Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(10):1191-1202. doi:10.7150/ijbs.16437 This issue Cite
Research Paper
Expression of Iron Regulatory Protein 1 Is Regulated not only by HIF-1 but also pCREB under Hypoxia
Qian-Qian Luo1,2, Zhong-Ming Qian1,2 ![]() , Yu-Fu Zhou2,3, Meng-Wan Zhang2,3, Dang Wang1, Li Zhu1
, Yu-Fu Zhou2,3, Meng-Wan Zhang2,3, Dang Wang1, Li Zhu1 ![]() , Ya Ke2,3
, Ya Ke2,3 ![]()
1. Department of Biochemistry, Institute for Nautical Medicine, Nantong University, Nantong, 226001, China
2. Laboratory of Neuropharmacology, FudanUniversity School of Pharmacy,826 Zhang Heng Road, Pu Dong, Shanghai201203, China
3. School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, NT, Hong Kong.
Received 2016-6-9; Accepted 2016-7-18; Published 2016-9-15
Abstract

The inconsistent of responses of IRP1 and HIF-1 alpha to hypoxia and the similar tendencies in the changes of IRP1 and pCREB contents led us to hypothesize that pCREB might be involved in the regulation of IRP1 under hypoxia. Here, we investigated the role of pCREB in IRP1 expression in HepG2 cells under hypoxia using quantitative PCR, western blot, immunofluorescence, electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP). We demonstrated that 1) Hypoxia increased pCREB levels inside of the nucleus; 2) Putative CREs were found in the IRP1 gene; 3) Nuclear extracts of HepG2 cells treated with hypoxia could bind to CRE1 and CRE3, and 100-fold competitor of putative CREs could abolish the binding activity to varying degrees; 4) pCREB was found in the CRE1 and CRE3 DNA-protein complexes of EMSA; 5) CRE1 and CRE3 binding activity of IRP1 depended on CREB activation but not on HIF-1; 6) Increased IRP1 expression under hypoxia could be prevented by LY294002; 7) ChIP assays demonstrated that pCREB binds to IRP1 promoter; and 8) HIF-1 and/or HIF-2 siRNA had no effect on the expression of pCREB and IRP1 proteins in cells treated with hypoxia for 8 hours. Our findings evidenced for the involvement of pCREB in IRP1 expression and revealed a dominant role of PI3K/Akt pathway in CREB activation under hypoxia and also suggested that dual-regulation of IRP1 expression by HIF-1 and pCERB or other transcription factor(s) under hypoxia might be a common mechanism in most if not all of hypoxia-inducible genes.
Keywords: Hypoxia, iron regulatory proteins 1, hypoxia-inducible factor-1 (HIF-1), cyclic AMP-responsive element-binding protein (CREB), HepG2 cells.
Introduction
The transcription factor hypoxia-inducible factor-1 (HIF-1) is a master regulator responsible for the induction of genes that facilitate adaptation and survival of cells and the whole organism from normoxia to hypoxia [1]. This factor consists of the HIF-1 alpha and HIF-1 beta subunits and is expressed in many cell types [2]. Under the conditions of hypoxia, HIF-1 alpha is relocated into the nucleus where it binds HIF-1 beta to form the active HIF-1 [3]. The heterodimer can bind to a specific DNA consensus sequence hypoxia response element (HRE), which is contained in the promoter or enhancer regions of the target genes [4,5].
A number of genes operated in all cells have been identified as regulated by HIF-1 in response to hypoxia [6,7]. Adaptation to hypoxia in cells and tissues leads to the transcriptional induction of a series of genes, including several with important functions in iron metabolism [8]. It has been demonstrated that hypoxia can increase iron uptake by cells as well as the expression of transferrin (Tf) and transferrin receptor 1 (TfR1), both of which have been identified to be hypoxia-inducible genes [9,10]. Furthermore, several other proteins that are involved in iron transport or regulation, such as ceruloplasmin [11], iron regulatory protein 1 (IRP1) [12] and 2 (IRP2) [13], hepcidin [14] and divalent metal transporter1 (DMT1) [15,16], have also been reported to be regulated by hypoxia.
Iron is an essential trace element for many aspects of the physiological activities of mammalian cells including oxygen transport, cell growth and cell survival. Both iron deficiency and iron excess can lead to cellular dysfunctions, therefore maintaining normal iron homeostasis is crucial [17,18]. The same as iron regulation hormone hepcidin, IRP1 is also a key protein involved in the regulation of iron homeostasis. The regulation and management of iron at the cellular level is primarily done by the two proteins TfR1 and ferritin (Ft). In most types of cells, the coordinated control of the two primary proteins TfR1 and Ft by cellular iron is mediated by IRP1 [19,20]. It has been reported that hypoxia inhibited degradation of HIF-1 alpha, leading to an increase in content of HIF-1 which down-regulated IRP1 expression [21]. However, we recently found that hypoxia induced a decrease first and then an increase, rather than a stable decrease, in IRP1 expression. Obviously, the increased expression of IRP1 in the late phase of hypoxia cannot be explained by HIF-1 because HIF-1 returns to almost normal level by this phase. This led us to speculate that IRP1 expression might be also controlled by other factor(s) in addition to HIF-1under hypoxia.
The transcription factor cyclic AMP response element-binding protein (CREB) is crucial for stimulus-transcription coupling [22]. This protein has been proposed to be involved in various cellular processes such as protein translation, ribosome biogenesis, proliferation, and differentiation by up-regulating transcription of a large number of genes [23]. Phosphorylation of CREB (pCREB) on the transcriptional regulatory site Ser133 has been demonstrated to be an important mechanism of ischemic tolerance in neurons [24-26]. The increased CREB phosphorylation has been associated with various pathological conditions including hypoxia [27-29] and also plays an important role in the protection of the cells against ischemia [30-31]. Our preliminary data showed that there is a very similar tendency in the changes of IRP1 levels and pCREB contents during the late phase of hypoxia. Based on the above, we hypothesized that pCREB might also play a role in the regulation of IRP1 expression in addition to HIF-1 under the conditions of hypoxia. In the present study, we tested this hypothesis and demonstrated for the first time that IRP expression is regulated not only by HIF-1 but also pCREB under hypoxia.
