our third aim was to explore the effect of TRPA1 channel activation on the tumor necrosis factor-α (TNF-α)-induced inflammatory response in macrophages.
Materials and methods
Reagents
Human LDL, HC030031, AITC, apoAI, HDL, Oil-red O, Gress's reagent, and mouse antibody for α-tubulin (clone DM1A, 1:10000) were from Sigma-Aldrich (St. Louis, MO, USA). Mouse antibody for TRPA1 (clone 6G8, 1:100 for immunohistochemistry or 1:1000 for western blot) was from Abnova (Taoyuan, Taiwan). Rabbit antibody for ABCG1 (E-20, sc-11150, 1:500) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse antibodies for ABCA1 (AB.H10, ab18180, 1:2000), F4/80 (CI:A3-1, ab6640, 1:100) and α-actin (1A4, ab7817, 1:100) were from Abcam (Cambridge, MA, USA). Macrophage colony stimulating factor (MCSF), TNF-α and ELISA kits for interleukin-1β (IL-1β), monocyte chemoattractant protein-1 (MCP-1), IL-6 and macrophage I nflammatory protein-2 (MIP-2) were from R&D systems (Minneapolis, MN, USA). The ELISA kit for NF-κB activity was from Cayman Chemical (Ann Arbor, MI, USA). Dil-labeled oxLDL was from Biomedical Technologies (Stoughton, MA, USA). NBD-cholesterol was from Cayman Chemical (Ann Arbor, MI, USA). Fluo-8 Ca2+ assay kit was from AAT Bioquest (Sunnyvale, CA, USA). Cholesterol and triglyceride assay kits were from Randox (Crumlin, Co. Antrim, UK). TurboFect was from Fermentas (Glen Burnie, MD, USA).
Mice
The investigation conformed to the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, eighth edition, 2011), and all animal experiments were approved by the Animal Care and Utilization Committee of National Yang-Ming University. Male wild-type (WT) C57BL/6 mice (8 week old) were purchased from the National Laboratory Animal Center, National Science Council (Taipei, Taiwan); male TRPA1-/- and apoE-/- mice (8 week old) were purchased from Jackson laboratory (Bar Harbor, ME). To generate TRPA1-/-apoE-/- mice, TRPA1-/- mice were crossed into the apoE-/- background, and PCR of genomic DNA was used to confirm apoE-/- and TRPA1-/- genotypes. Mice (16 week old) received oral daily treatment with HC030031 (10 mg/kg body weight), AITC (10 mg kg-1 body weight) or vehicle by gastric gavage for 4 weeks. At the end of the experiment, mice were killed by use of CO2. Hearts, aortas were isolated for histological examination and western blot analysis.
Histology and immunohistochemistry
Hearts harvested from mice were fixed with 4% paraformaldehyde, embedded in paraffin, and serially sectioned at 8 μm. To detect the intralesional expression of TRPA1, F4/80 and α-actin, sections underwent immunohistochemical staining. Briefly, the deparaffinized, rehydrated sections were covered with 3% H2O2 for 10 min, then blocked with bovine serum albumin. After blocking, slides were incubated with primary antibodies overnight at 4°C, then with corresponding secondary antibodies for 1 h. DAB was added as chromogen, and slides were viewed under a Motic TYPE 102M microscope. For quantification of atherosclerotic lesions, the lesion size was calculated from the mean of the area sampled from 10 sections as described [20].
Western blot analysis
Bone-marrow-derived macrophages (BMDMs) or aortas were rinsed with PBS, then lysed in immunoprecipitation lysis buffer (50 mmol/L Tris pH 7.5, 5 mmol/L EDTA, 300 mmol/L NaCl, 1% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin and 10 μg/mL aprotinin). Aliquots (50 μg) of cell or aortic lysates were separated on 8% SDS-PAGE. After transfer to membranes, samples were immunoblotted with primary antibodies, then horseradish peroxidase-conjugated secondary antibodies. Bands were revealed by use of an enzyme-linked chemiluminescence detection kit (PerkimElmer, Waltham, MA, USA), and density was quantified by use of Imagequant 5.2 (Healthcare Bio-Sciences, Philadelphia, PA, USA).
Measurement of inflammatory cytokines
The serum concentrations of pro-inflammatory cytokines, including TNF-α, IL-1β, MCP-1, IL-6 and MIP-2, were measured by use of ELISA kits.
Serum lipid profile analysis
Blood was collected by cardiac puncture. After clotting and centrifugation, serum was isolated and serum levels of total cholesterol, high-density lipoprotein cholesterol (HDL-c) and triglycerides were measured by use of the blood biochemical analyzer Spotchem EZ SP 4430 (ARKRAY, Inc., Kyoto, Japan).
Cell culture
BMDMs were prepared as described [20]. Briefly, wild-type (WT) mice were killed by CO2 exposure, and mononuclear cells from femurs were harvested by Percoll (1.073 gcm-3) density gradient centrifugation. The cells were seeded in minimum essential medium α (MEMα) supplemented with 50 ng/mL MCSF, 10% fetal bovine serum (FBS), and penicillin (100 U/mL)/streptomycin (100 μg/mL) (HyClone, Logan, UT) at 37°C for 5 d. Human embryonic kidney 293 cells (HEK293, ATCC® CRL-1573™) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, penicillin (100 U/mL)/streptomycin (100 μg/mL) at 37ºC.
LDL modification
LDL was exposed to CuSO4 (5 μmol/L) for 24 h at 37°C and Cu2+, then removed by extensive dialysis. The extent of modification was determined by measuring thiobarbituric acid-reactive substances (TBARs). OxLDL containing approximately 30-60 nmol TBARs defined as malondialdehyde equivalents per milligram LDL protein was used for experiments.
Plasmid construction and transient transfection
The coding region for the human TRPA1 DNA fragment was cloned into a pCMV5 N-Flag vector with MluI and HindIII restriction sites. The sequence of isolated DNA fragments was confirmed by sequence analysis. TurboFect was used for transient transfection experiments according to the manufacturer's instructions. Briefly, 1 μg of vector or TRPA1 plasmid was transfected into HEK293 cells, which were used in further experiments.