Materials and methods
Reagents and antibodies
Unless otherwise stated, all chemicals including β-actin were obtained from the Sigma Chemical Company, St. Louis, MO, USA. Primary anti-rabbit IRP1 antibody was bought from Abcam, Cambridge, UK; anti-rabbit pCREB (Ser133) and anti-rabbit CREB from Cell Signaling Technology, Danvers, Massachusetts, USA; and primary anti-HIF-1 alpha antibody from Santa Cruz Biotechnology, Dallas, Texas, USA. Protein extraction Kit, H-89 (PKA inhibitor) and LY294002 (PI3K inhibitor) were obtained from Beyotime, Nantong, JS, China; RNA Extraction Kit from Qiagen, Dusseldorf, Nordrhein-Westfalen, Germany; RevertAidTM First Strand cDNA Synthesis Kit from Fermentas, Wien, Austria; AlexaFluor488-conjugated secondary antibody from Invitrogen, Carlsbad, CA, USA; goat anti-rabbitor anti-mouse IgG-HRP secondary antibody and BCA kit from Pierce, Rockford, Illinois, USA.pGL3-Basic and pcDNA 3.1(+) vectors were purchased from Promega, Madison, Wisconsin, USA; CHIP kits from Millipore, Billerica, MA, USA; and biotin-labeled probes from IDT Company, San Jose, CA, USA.
Cell culture and hypoxia treatment
All cell culture media and reagents were purchased from Gibco, Grand Is., NY, USA. The human hepatoma cell line HepG2 cells (HB 8065), purchased from American Type Culture Collection, Rockville, MD, USA, were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum in a humidified 5% CO2 incubator (Forma 3111, Thermo, Marietta, Georgia, USA) at 37oC. The cells were placed in Hank's medium without glucose, and then exposed to hypoxia (94% N2, 5% CO2, and 1% O2) for 4, 8, 12, or 24 hours at 37oC in an incubator (Ruskin, Biotrace, Bridgend, UK) as previously described [32-33]. Control cultures were exposed to normoxia only. In some experiments, the cells were pre-treated with 20 mMH-89 or 20 mM ofLY294002, and then incubated in DMEM medium containing 10% serum for 1 hour before hypoxia.
Assessment of cell viability
The cell viabilities of the HepG2 cells that received different treatments were measured using an MTT (3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolinum bromide) assay as previously described [34]. Briefly, 25 μL MTT (1 g/L in PBS) was added to each well before incubation at 37 °C for 4 hours. The assay was stopped by the addition of a 100 μl lysis buffer (20% SDS in 50% N'Ndimethylformamide, pH 4.7). Optical density (OD) was measured at the 570 nm and 630 nm wavelengths by the use of an ELX-800 microplate assay reader (Bio-Tek, Winooski, VT, USA) and the results were expressed as a percentage of the absorbance measured in the control cells.
Western blot analysis
The nuclear and cytosolic proteins were extracted using a protein extraction kit and the protein contents were determined using a BCA kit. Equivalent amounts of protein (40 μg) were loaded into each well and separated by 10% SDS-polyacrylamide gels, then transferred to 0.45 μm polyvinylidenedifluoride membranes (Millipore), blocked with 5% nonfat milk in TBST, and incubated overnight at 4oC with the primary antibodies: IRP1 (1:1000), HIF-1 alpha (1:200), pCREB (Ser133) (1:1000), CREB (1:1000), β-actin (1:10000), before being incubated with goat anti-rabbitor anti-mouse IgG-HRP secondary antibody for 2 hours at room temperature. Finally, the complexes were visualized by enhanced chemiluminescence (Pierce) and exposed to X-ray film (Kodak).
Total RNA extraction and quantitative PCR
Total RNA was extracted from HepG2 cells using the RNA extraction Kit. First-strand cDNA was generated with the RevertAidTM First Strand cDNA Synthesis Kit. IRP1 mRNA expression was quantified by real-time PCR. The reaction was carried out with the FAM/Sybr system (Rotor-Gene 3000, Rcorbeet Research, Mortlake, NSW, Australia). IRP1 mRNA were determined by the 2ΔΔCt method according to Livak and Schmittgen [35] and normalized to normal IRP1 mRNA levels. The primer sequences used in PCR were described in a previous study [16].
Immunofluorescence
HepG2 cells were plated at a density of 1.0 x 105cells/cm2 on gelatin/poly-D-lysine-coated glass cover slips. The cells that received different treatments were fixed in 4% paraformaldehyde for 30 min. After being washed with 0.01M phosphate-buffered saline (PBS) for three times, the cells were incubated in PBS containing 3% bovine serum albumin (BSA) for 2 hours and then with primary antibody against pCREB (1:1000) overnight at 4oC. The immunoreactivity was detected using the AlexaFluor488-conjugated secondary antibody (1:1000) for 2 hours at room temperature. A nucleus staining was performed by using Hoechst 33342 (5 mg/ml) as described previously [36]. The glass covers were mounted with 50% glycerin. The images were observed under a confocal laser microscope (Leica, Germany).
Electrophoretic mobility shift assay and supershift(EMSA)
EMSA was used to determine whether nuclear extracts of HepG2 cells could bind to the putative cAMP response element (CRE) sequences of IRP1. The positive probe (CRE consensus sequence) and the putative CRE sequences (CRE1, CRE2, and CRE3) in the 5'-flanking region of IRP1 promoter were synthesized and used as oligonucleotide probes for EMSA (Table 1). CRE2 and CRE3 both contain three putative CREs in the probes sequences. Equal amounts of nuclear proteins (13.0 μg) for each sample were incubated with 45 pmol of biotin-labeled probe. Finally, a picture was obtained in the chemical system (Alpha Innotech Fluorchem, Silicon Valley, CA, USA). Cold competition control was performed by adding 100-fold excess of each unlabeled DNA fragment along with the biotin-labeled probe. The DNA-protein complexes were separated by electrophoresis on 5% native polyacrylamide gels. For supershift assays, nuclear extracts were co-incubated with the indicated specific anti-CREB antibodies (7 μg/μL) and biotin-labeled oligonucleotide probes.
Nucleotide sequences of oligonucleotide probes used in EMSA & supershift analysis.
| Element (abbreviation) | Sequences (5'-3') |
|---|---|
| Positive probe (Consensus CRE) | 5'-AGAGATTGCCTGACGTCAGACAGCTAG-3' |
| CRE1 | 5'-AACTGAATAACGTGGAAAGAGTC-3' |
| CRE2 | 5'-AGTACCGACGCGGCCCGACCCACGTCCCC-3' |
| CRE3 | 5'-GGGGACGTGTTTCTGGCGACTGACGTCTCC-3' |
NOTE: Positive CRE and the putative IRP1 promoter 5'-flanking region CRE nucleotides are represented in bold.
Plasmids construction
Seven different fragments in IRP1 promoter were amplified from genomic DNA (GenBank accession number AL161783.20) by PCR using the primers given in Table 2 and cloned into the pGL3-Basic vectors. The constructs pBI-CRE1 and pBI-CRE3 contained 194 bp and 637 bp fragments respectively, which included the putative CREs from -852 up to -832, +146 to +321 (set the translation initiation site of NM_001278352.1 as +1). CREB CDs region was amplified from CREB mRNA by the primers given in Table 2 and cloned into the pcDNA 3.1(+) vectors which formed the pcDNA-CREB plasmid. pcDNA-HIF-1 plasmid was constructed as described previously [21].
Primer sequences.
| Name | A. Primers used for cloning of IRP1 CRE reporter constructs |
|---|---|
| pBI-CRE1 sense | 5'GGGGTACCCCAGGTGGTTGGAAGAGC3' |
| pBI-CRE1 antisense | 5'CCGCTCGAGTGAAAGGTGGTGAGGTT3' |
| pBI-CRE3 sense | 5'GGGGTACCCCCTGCTTGGGTCAGGTT3' |
| pBI-CRE3 antisense | 5'CCGCTCGAGATGCCATTATGATGTGCT3' |
| Name | B. Primers used for cloning of CREB expression plasmid |
| CREB sense | 5'CGCGGATCCATGACCATGGAATCTGG3' |
| CREB antisense | 5'TGCTCTAGATTAATCTGATTTGTGGC3' |
Chromatin immunoprecipitation (ChIP)
ChIP assays were performedin HepG2 using CHIP kits (Millipore). The cells were treated with or without hypoxia for 8 hours. After stimulation, cells were cross-linked by being added directly to the medium formaldehyde to a final concentration of 1% at room temperature. After 10 min, ice-cold PBS was added and plates were placed on ice, washed extensively with PBS, and scraped. After centrifugation, cells were lysed in SDS Lysis buffer supplemented with proteases inhibitor II. Chromatin was sheared by sonication, resulting in DNA fragments between 150 and 200 bp in size. After centrifugation, 1% of the sample was kept as Input and then diluted 10 times in dilution buffer supplemented with protease inhibitor II. Extracts were precleared for 1 hour at 4°C using 200 µl sheared sample DNA and 60 μL of protein G Agarose. Immunoprecipitations were carried out overnight at 4°C with 1 μg of each antibody. In parallel, supernatants were incubated with normal rabbit IgG as negative controls. Immune complexes were collected with protein G Agarose and washed three times (5 min each) with low salt buffer, high salt buffer, LiCL buffer and two times with TE buffer. Immune complexes were extracted with 1% SDS (v/v), 0.1 M NaHCO3, and heated for 5 hours at 65°C to reverse the cross-linking. After proteinase K digestion (2 hours at 45°C), DNA fragments were purified on Spin columns in 50 μL of EB (elution buffer) and 2 μL was used in each Q-PCR. Sequences of promoter-specific primers are available in Table 3 and were designed to specifically amplify proximal promoter regions containing three binding sites for pCREB discussed in the text.
Primer sequences.
| Name | A. Primers used for CHIP assay of IRP1 promoter regions |
|---|---|
| CRE1 sense | 5' GGTGTAGGCTTTGGTGTC 3' |
| CRE1 antisense | 5' ACTGCCCTCCTATTCTGC 3' |
| CRE2 sense | 5' TGCTTGGGTCAGGTTC3' |
| CRE2 antisense | 5' GAGGGATACAGGGTGG3' |
| CRE3 sense | 5' TCCACCCTGTATCCCTC3' |
| CRE3 antisense | 5' CCAATCCCTGGTTTCTC 3' |
HIF-1 and HIF-2 siRNA
HepG2 cells were plated into 6-well plates 1 day before transfection. The cells were grown to 50% confluence and then transfected with human HIF1-1, 5'-r(CUGGACACAGUGUGUUUGA)d(TT)-3'; HIF1-2, 5'-r(CUGAUGACCAGCAACUUGA)d(TT)-3'; HIF2-1, 5'-r(GCUCUUCGCCAUGGACACA)d(TT)-3'; HIF2-2, 5'-r(GCGACAGCUGGAGUAUGAA)-d(TT)-3' specific siRNA duplex, or negative control siRNA duplex, 5'-r(UUCUCCGAACGUGUCACGU)d(TT)-3' and 5'-r(ACGUGACACGUUCGGAGAA)d(TT)-3' (GenePharma, Shanghai, China) using the micropolysiRNA transfection reagent (Maikerui, Nantong, JS, China) according to the manufacturer's instruction.
Statistical analysis
All data were presented as mean± standard error (SEM). Statistical analyses were performed using SPSS software for Windows (version 13.0) (SPSS, Inc., Chicago, IL). The differences between the means were all determined by two-way analysis of variance (ANOVA). A probability value of p < 0.05 was taken to be statistically significant.