Measurement of [Ca2+]i level
Ca2+ assay was performed according to the manufacturer's protocol. Briefly, BMDMs were seeded in 24-well plates and grown for 24 h. Cells were then washed and Fluo-8 NE dye-loading solution was added for 1 h at room temperature. Medium was replaced with fresh medium containing test compounds. Fluorescence was measured by fluorometry (Molecular Devices, Sunnyvale, CA, USA) with 490-nm excitation and 525-nm emission.
Oil-red O staining
BMDMs were fixed with 4% paraformaldehyde and stained with 0.5% Oil-red O. Hematoxylin was used for counterstaining.
Cholesterol and triglycerides measurement
Cellular cholesterol and triglycerides were extracted by use of hexane:isopropanol (3:2, vol/vol). After cellular debris was removed, the supernatant was dried under nitrogen flush. The levels of cholesterol and triglycerides were measured with use of cholesterol and triglyceride assay kits.
Dil-oxLDL binding assay
Dil-oxLDL, labeled with red fluorescence, is used to measure oxLDL binding to SRs of macrophages [20]. Briefly, BMDMs were treated with concentrations of evodiamine or capsaicin for 24 h, then incubated with Dil-labeled oxLDL (10 μg/ml) for an additional 4 h at 4°C. After a washing with phosphate-buffered saline (PBS), cell lysates were analyzed by fluorometry (Molecular Devices, Sunnyvale, CA) at 540-nm excitation and 590-nm emission.
Cholesterol efflux assay
BMDMs were treated with concentrations of HC030031 or AITC for 12 h, then underwent equilibration with NBD-cholesterol (1 μg/mL) for an additional 6 h. NBD-cholesterol-labeled cells were washed with PBS and incubated in MEMα for 6 h with apoAI (10 μg/ml) or HDL (50 μg/ml). Fluorescence-labeled cholesterol released from cells into the medium was measured by use of a multilabel counter (PerkinElmer, Waltham, MA, USA) at 485-nm excitation and 535-nm emission. Cholesterol efflux was expressed as percentage fluorescence in the medium relative to total fluorescence (cells and medium). In vivo cholesterol efflux was examined as described [20]. Briefly, after removing the apoB-containing lipoproteins from plasma, the supernatant as the HDL fraction was diluted to 2.8% (equivalent to 2% serum) in cultured medium. Cholesterol efflux was measured as the difference in release of fluorescence-labeled cholesterol from vehicle-treated BMDMs or TO901317-treated BMDMs.
Measurement of nitrite production
The level of nitrite (NO2-) in cultured medium was evaluated by incubating an equal volume of Griess reagent for 15 min. The color intensity of Azo dye was examined by SP-8001 UV/VIS spectrophotometry (Metertech, Taipei, Taiwan) with absorbance at 540 nm.
Measurement of DNA-binding activity of NF-κB
The DNA-binding activity of NF-κB was measured by use of non-radioactive ELISA kit. Briefly, the cell lysates were prepared and added into wells coated with specific dsDNA sequence bound with NF-κB. After incubation for overnight at 4°C, specific antibody against NF-κB was added and incubated for overnight at 4°C, then with corresponding secondary antibodies for 1 h. After colorimetric reaction, the color intensity was examined by SP-8001 UV/VIS spectrophotometry with absorbance at 450 nm.
Statistical analysis
Results are presented as mean ± SD from 10 mice or 5 independent experiments. Mann-Whitney U test was used to compare 2 independent groups. The Kruskal-Wallis test followed by Bonferroni post-hoc analysis was used to account for multiple testing. SPSS 20.0 (SPSS Inc, Chicago, IL) was used for analysis. Differences were considered statistically significant at P < 0.05.
Results
Role of TRPA1 activation in development of atherosclerosis in apoE-/- mice
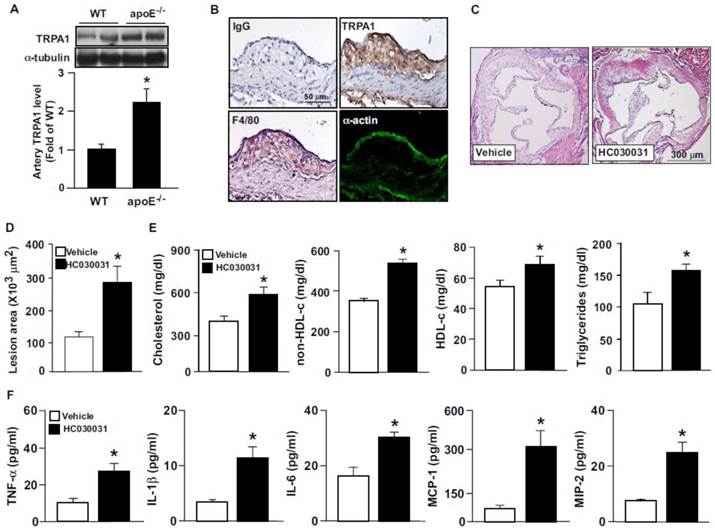
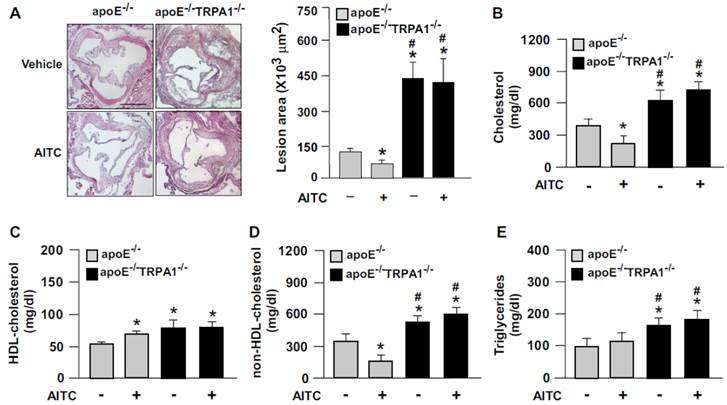
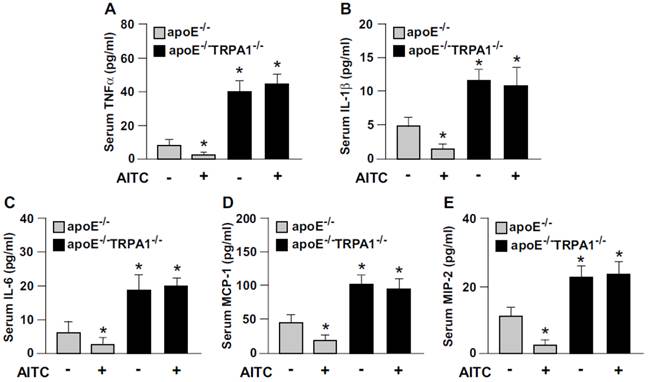