Results
Effects of hypoxia on cell viabilities, expression of IRP1 mRNA and protein, and contents of HIF-1alpha and pCREB
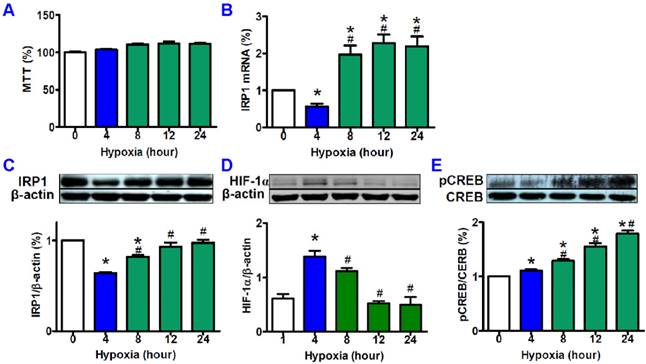
MTT assay showed that treatment with hypoxia (1% O2) for the different times (0, 4, 8, 12, and 24 hours) did not induce any significant differences in cell viabilities (Fig. 1A). Quantitative PCR demonstrated that hypoxia could induce a significant reduction first and then a remarkable increase in IRP1 mRNA expression. IRP1 mRNA levels in HepG2 cells treated with 4 hours of hypoxia were markedly lower than those in the control cells as well as the cells treated with hypoxia for 8, 12 and 24 hours (Fig. 1B). Western blot analysis found that the tendencies in the changes of IRP1 protein expression induced by different times of hypoxia were very similar to those of IRP1 mRNA expression, and IRP1 protein levels in HepG2 cells treated with 4 hours of hypoxia also being significantly lower than those in the control cells as well as the cells treated with hypoxia for 8, 12 and 24 hours (Fig. 1C). Western blot analysis also demonstrated that hypoxia led to a significant increase at 4 hours of hypoxia and then a decrease with the time of treatment in HIF-1 alpha contents in HepG2 cells (Fig. 1D). HIF-1 alpha contents treated with 4 hours of hypoxia were significantly higher than those in the control cells as well as the cells treated with hypoxia for 8, 12 and 24 hours. There were no differences in the contents of HIF-1 alpha between the cells treated with 12 or 24 hours of hypoxia and the control cells. In addition, it was found that pCREB (pCREB/CREB) level progressively increased with the treatment times of hypoxia until 24 hours (Fig. 1E).
Effects of hypoxia on cell viabilities, expression of IRP1 mRNA and protein, and contents of HIF-1α and pCREB. HepG2 cells were exposed to hypoxia (1% O2) for different times (0, 4, 8, 12, and 24 hours) and then cell viabilities (A, n=6) and IRP1 mRNA (B, n=3) expression were measured by MTT assay and Quantitative PCR respectively, and the contents of IRP1 protein (C, n=3), HIF-1α (D, n=3) and pCREB (E, n=3) were determined by Western blotting analysis as described in “Methods”. The data were presented as mean ± SEM (n=3). *P<0.05 versus the control (0 hour hypoxia), # P<0.05 versus 4 hours of hypoxia.
The increased pCREB induced by hypoxia is mainly located inside of the nucleus
The inconsistency in the responses of IRP1 (mRNA and protein) and HIF-1 alpha to hypoxia suggested that IRP1 (mRNA and protein) expression is not controlled by HIF-1 alpha only. The very similar tendencies in the changes of IRP1 mRNA and protein levels and also pCREB contents during the later period of hypoxia (8-24 hours) implied that pCREB might play a role in regulation of IRP1 expression at the transcriptional level under the conditions of hypoxia. To test this possibility, we then investigated the sub-cellular distribution of the increased pCREB induced by hypoxia. This investigation was conducted because the proposed transcriptional regulation of IRP1 by the increased pCREB should be located inside of the nucleus where pCREB could bind to its possible target gene IRP1. Immunostaining analysis showed that treatment with hypoxia for 8 hours enhanced the staining of pCREB (green) in the nuclei (blue) in HepG2 cells (Fig. 2), indicating a significant increase in the level of pCREB inside of the nucleus. This finding provided the first piece of evidence for the possible involvement of pCREB in the regulation of IRP1 expression at the transcriptional level under the conditions of hypoxia.
The increased pCREB induced by hypoxia is mainly located inside of the nucleus. HepG2 cells were treated with hypoxia for 0, 4 or 8 hours and then incubated with primary antibody against pCREB (1:1000) overnight at 4oC. The immunoreactivity(green) was detected using the AlexaFluor488-conjuagated secondary antibody (1:1000) for 2 hours at room temperature. A nucleus staining (blue)was performed by using Hoechst 33342 (5 mg/ml) as described in “Methods”. Bar =50 µm.