To elucidate the possible contribution of TRPA1 channels in atherogenesis, we first investigated the expression of TRPA1 channels in normal and atherosclerotic aortas. The protein level of TRPA1 channels was markedly higher in apoE-/- than wild-type mouse aortas (Fig. 1A). Immunohistochemical staining revealed the expression of TRPA1 channels in aortic endothelial cells, with positive signals restricted mainly to macrophage areas in atherosclerotic lesions of apoE-/- mouse aortas (Fig. 1B). To determine the functional significance of TRPA1 channels in atherogenesis, 4-month-old apoE-/- mice received daily oral treatment with the TRPA1 antagonist HC030031 for 4 weeks. As compared with vehicle treatment, HC030031 treatment enlarged atherosclerotic lesion area in aortas (Fig. 1C-D). In addition, serum levels of total cholesterol, non-HDL-c, HDL-c, and triglycerides (Fig. 1E) as well as pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, MCP-1 and MIP-2 (Fig. 1F) were increased. Similarly, genetic deletion of TRPA1 in apoE-/- mice exacerbated atherosclerotic lesion area, hyperlipidemia and systemic inflammation as compared with apoE-/- mice alone (Fig. 2). Moreover, with agonist AITC treatment to activate TRPA1 channels, apoE-/- mice but not apoE-/-TRPA1-/- mice showed significantly reduced atherosclerotic lesion area (Fig. 2A) and serum levels of total cholesterol, non-HDL-c and triglycerides (Fig. 2B-E), TNF-α, IL-1β, IL-6, MCP-1 and MIP-2 (Fig. 3A-E). Collectively, these findings suggest that TRPA1 activation may play a crucial role in the development of atherosclerosis.
TRPA1 is expressed in atherosclerotic lesions and inhibition of TRPA1 activity with HC030031 exacerbates hyperlipidemia, inflammation and atherosclerosis in apoE-/- mice. Aortas were collected from apolipoprotein E knockout (apoE-/-) mice and subjected to (A) western blot analysis to evaluate the protein level of TRPA1 and α-tubulin or (B) immunohistochemistry. Immunostaining was with normal rabbit IgG, anti-TRPA1, anti-F4/80 antibody, a macrophage marker, or anti-α-actin, a vascular smooth muscle cell marker. Cell nuclei were stained with hematoxylin. Bar = 50 μm. Four-month-old apoE-/- mice received daily treatment with HC030031 (TRPA1 antagonist, 10 mg/kg body weight) or DMSO (vehicle control) by gastric gavage for 4 weeks. (C and D) Representative sections of aortic roots with H&E staining from each group of mice and quantification of atherosclerotic lesion areas. (E) Quantification of serum levels of total cholesterol, high-density lipoprotein (HDL) cholesterol, non-HDL cholesterol and triglycerides. (F) ELISA of serum levels of tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), IL-6, monocyte chemotactic protein 1 (MCP-1) and macrophage inflammatory protein 2 (MIP-2). Data are mean ± SD from 10 mice. *, P < 0.05 vs. WT mice or vehicle treatment.
Genetic ablation of TRPA1 channel function abolishes the atheroprotective effect of allyl isothiocyanate (AITC) in apoE-/- mice. Four-month-old apoE-/- or apoE-/-TRPA1-/- mice received daily treatment with AITC (TRPA1 agonist, 10 mg/kg body weight) or DMSO (vehicle control) by gastric gavage for 4 weeks. (A) Immunohistochemistry and quantification of atherosclerotic lesion area in mice. (B-E) Quantification of serum levels of total cholesterol, HDL cholesterol, non-HDL cholesterol and triglycerides. Data are mean ± SD from 10 mice. *, P < 0.05 vs. vehicle-treated apoE-/- mice, #, P < 0.05 vs. AITC-treated apoE-/- mice.
Loss of function of TRPA1 channels reverses the anti-inflammatory effect of AITC in apoE-/- mice. ELISA of serum levels of (A) TNF-α, (B) IL-1β, (C) IL-6, (D) MCP-1, and (E) MIP-2. Data are mean ± SD from 10 mice. *, P < 0.05 vs. vehicle-treated apoE-/- mice.
OxLDL activates TRPA1 channels in macrophages
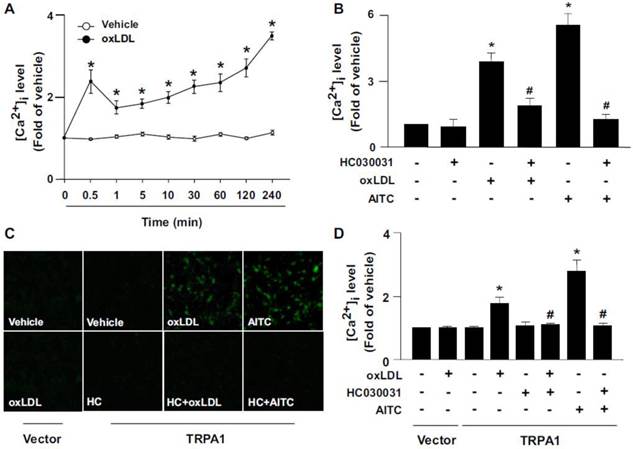
TRPA1 is activated by several oxidative stimuli [4,5], so we next examined the effect of oxLDL, the most important player in the initiation and progression of atherogenesis, on the expression and activation of TRPA1 in BMDMs. In response to oxLDL exposure, the intracellular level of Ca2+ ([Ca2+]i) in BMDMs, as reflected by the intensity of Ca2+-sensitive Fluo-8 fluorescence, rapidly peaked at 30 sec, slightly dropped at 1 min and gradually increased to peak again at 4 h post-stimulation (Fig. 4A). Importantly, the oxLDL-induced increase in [Ca2+]i at 30-sec post-stimulation was prevented by pretreatment with HC030031 (Fig. 4B). We then investigated the specificity of TRPA1 channels and found that exposing BMDMs to AITC also increased [Ca2+]i at 30 sec, which was abolished by HC030031 pretreatment (Fig. 4B). To further study whether oxLDL could directly activate TRPA1, we used a gain-of-function strategy to measure Ca2+ permeability of TRPA1 channels by re-expressing full-length TRPA1 in HEK293 cells, which lack TRP channels. Treatment with oxLDL or AITC significantly increased the intracellular level of Ca2+ in TRPA1-re-expressed HEK293 cells but not vector-transfected HEK293 cells (Fig. 4C-D). Blocking TRPA1 activation by HC030031 abrogated oxLDL- or AITC-induced Ca2+ influx in TRPA1-re-expressed HEK293 cells (Fig. 4C-D). Collectively, these findings indicate that TRPA1-mediated Ca2+ influx might be an important event in the formation of macrophage foam cells.
Oxidized low-density lipoprotein (oxLDL) triggers TRPA1-dependent Ca2+ influx in macrophages or TRPA1 re-expressed HEK293 cells. (A) Bone marrow-derived macrophages (BMDMs) were treated with or without oxLDL (50 μg ml-1) for the indicated times. (B) BMDMs were pretreated with or without HC010031 (HC, 10 μM) for 1 h, then oxLDL (50 μg/ml) or AITC (10 μM) for an additional 0.5 min. Intracellular Ca2+ level was measured by Fluo-8 calcium assay kit with green fluorescence detection. (C and D) HEK293 cells were transfected with vector or TRPA1 plasmid for 24 h, then treated with or without oxLDL (50 μg/ml), HC030031 (HC, 10 μM) or AITC (10 μM) for 10 min. Images were photographed by fluorescence microscopy and quantified. Data are mean ± SD from 5 independent experiments. *, P < 0.05 vs. vehicle-treated group, #, P < 0.05 vs. oxLDL-treated group.
Role of TRPA1 in oxLDL-induced lipid accumulation in macrophage foam cells
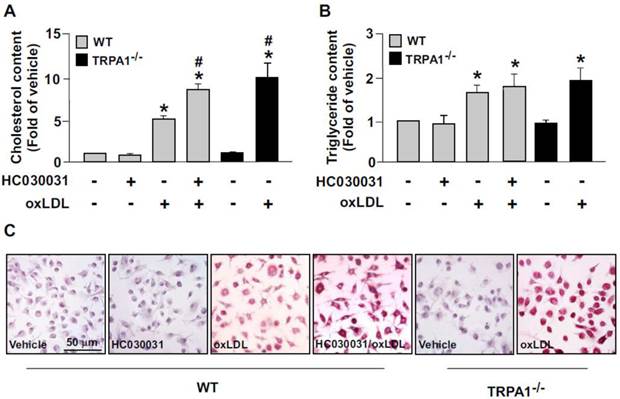
We then determined the functional significance of TRPA1 in foam-cell formation in BMDMs. Pretreatment with HC030031 or deletion of TRPA1 channels in macrophages aggravated the oxLDL-induced lipid accumulation and cellular levels of cholesterol and triglycerides (Fig. 5). These results suggest that activation of TRPA1 channels by an agonist may protect against the formation of macrophage foam cells.
Ablation of TRPA1 channel function increases oxLDL-induced cholesterol accumulation in macrophages. Wild-type (WT) or TRPA1-/- BMDMs were pretreated with or without HC030031 (10 μM) or for 1 h, then with 50 μg/ml oxLDL for 24 h. (A and B) Intracellular levels of cholesterol and triglycerides were extracted by use of hexane/isopropanol (3/2, v/v) and analyzed by using colorimetric assay kits. (C) Cells were fixed with 4% paraformaldehyde, then stained with Oil-red O. Cellular nuclei were stained with hematoxylin. Bar = 50 μm. Data are mean ± SD from 5 independent experiments. *, P < 0.05 vs. vehicle-treated group, #, P < 0.05 vs. oxLDL-treated group.
Role of TRPA1 channels in cholesterol metabolism
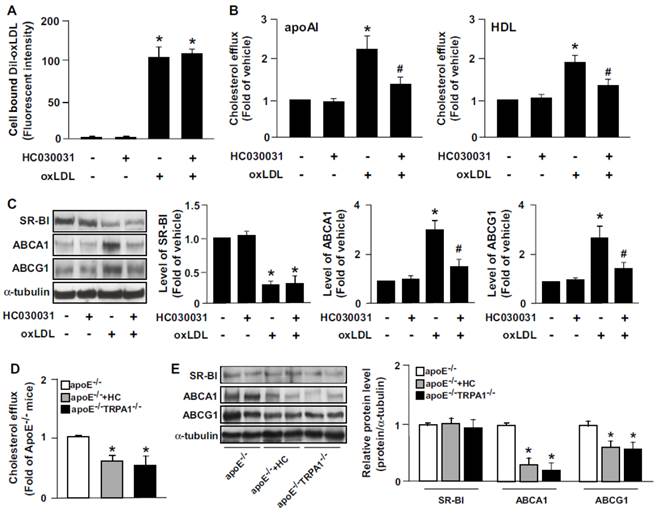
We then elucidated the role of TRPA1 channels in oxLDL internalization and cholesterol efflux. Pretreatment with the TRPA1 antagonist HC030031 in BMDMs did not alter oxLDL binding (Fig. 6A) but decreased apoAI- or HDL-dependent cholesterol efflux (Fig. 6B). SR-BI, ABCA1 and ABCG1 have a crucial role in reverse cholesterol transport during foam-cell formation [12,13]. Therefore, we delineated the mechanisms by which inhibiting TRPA1 activity increased lipid accumulation by examining the alterations in levels of these transporters. Treatment with HC030031 decreased the oxLDL-increased protein levels of ABCA1 and ABCG1, but oxLDL-decreased protein level of SR-BI was not affected (Fig. 6C). Our in vivo results demonstrate that inhibiting TRPA1 with HC030031 or deleting TRPA1 channels in apoE-/- mice impaired the capacity for reverse cholesterol efflux (Fig. 6D). In addition, inhibiting TRPA1 by HC030031 or deleting TRPA1 in apoE-/- mice did not affect the protein expression of SR-BI but decreased the aortic protein levels of ABCA1 and ABCG1 (Fig. 6E). Collectively, these findings suggest that the suppression of intracellular lipid accumulation by TRPA1 activation is likely due to increased ABC-dependent cholesterol efflux.