Activated CREB binds to IRP1 gene in HepG2 cells
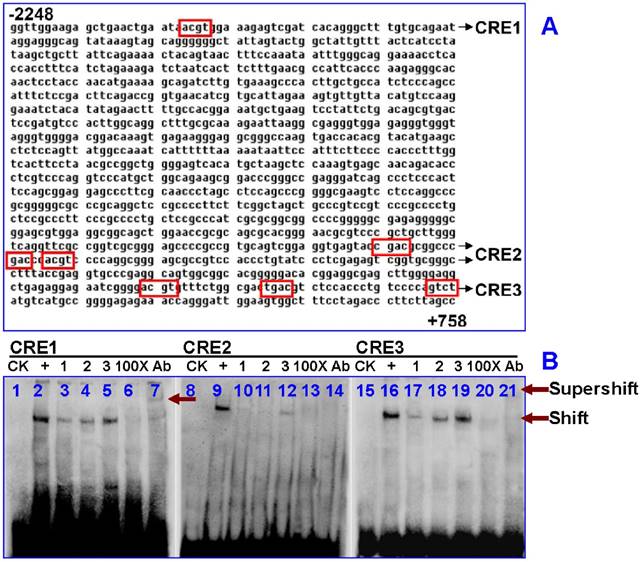
Next, we analyzed the IRP1 gene using several pieces of software available online. The longest sequence of promoter 5'-flanking region (from -2248 to +752 sites) was identified. Using genomatixmat Inspector software (37), seven putative CREs (TGACGTCA) were found (-852/-832, +146/+166, +149/+169, +283/+303, +298/+318, +300/+320, +301/+321) in the IRP1 gene. Here, we refer to -852/-832 as CRE1, +146/+169 as CRE2, +283/+321 as CRE3 (Fig. 3A). We then assessed whether nuclear extracts of HepG2 cells treated with hypoxia could bind to these three putative CREs sequences of IRP1 using EMSA. The shifted complexes of CRE1 or CRE3 probes were observed in the cells after 4 or 8 hours of hypoxia treatment (Fig. 3B, line 4, 5, 18, 19). The intensities of the shifted bands of CRE1 (Fig. 3B, line 5) or CRE3 (Fig. 3B, line 19) probes in the cells treated with hypoxia for 8 hours were very similar to those of the positive control (Fig. 3B, line 2, 9, 16). The binding specificity of the three CREs probes was determined by competition experiments. Addition of 100-fold competitor (unlabeled DNA fragment) to the three putative CREs (CRE1, CRE2, and CRE3) abolished the binding activities to varying degrees (Fig. 3B, line 6, 13, 20). To further investigate whether there was pCREB in the DNA-protein complexes of EMSA, we tested to see if pCREB antibodies could specifically combine to the DNA-protein complexes. The results showed that pCREB antibodies could induced a supershift of the DNA-protein complexes in the nuclear extracts of HepG2 cells treated with hypoxia for 8 hours (Fig. 3B, line 7, 14, 21). The results provide evidence for the existence of pCREB in the CRE1 and CRE3 DNA-protein complexes.
Analysis of the binding activity of the putative CREs in the IRP1 gene in HepG2 cell. A. Schematic diagram of the seven putative CREs (box) in 5'-flanking region of IRP1 promoter region (CRE1: -852/-832, CRE2: +146/+169, CRE3: +283/+321). B. EMSA was done with biotin-labeled CRE, CRE1, CRE2 and CRE3 oligonucleotides in the presence of nuclear extracts from HepG2 cells treated with 1%O2for 4 or 8 hours. CK = control check, + = positive control which used bio-labeled CRE consensus sequences as a positive probe, 1 = normoxia control, 2 = hypoxia for 4 hours, 3 = hypoxia for 8 hours, 100× = cold competition control, and Ab = Supershift with pCREB antibody at 8 hours of hypoxia.
Hypoxia increases transcriptional activity of IRP1
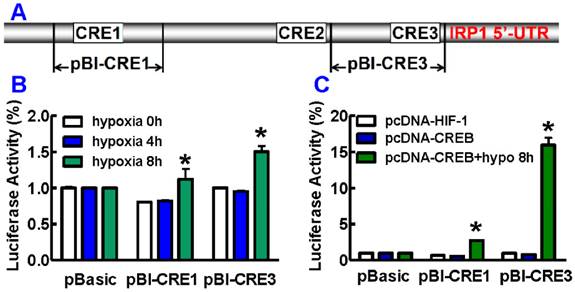
We also investigated the effect of pCREB on the regulation of IRP1 transcription under hypoxia by transfecting HepG2 cells with the reporter plasmids containing CRE1 or CRE3 (Fig. 4A). It was found that hypoxia for 8 hours induced a significant increase in luciferase reporter activity in cells transfected with pBI-CRE1 or pBI-CRE3 as compared with the normoxic control (hypoxia 0 hour), while hypoxia for 4 hours did not lead to such an increase in activity (Fig. 4B). In addition, we did not find any changes in luciferase reporter activity in the cells co-transfected with pBI-CRE1 or 3 and pcDNA-CREB or pcDNA-HIF-1 under normoxia. However, hypoxia for 8 hours resulted in a remarkable enhancement in luciferase reporter activity in cells co-transfected with pBI-CRE1 or 3 and pcDNA-CREB (Fig. 4C). The luciferase reporter activity was significantly higher in cells (co-transfected with pBI-CRE1 or 3 and pcDNA-CREB) treated with hypoxia than that in cells treated without hypoxia (Fig. 4C).
Promoter location of CREs region in the human IRP1 promoter. A. Schematic diagram of the test DNA plasmids. B. HepG2 cells were transfected with test DNA combined with pRL-TK control vector DNA. 36 or 44 hours after transfection, cells were cultured under 21% or 1% oxygen for 4 or 8 hours. Luciferase assays were measured using the dual luciferase reporter system. C. CREB over-expression under hypoxia for 8 hours induces pCREB-induced IRP1 promoter activity. The values represent means ± SEM (n=3). *P<0.05 versus normoxia control.
The increased IRP1 expression induced by pCREB under hypoxia could be prevented by LY294002
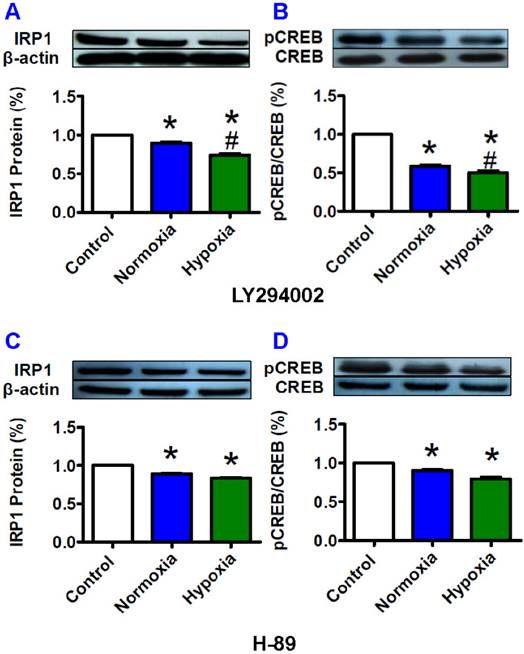
Furthermore, we investigated the effects of H-89 (PKA inhibitor) and LY294002 (PI3K inhibitor) on the expression of IRP1 protein to find out whether inhibition of pCREB could prevent the increase in expression of IRP1 protein under hypoxia. The HepG2 cells were pre-treated with the two inhibitors for 1 hour before being exposed to hypoxia for 8 hours. We demonstrated that LY294002 (Fig. 5A and B) and H-89 (Fig. 5C and D) both induced a significant reduction in IRP1 protein and pCREB under the conditions of not only normxia but also hypoxia. We also found that hypoxia led to a further reduction in IRP1 protein (Fig. 5A) and pCREB (Fig. 5B) levels in the cells treated with LY294002 but not with H-89 (Fig. 5C and D). The levels of IRP1 protein and pCREB both were significantly lower in the cells treated with LY294002 plus hypoxia than those in the cells treated with LY294002 plus normoxia. In the case of H-89, such a difference was not found. Total amounts of CREB were found to remain largely stable under different conditions (Fig. 5B and D).