Inhibition of TRPA1 activity decreases oxLDL-induced cholesterol efflux and upregulation of ABCA1 and ABCG1. (A) For Dil-oxLDL binding assay, WT BMDMs were treated with or without HC030031 (10 μM) for 18 h, followed by 10 μg/ml Dil-oxLDL for an additional 4 h. Cellular lysates were analyzed by fluorometry. (B) Macrophages were treated with HC030031 (10 μM) for 12 h, then NBD-cholesterol (1 μg/ml) for an additional 6 h in the presence of oxLDL, apoAI (10 μg/ml) or HDL (50 μg/ml). Cholesterol efflux was expressed as a percentage of fluorescence in the medium relative to the total amount of fluorescence. Data are mean ± SD from 5 independent experiments. (C) Western blot analysis of SR-BI, ABCA1 and ABCG1 protein levels. Data are mean ± SD from 5 independent experiments. *, P < 0.05 vs. vehicle-treated group, #, P < 0.05 vs.oxLDL-treated group. (D) The capacity of reverse cholesterol efflux of apoE-/-, HC-treated apoE-/- and apoE-/-TRPA1-/- mice. (E) Western blot analysis and quantitation of protein levels of SR-BI, ABCA1 and ABCG1 in aortas of apoE-/-, HC-treated apoE-/- and apoE-/-TRPA1-/- mice. Data are mean ± SD from 10 mice. *, P < 0.05 vs. apoE-/- mice.
Activation of TRPA1 channels confers protection against the TNFα-induced inflammatory response
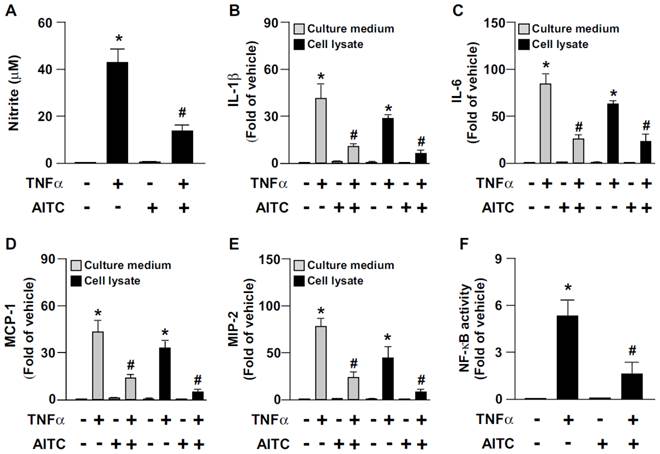
TNF-α-induced inflammation is a key event in the progression of atherosclerosis [21]. We next examined the effect of AITC activation of TRPA1 on the TNFα-induced inflammatory response in macrophages. The TNFα-increased production of NO, IL-1β, IL-6, MCP-1, MIP-2 and NF-κB in BMDMs was significantly decreased by AITC treatment (Fig. 7). These results suggest that the TRPA1 activation has an anti-inflammatory action in macrophages.
AITC activation of TRPA1 channels prevents TNF-α-induced inflammation in macrophages. WT BMDMs were pretreated with or without AITC (10 μM) for 1 h, then incubated with TNF-α (10 ng/mL) for an additional 18 h (A-E) or 30 min (F). (A) Griess's assay of levels of nitrite, (B-E) ELISA of IL-1β, MCP-1, IL-6 and MIP-2 in cultured medium and cell lysates. (F) ELISA of cellular NF-kB activity. Data are mean ± SD from 5 independent experiments. *, P < 0.05 vs. vehicle-treated group, #, P < 0.05 vs. TNF-α-treated group.
Discussion
In this study, we characterized a new function of TRPA1 and its potential mechanism in atherosclerosis and in the biology of macrophage foam cells. Our in vivo study demonstrated that chronic treatment with the TRPA1 antagonist HC030031 significantly increased the size of atherosclerotic lesions, hyperlipidemia and systemic inflammation of apoE-/- mice. Moreover, genetic deletion of TRPA1 increased the serum levels of cholesterol, pro-inflammatory cytokines and chemokines and aggravated the progression of atherosclerosis in apoE-/- mice. Thus, TRPA1 has an intrinsic function in limiting the progression of atherosclerosis. As well, the ability of TRPA1 to restrain atherogenesis appeared to be amplified by chronic treatment of apoE-/- mice with the TRPA1 agonist AITC, because this exogenous agonist reduced the pathological onset of atherosclerosis features measured above. However, the therapeutic effect of AITC in atherosclerosis was not seen in TRPA1-/-apoE-/- mice, which suggests that TRPA1 activation may have therapeutic value for treating atherosclerosis.
However, our findings regarding the crucial role of TRPA1 in atherogenesis are inconsistent with those of Engel et al. and Nassini et al. who found that inhibition of TRPA1 channel activity by genetic ablation or treatment with specific pharmacological antagonists decelerated the development of inflammatory diseases such as dextran sulfate sodium-induced colitis and cigarette smoke-induced lung inflammation [22, 23]. Nevertheless, much remains to be learned about the detailed cellular and molecular mechanisms underlying TRPA1-mediated regulation of lipid metabolism and the inflammatory response during the development of atherosclerosis.