Effects of LY294002 (PI3K inhibitor) and H-89 (PKA inhibitor) on IRP1 protein expression and pCREB content in HepG2 cells under hypoxia. HepG2 cells were pretreated with LY294002 (20 mM) or H-89 (20 mM) for 1 hour and then treated with hypoxia for 8 hours. The levels of IRP1 (A and C) and pCREB (B and D) were measured by Western blotting analysis as described in “Methods”. The data were presented as mean ± SEM (n=3). *P<0.05 versus the control, #P<0.05 versus normoxia.
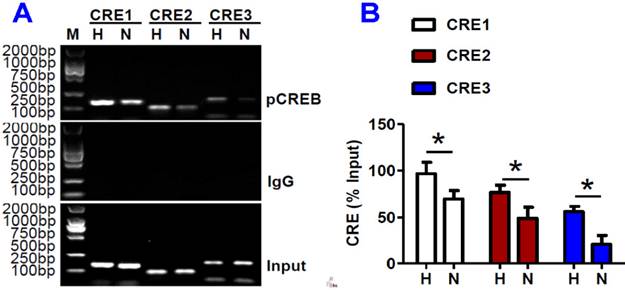
ChIP assays demonstrated that pCREB binds to IRP1 promoter
We further assessed the binding activity of the three putative CREs with pCREB in HepG2 cells under hypoxia by ChIP analysis. The interaction of pCREB and CRE1 (-852/-832), CRE2 (+146/+169) or CRE3 (+283/+321) in IRP1 promoter regions was all found to be significantly higher in HepG2 cells treated with hypoxia for 8 hours than in the controls (Fig. 6). This showed that hypoxia could lead to a marked increase in the binding activity of the three putative CREs with pCREB, and also provided evidence that pCREB can bind to IRP1 promoter to increase IRP1 mRNA transcription.
ChIP assays demonstrated that pCREB binds to IRP1 promoter. HepG2 cells were treated with hypoxia (H) or normoxia (N) for 8 hours and ChIP assays were conducted as described in Materials and Methods. A: ChIP assays to show that pCREB binds to IRP1 promoter (M = DNA marker). The bands of 168 bp, 112bp and 185bp contain CRE1, CRE2 and CRE3 in the IRP1 promoter gene; respectively. B. Data are presented as mean±SEM (% Input), n=3. *P <0.05 versus H (normoxia).
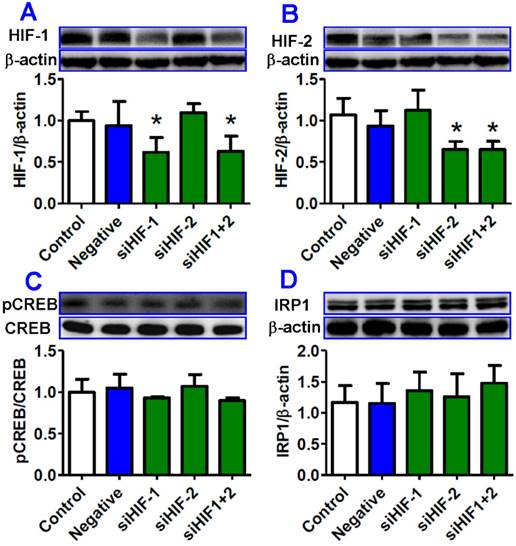
Transfection with HIF-1 and/or HIF-2 siRNA had no significant effect on the expression of pCREB and IRP1 proteins in HepG2 cells treated with hypoxia
Finally, we investigated the effects of transfection with HIF-1and/or HIF-2 interference sequences on pCREB and IRP1 expression in HepG2 cells under hypoxia for 8 hours. Western blot analysis showed that HIF-1 and HIF-2 expression in cells in siHIF-1, siHIF-2 or siHIF-1+2 groups was significantly lower than that in the cells in the control or negative groups (Fig. 7A and B). There were no significant differences in the expression of pCREB protein between the cells transfected with siHIF-1, siHIF-2 or siHIF-1+2 and the control or negative cells (Fig. 7C), indicating that inhibition of HIF-1 and/or HIF-2 has no effect on the expression of pCREB protein. Expression of IRP1 protein was found to be slightly higher in cells transfected with siHIF-1, siHIF-2 or siHIF-1+2 than that in the cells in the control or negative groups (Fig. 7D), though there were no significant differences among them.
Transfection with HIF-1 and/or HIF-2 siRNA had no significant effect on expression of pCREB and IRP1 proteins in HepG2. cells treated with hypoxia. HepG2 cells were transfected with interference sequences of HIF-1 (siHIF-1), HIF-2 (siHIF-2), HIF-1 and HIF-2 (siHIF-1+2) or negative control (Negative) under normoxia for 36 hours and then treated by hypoxia for 8 hours. The expression of HIF-1 (A), HIF-2 (B), PCREB and CREB (C), and IRP1 (D) proteins was measured by Western blotting. *P <0.05 versus Control.