TRPA1 expression in atherosclerotic lesions mainly appeared in macrophage foam cell areas. We used a cell culture system to investigate how macrophage-associated TRPA1 level acts as a key regulator in the transformation of foam cells. The most important atherogenic molecule, oxLDL, increased intracellular Ca2+ level via TRPA1 in macrophages. This increased intracellular Ca2+ level then contributed to upregulation of ABCA1 and promotion of cholesterol efflux, which in turn regulated the cholesterol homeostasis of macrophage foam cells. This notion was further supported by HC030031-inhibited TRPA1 channels downregulating the expression of ABCA1, decreasing cholesterol efflux and thereby promoting oxLDL-induced lipid accumulation in macrophages. In addition, pretreatment with AITC attenuated the TNF-α-induced inflammation. These observations suggest that macrophage TRPA1 may participate in the atherogenic molecule-induced deregulation of cholesterol metabolism and inflammation as well as the development of atherosclerosis via the function of influx Ca2+.
Atherosclerosis is considered a disorder of cholesterol metabolism and a chronic inflammatory disease resulting from LDL oxidation induced by ROS [24, 25]. With atherogenic stimuli, ROS are released from vascular cells and further cause lipid peroxidation and the inflammatory response, thereby leading to the progression of atherosclerosis [26, 27]. TRPA1 is a sensor of ROS and endogenous inflammatory mediator in various cell types [4, 5, 28]. These stimuli can directly activate TRPA1 channels by covalent modification of cysteine residues located in the ankyrin repeat domain of the N-terminus of these channels [29, 30]. In addition, several oxidants or lipid metabolites such as hydrogen peroxide and 15-deoxy-Δ12,14-prostaglandin J2 can target these sites and activate TRPA1 channels [31]. ROS can promote the oxidative modification of both lipid and protein composition of LDL and thus generate various oxidative products within oxLDL [24-27]. In this study, we found that oxLDL activated macrophage TRPA1 channels; however, we cannot verify the potential contribution of the oxidative products to oxLDL-induced activation of TRPA1 channels observed in this study.
TRPA1 channel was originally thought to be uniquely expressed in primary nociceptive sensory neurons and mediated inflammatory pain in response to chemical irritants [32, 33]. Recently, emerging evidence has suggested that Ca2+ signaling mediated by TRPA1 channels in non-neuronal cells regulates the physiological functions or pathological response of the cardiovascular system under different conditions [7-10]. For instance, the TRPA1 channel involved in the regulation of vascular tone was originally thought to be mediated mainly through the release of vasodilator neuropeptides from these sensory fibers after activation of TRPA1 channels located at these fiber terminals [7, 8]. However, an elegant study by Earley et al. reported that endothelial TRPA1 channels may be an important sensor and regulator in AITC-induced artery dilation [7]. Furthermore, Yanaga et al. demonstrated cinnamaldehyde (a TRPA1 agonist)-induced vasorelaxant action via endothelium-dependent or -independent mechanisms [34]. Nevertheless, whether TRPA1 is also engaged in the atheroprotective effects of endothelium-derived nitric oxide is largely unknown.
In this study, we discovered a link between TRPA1, inflammation, cholesterol metabolism and atherosclerosis; inhibiting TRPA1 channels by HC030031 decreased the efficacy of cholesterol efflux, thereby promoting macrophage foam cell formation. Moreover, AITC-activated TRPA1 prevented TNF-α-induced inflammation in macrophages. This notion was further supported by our in vivo studies that loss of TRPA1 channel function aggravated hyperlipidemia and systemic inflammation and ultimately, accelerated the progression of atherosclerosis, which broadens the cardiovascular disease-related implication of TRPA1. The therapeutic effects with activation of TRPA1 channels in vivo seem to be widespread, but the target cells for AITC have not yet been identified. Therefore, our in vivo data cannot exclude that nerve system-mediated regulation is also involved in AITC-conferred protection against atherosclerosis. According to the findings from our and other studies, the collaboration of multiple physiological pathways in different organs might be required for the TRPA1-dependent anti-atherogenic effects.
Deregulation of cholesterol metabolism is the most critical factor for the development of atherosclerosis [11-14]. ABC transporters are known to play a crucial role in regulating cholesterol homeostasis [35, 36]. For instance, ABCA1 and ABCG1 promote cholesterol efflux and reverse cholesterol transport (RCT) by exporting lipid to apoAI or HDL to maintain cholesterol homeostasis and protect arteries against atherosclerosis [17-19]. Genetic deletion of ABCA1 or ABCG1 in mice leads to the accumulation of excessive cholesterol in peripheral tissues and macrophages and promotes the progression of atherosclerosis [37, 38]. In contrast, physiological upregulation of ABCA1 or ABCG1 expression by cytokines, nutrition or pharmacological reagents impedes the progression of atherosclerosis [35, 36, 39]. Recently, emerging evidence indicates that ABCA1 and ABCG1 have functions independent of lipid export into the RCT pathway. These two ABC transporters have critical roles in regulating the inflammatory response and mobilization of hematopoietic stem and multipotential progenitor cells (HSPCs) [40, 41]. Deficiency of ABCA1 and ABCG1 in macrophages increased inflammation and accelerated atherosclerosis in LDLR-/- mice and ABCA1-/-ABCG1-/- mice showed increased HSPC mobilization from bone marrow to spleen [41]. Additionally, ABCA1-/-, ABCG1-/- or ABCA1-/-ABCG1-/- macrophages showed increased levels of pro-inflammatory mediators in response to toll-like receptor ligands [42]. Moreover, the Ca2+ signaling pathway is reported to be involved in regulating gene expression and protein stability of ABCA1 in macrophages [43, 44]. Indeed, we demonstrated that inhibiting TRPA1 channel activation by HC030031 or genetic ablation increased the lipid accumulation of macrophage foam cells by decreasing ABC transporter-mediated cholesterol efflux, thereby aggravating systemic inflammation and atherosclerosis in apoE-/- mice. In view of their function, the upregulation of ABCA1 and ABCG1 by AITC observed in this study may be attributed to the anti-atherogenic effects conferred by activation of TRPA1 channels.