Discussion
Studies have concluded that IRP1 is one of the hypoxia-inducible genes [12,13,21]. In the present study, we showed that the contents of HIF-1 alpha are significantly higher and levels of IRP1 mRNA and protein lower in the cells treated with hypoxia than those in the controls in the early phase of hypoxia (4 hours). The findings are consistent with what has reported by others [12,21] and also provided further evidence for the conclusion that hypoxia could inhibit degradation of HIF-1 alpha, leading to an increase in HIF-1 content which down-regulates IRP1 expression. However, same as what we found in our preliminary investigation, we also demonstrated that levels of IRP1 mRNA and protein both gradually increased with time during the period from 8 to 24 hours of hypoxia, while the contents of HIF-1 alpha significantly decreased at 8 hours of hypoxia as compared with the levels at 4 hours of hypoxia, almost returning to the normal levels at 12 and 24 hours of hypoxia. These findings implied that the increased expression of IRP1 in the late phase (8-24 hours) of hypoxia should not be relevant to HIF-1.
At the same time, it was found that the tendencies in the changes of IRP1 mRNA and protein levels are very similar to those of pCREB contents during the late phase of hypoxia, and pCREB contents also gradually increased with time during the period from 8 to 24 hours of hypoxia. The very similar tendencies in the changes of IRP1 and pCREB contents suggested that pCREB, in addition to HIF-1, might also be involved in the control of IRP1 expression under the conditions of hypoxia. The findings obtained from the present study provide solid evidence for this view. First, immunostaining analysis demonstrated that treatment with hypoxia for 8 hours dramatically enhanced pCREB levels inside of the nucleus. Second, analysis by using genomatix-mat Inspector software revealed the existence of the putative CREs in the IRP1 gene. Third, EMSA indicated that nuclear extracts of HepG2 cells treated with hypoxia could bind to CRE1 and CRE3, and the competition experiment showed that 100-fold competitor of the putative CREs could abolish the binding activities to varying degrees. Fourth, the supershift experiment evidenced the existence of pCREB in the CRE1 and CRE3 DNA-protein complexes of EMSA. Fifth, dual-luciferase assay demonstrated that CRE1 and CRE3 binding activity of IRP1 depended on CREB activation but not on HIF-1 at the later phase of hypoxia, with CRE3 which contains three CREs being more active than CRE1. Sixth, ChIP assays demonstrated that pCREB bound to IRP1 promoter. Finally, transfection with HIF-1 and/or HIF-2 siRNA had no significant effect on expression of pCREB and IRP1 proteins in HepG2 cells treated with hypoxia for 8 hours. These findings plus studies on the role of HIF-1 in IRP1 expression support the notion that expression of IRP1 is regulated by not only HIF-1 but also pCREB under hypoxia. HIF-1 plays its role in the early phase, while pCREB plays its role in the late phase of hypoxia.
It has been documented that CREB could bind to HRE of some target genes of HIF-1, for example plasminogen activator inhibitor-1 [38]. In addition, the core sequence of HIF-1 binding site HRE (RCGTG) was similar to that of CREB binding site (CRE) (TGACGTCA). In the present study, we investigated the effect of HIF-1 on the transcription of pBI-CRE1 and pBI-CRE3, which have a functional HRE, and demonstrated that over-expression of HIF-1 could not affect the binding activities of pBI-CRE1 and pBI-CRE3. We also constructed an over-expression plasmid of CREB and found that the luciferase activity did not changed between pGL-3Basic control and pBI-CRE1/3 under normoxic condition. However, 8 hours of hypoxia was found to induce a significant increase in the transcription activities of pBI-CRE1 and pBI-CRE3. These findings support the possibility that CRE might also function as HRE in the IRP gene, thus preventing HIF-1 from binding to its responsive element HRE after CREB binds to CRE. If true, this is likely why the over-expression of HIF-1 could not affect the binding activities of pBI-CRE1 and pBI-CRE3 in the cells.
It has been reported that CREB will be activated when being phosphorylated at Ser133 by various kinases, including protein kinase A (PKA) [39], protein kinase C [40], calmodulin kinases II and IV [41,42], pp90RSK [43], microtubule-activated protein kinase-activated protein 2 [44], protein kinase Akt/PKB [45] and phosphoinositide 3-kinase (PI3K)/Akt [46]. In order to identify the signaling pathway involved in the hypoxia-induced increase in pCREB under our experimental conditions, we also investigated the effects of LY294002 (PI3K inhibitor) and H-89 (PKA inhibitor) on pCREB contents as well as IRP1 protein expression as PI3K and PKA might link hypoxia with CREB phosphorylation [47-50]. We demonstrated that both PI3K and PKA inhibitors could significantly reduce pCREB content and also IRP1 protein expression in the cells treated with not only normoxia but also hypoxia. However, treatment with LY294002, but not H-89, markedly decreased pCREB and IRP1 protein levels in the cells exposed to 8 hours of hypoxia. These findings showed that the increased IRP1 expression induced by the enhanced pCREB under hypoxia could be prevented by LY294002 and also implied that the PI3K/Akt pathway, rather than PKA, might play a dominant role in CREB activation in the late phase of hypoxia.
It was noticed that the levels of IRP mRNA in cells treated with hypoxia for 8, 12 and 24 hours were about 2-fold of that of the controls, while the contents of IRP1 were lower than (8 and 12 hours) or equal to (24 hours) those of the controls during this period. Currently, the associated causes are unknown. However, the difference in the induction of IRP-1 mRNA and protein by hypoxia might imply the existence of some unknown factors that inhibit translational expression of IRP1 during the late phase of hypoxia, and further work is clearly required to substantiate this possibility.
A significantly increased HIF-1 alpha and a reduction in IRP mRNA and protein was found in the cells treated with hypoxia for 4 hours. This provided further evidence that hypoxia could inhibit degradation of HIF-1 alpha, leading to an increase in HIF-1 content which could bind to the three HREs in the 5'-regulatory region of IRP1 and then down-regulated IRP1 expression by a transcriptional mechanism [21]. This conclusion contradicts that of Hanson and Leibold [12]. They demonstrated that hypoxia inactivates the IRE-binding activity of IRP1 by a post-translational mechanism, without affecting IRP1 mRNA expression. The conflicting results may be partly due to the different degrees of hypoxia (3% vs. 1%) and differences in cell models (a rat hepatoma cell line, FTO2B vs. a human hepatoma cell line, HB 8065) used although the precise causes are unknown at present.