The TRP family has at least 28 isoforms and many serve as a cellular sensor in response to a wide spectrum of physical or chemical stimili and play a central role in regulating pathophysiological functions [1, 2]. For example, TRPV1 channels are an important integrator in vascular physiology and in the pathogenesis of cardiovascular diseases [43, 45, 46]. Activation of TRPV1 permits Ca2+ entry, which increases intracellular Ca2+ level, then activates signaling pathway cascades, thereby leading to the adaptive response of endothelial nitric oxide synthase (eNOS) and cholesterol metabolism in endothelial cells or macrophages [44-46]. Additionally, TRPV1 channels mediate simvastatin-, 14,15-epoxyeicosatrienoic acid- or epigallocatechin-3-gallate-induced eNOS activation and angiogenesis in ECs [47-49]. Previously, activation of TRPV1 channels was found to retard the development of hypertension and atherosclerosis, and their inhibition accelerated the progression of these diseases [45]. To be functional cation channels, TRP subunits have to form homo- or hetero-tetramers in response to multiple triggers for initiating activation of TRP channels, thereby increasing intracellular Ca2+ level and activating downstream molecules including kinases, phosphatase or other TRP channels [1, 2]. As well, the N-terminus of TRPV1 or TRPA1 channels contains several ankyrin repeats that would bind with intracellular proteins such as calmodulin and AMP-activated protein kinase to activate the downstream signaling pathways upon stimulation [45, 50]. Recently, we reported that simvastatin activates the TRPV1-TRPA1 signaling pathway, thereby promoting the formation of the CaMKII-AMPK-eNOS complex and activating eNOS, NO production and, ultimately, angiogenesis in endothelial cells [47]. However, whether these molecules and TRPA1 work in concert to modulate cholesterol metabolism, inflammation and development of atherosclerosis remains to be investigated.
In conclusion, we demonstrate a novel TRPA1-dependent protective property in atherosclerosis. Inhibition of TRPA1 channel activity exacerbated the progression of atherosclerosis, and activation of TRPA1 channels suppressed atherosclerosis in hyperlipidemic mice by decreasing hyperlipidemia and systemic inflammation. We discover a link between TRPA1, cholesterol metabolism, inflammation, foam-cell formation and atherosclerosis. Our findings suggest a new therapeutic target for treating or preventing atherosclerosis and related metabolic disorders.
Acknowledgements
The authors thank Laura Smales for help in language editing. We thank Dr. Chien-Yu Chen for technical assistance.
Sources of Funding
This study was supported by grants from Ministry of Science and Technology (102-2628-B-010-001-MY3, 103-2628-B-010-040-MY3 and 104-2811-B-010-020) and Ministry of Education, Aim for the Top University Plan, Taiwan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bodkin JV, Brain SD. Transient receptor potential ankyrin 1: emerging pharmacology and indications for cardiovascular biology. Acta Physiol (Oxf). 2011;203:87-98
2. Koivisto A. Sustained TRPA1 activation in vivo. Acta Physiol (Oxf). 2012;204:248-254
3. Jaquemar D, Schenker T, Trueb B. An ankyrin-like protein with transmembrane domains is specifically lost after oncogenic transformation of human fibroblasts. J Biol Chem. 1999;274:7325-7333
4. Sawada Y, Hosokawa H, Matsumura K. et al. Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci. 2008;27:1131-1142
5. Julius D. TRP channels and pain. Annu Rev Cell Dev Biol. 2013;29:355-384
6. Steinberg X, Lespay-Rebolledo C, Brauchi S. A structural view of ligand-dependent activation in thermoTRP channels. Front Physiol. 2014;5:171
7. Earley S, Gonzales AL, Crnich R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ Res. 2009;104:987-994
8. Inoue R, Jian Z, Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther. 2009;123:371-385
9. Pozsgai G, Bodkin JV, Graepel R. et al. Evidence for the pathophysiological relevance of TRPA1 receptors in the cardiovascular system in vivo. Cardiovasc Res. 2010;87:760-768
10. Earley S. Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels. J Cardiovasc Pharmacol. 2011;57:148-153
11. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801-809
12. Peng N, Meng N, Wang S. et al. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E⁻/⁻ mice. Sci Rep. 2014;4:5519
13. Yan P, Xia C, Duan C. et al. Biological characteristics of foam cell formation in smooth muscle cells derived from bone marrow stem cells. Int J Biol Sci. 2011;7:937-946
14. Chang MY, Yang YS, Tsai ML. et al. Adenovirus-mediated prothymosin α gene transfer inhibits the development of atherosclerosis in ApoE-deficient mice. Int J Biol Sci. 2014;10:358-366
15. Zhong Q, Zhao S, Yu B. et al. High-density lipoprotein increases the uptake of oxidized low density lipoprotein via PPARγ/CD36 pathway in inflammatory adipocytes. Int J Biol Sci. 2015;11:256-265
16. Zhao JF, Shyue SK, Lee TS. Excess Nitric Oxide Activates TRPV1-Ca(2+)-Calpain Signaling and Promotes PEST-dependent Degradation of Liver X Receptor α. Int J Biol Sci. 2016;12:18-29
17. Tsai JY, Su KH, Shyue SK. et al. EGb761 ameliorates the formation of foam cells by regulating the expression of SR-A and ABCA1: role of haem oxygenase-1. Cardiovasc Res. 2010;88:415-423
18. Song X, Fischer P, Chen X. et al. An apoA-I mimetic peptide facilitates off-loading cholesterol from HDL to liver cells through scavenger receptor BI. Int J Biol Sci. 2009;5:637-646
19. Daniels TF, Killinger KM, Michal JJ. et al. Lipoproteins, cholesterol homeostasis and cardiac health. Int J Biol Sci. 2009;5:474-488
20. Chen CY, Ching LC, Liao YJ. et al. Deficiency of glycine N-methyltransferase aggravates atherosclerosis in apolipoprotein E-null mice. Mol Med. 2012;18:744-752
21. Liu YC, Zou XB, Chai YF. et al. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10:520-529
22. Engel MA, Leffler A, Niedermirtl F. et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology. 2011;141:1346-1358
23. Nassini R, Pedretti P, Moretto N. et al. Transient receptor potential ankyrin 1 channel localized to non-neuronal airway cells promotes non-neurogenic inflammation. PLoS One. 2012;7:e42454
24. Ding Z, Liu S, Wang X. et al. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep. 2013;3:1077
25. Nomura J, Busso N, Ives A. et al. Xanthine oxidase inhibition by febuxostat attenuates experimental atherosclerosis in mice. Sci Rep. 2014;4:4554
26. Graham A, Hogg N, Kalyanaraman B. et al. Peroxynitrite modification of low-density lipoprotein leads to recognition by the macrophage scavenger receptor. FEBS Lett. 1993;330:181-185
27. Zhao JF, Shyue SK, Lin SJ. et al. Excess nitric oxide impairs LXR(α)-ABCA1-dependent cholesterol efflux in macrophage foam cells. J Cell Physiol. 2014;229:117-125
28. Leeuwenburgh C, Hardy MM, Hazen SL. et al. Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J Biol Chem. 1997;272:1433-1436
29. Trebak M, Ginnan R, Singer HA. et al. Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid Redox Signal. 2010;12:657-674
30. Macpherson LJ, Dubin AE, Evans MJ. et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541-545
31. Ogawa H, Takahashi K, Miura S. et al. H(2)S functions as a nociceptive messenger through transient receptor potential ankyrin 1 (TRPA1) activation. Neuroscience. 2012;218:335-343
32. Pokorski M, Takeda K, Sato Y. et al. The hypoxic ventilatory response and TRPA1 antagonism in conscious mice. Acta Physiol (Oxf.). 2014;210:928-938
33. Takahashi N, Mori Y. The O2-sensing TRPA1 channel illustrates the significance of vagal nerves in cardio-respiratory adaptation to hypoxia. Acta Physiol (Oxf.). 2014;210:705-707
34. Yanaga A, Goto H, Nakagawa T. et al. Cinnamaldehyde induces endothelium-dependent and -independent vasorelaxant action on isolated rat aorta. Biol Pharm Bull. 2006;29:2415-2418
35. Lee TS, Lu KY, Yu YB. et al. β common receptor mediates erythropoietin-conferred protection on oxLDL-induced lipid accumulation and inflammation in macrophages. Mediators Inflamm. 2015;2015:439759
36. Tsou CY, Chen CY, Zhao JF. et al. Activation of soluble guanylyl cyclase prevents foam cell formation and atherosclerosis. Acta Physiol (Oxf.). 2014;210:799-810
37. Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156-1166
38. Yvan-Charvet L, Ranalletta M, Wang N. et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900-3908
39. Zhao JF, Ching LC, Huang YC. et al. Molecular mechanism of curcumin on the suppression of cholesterol accumulation in macrophage foam cells and atherosclerosis. Mol Nutr Food Res. 2012;56:691-701
40. Lu KY, Ching LC, Su KH. et al. Erythropoietin suppresses the formation of macrophage foam cells: role of liver X receptor alpha. Circulation. 2010;121:1828-1837
41. Murphy AJ, Akhtari M, Tolani S. et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138-4149
42. Westerterp M, Gourion-Arsiquaud S, Murphy AJ. et al. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012;11:195-206
43. Yvan-Charvet L, Welch C, Pagler TA. et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837-1847
44. Zhao JF, Ching LC, Kou YR. et al. Activation of TRPV1 prevents oxLDL-induced lipid accumulation and TNF-α-induced inflammation in macrophages: role of liver X receptor α. Mediators Inflamm. 2013;2013:925171
45. Wei J, Ching LC, Zhao JF. et al. Essential role of transient receptor potential vanilloid type 1 in evodiamine-mediated protection against atherosclerosis. Acta Physiol (Oxf). 2013;207:299-307
46. Ching LC, Kou YR, Shyue SK. et al. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc Res. 2011;91:492-501
47. Su KH, Lin SJ, Wei J. et al. The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta Physiol (Oxf). 2014;212:191-204
48. Su KH, Lee KI, Shyue SK. et al. Implication of transient receptor potential vanilloid type 1 in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. Int J Biol Sci. 2014;10:990-996
49. Guo BC, Wei J, Su KH. et al. Transient receptor potential vanilloid type 1 is vital for (-)-epigallocatechin-3-gallate mediated activation of endothelial nitric oxide synthase. Mol Nutr Food Res. 2015;59:646-657
50. Ching LC, Chen CY, Su KH. et al. Implication of AMP-activated protein kinase in transient receptor potential vanilloid type 1-mediated activation of endothelial nitric oxide synthase. Mol Med. 2012;18:805-815
Author contact
![]() Corresponding authors: Dr. Tzong-Shyuan Lee, DVM, PhD, Department of Physiology, School of Medicine, National Yang-Ming University, Taipei, 11221 Taiwan; Tel: +886-2-2826-7365; Fax: +886-2-2826-4049; E-mail: tsleeedu.tw or Dr. Tse-Min Lu, MD, PhD, Division of Cardiology, Department of Internal Medicine, Taipei Veterans General Hospital, Taipei, 11221, Taiwan; Tel: +886-2-28757511, Fax: +886-2-28753580, E-mail: tmlugov.tw
Corresponding authors: Dr. Tzong-Shyuan Lee, DVM, PhD, Department of Physiology, School of Medicine, National Yang-Ming University, Taipei, 11221 Taiwan; Tel: +886-2-2826-7365; Fax: +886-2-2826-4049; E-mail: tsleeedu.tw or Dr. Tse-Min Lu, MD, PhD, Division of Cardiology, Department of Internal Medicine, Taipei Veterans General Hospital, Taipei, 11221, Taiwan; Tel: +886-2-28757511, Fax: +886-2-28753580, E-mail: tmlugov.tw