The findings obtained from the present study supported the idea that a so called hypoxia-responsive gene or hypoxia-inducible gene is not regulated by HIF-1 only under the conditions of hypoxia. Dual-regulation by HIF-1 and pCREB or other transcription factor(s) in hypoxia might be a common mechanism in most if not all of hypoxia-responsive or -inducible genes. The Macrophage migration inhibitory factor (MIF) has been well described as a pro-inflammatory cytokine and regulator of neuro-endocrine function [51-53]. Several recent studies have also implicated MIF as a hypoxia-responsive gene in cancer, cardiac ischemic injury, and inflammatory responses [54]. A recent study showed that hypoxia, and specifically HIF-1alpha, is a potent and rapid inducer of MIF expression and demonstrated that hypoxia-induced MIF expression is dependent upon a HRE in the 5'UTR of the MIF gene but is further modulated by CREB expression. This study strongly supports the above possibility. It is also highly likely that different transcription factors might play a role in the regulation of the target gene in different phases of hypoxia. In the case of IPR1, HIF-1 might play its role only in the early phase, while pCREB in the late phase of hypoxia. Therefore, the transcription factor(s) and mechanisms involved in the regulation of hypoxia-inducible genes in addition to HIF-1 under hypoxia need to be investigated further. Currently, this important issue has not received much attention, however, the relevant studies are of great significance in fully understanding not only the mechanisms of the regulation of hypoxia-inducible gene expression but also the potential application of hypoxic preconditioning in clinical practice.
Abbreviations
CRE: Cyclic AMP-responsive element
CREB: Cyclic AMP-responsive element-binding protein
DMT1: divalent metal transporter1
HRE: hypoxia response element
HIF-1: hypoxia-inducible factor-1
IRE: iron-responsive element
IRP 1: iron regulatory protein 1
pCREB: Phosphorylated CREB
PI3K: phosphatidylinositol 3-kinase
PKA: protein kinase A
Tf : transferrin
TfR1: transferrin receptor 1
Acknowledgements
We thank Christopher Qian for assistance with preparation and English revision of the manuscript. The studies in our laboratories were financially supported by National Natural Science Foundation of China (31300973, 31171143, 31271132, 31371092, 31571195, 31330035), and Hong Kong Health and Medical Research Fund (01120146).
Author contributions
Q.Q.L., Y. K. and Z.M.Q. conceived, organized and supervised the study; Q.Q.L., Y.F.Z., M.W.Z. and D.W. performed the experiments; L.Z. contributed to the analysis of data. Y. K. and Z.M.Q. prepared and wrote the manuscript.
Competing Interests
The authors indicate no potential conflicts of interest.
References
1. Wang GL, Jiang BH, Rue EA. et al. Hypoxia-inducible factor 1 is a basic-helix- loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl AcadSci USA. 1995;92:5510-5514
2. Lahiri S, Roy A, Baby SM. et al. Oxygen sensing in the body. ProgBiophysMol Biol. 2006;91:249-286
3. Jewell UR, Kvicukova T, Scheid A. et al. Induction of HIF-1a in response to hypoxia is simultaneous. FASEB J. 2001;15:1312-1314
4. Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor1. J Biol Chem. 1995;270:1230-1237
5. Huang LE, Gu J, Schau M. et al. Regulation of hypoxia-inducible factor 1a is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl AcadSci USA. 1998;95:7987-7992
6. Zarember KA, Malech HL. HIF-1a: A master regulator of innate host defenses? J Clin Invest. 2005;115:1702-1704
7. Peyssonnaux C, Zinkernagel AS, Schuepbach RA. et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. 2007;117:1926-1932
8. Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). MolPharmacol. 2006;70:1469-1480
9. Rolfs A, Kvietikova I, Gassmann M. et al. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. 1997;272:20055-20062
10. Tacchini L, Bianchi L, Bernelli-Zazzera A. et al. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem. 1999;274:24142-24146
11. Mukhopadhyay CK, Mazumder B, Fox PL. Role of Hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem. 2000;275:21048-21054
12. Hanson ES, Leibold EA. Regulation of iron regulatory protein 1 during hypoxia and hypoxia/reoxygenation. J Biol Chem. 1998;273:7588-7593
13. Hanson ES, Foot LM, Leibold EA. Hypoxia post-translationally activates iron-regulatory protein 2. J Biol Chem. 1999;274:5047-5052
14. Nicolas G, Chauvet C, Viatte L. et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037-1044
15. Zhu L, Zhang L, Ke Y. et al. Correlation between the expression of divalent metal transporter 1 and the content of hypoxia-inducible factor-1 in hypoxic HepG2 cells. J Cell Mol Med. 2008;12:569-579
16. Qian ZM, Wu XM, Fan M. et al. Divalent metal transporter 1 is a hypoxia-inducible gene. J Cell Physiol. 2011;226:1596-1603
17. Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341:1986-1995
18. Fleming RE, Sly WS. Mechanisms of iron accumulation in hereditary hemochromatosis. Annu Rev Physiol. 2002;64:663-680
19. Kühn LC. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics. 2015;7:232-243
20. Anderson CP, She M, Eisenstein RS. et al. Mammalian iron metabolism and its control by iron regulatory proteins. BiochimBiophysActa. 2012;1823:1468-1483
21. Luo QQ, Wang D, Yu MY. et al. Effect of Hypoxia on the Expression of Iron Regulatory Proteins 1 and the Mechanisms Involved. IUBMB Life. 2011;63:120-128
22. Lee HT, Chang YC, Tu YF. et al. CREB activation mediates VEGF-A's protection of neurons and cerebral vascular endothelial cells. J Neurochem. 2010;113:79-91
23. Siu YT, Jin DY. CREB-a real culprit in oncogenesis. FEBS J. 2007;274:3224-3232
24. Mabuchi T, Kitagawa K, Kuwabara K. et al. Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci. 2001;21:9204-9213
25. Lee HT, Chang YC, Wang LY. et al. cAMP response element-binding protein activation in ligation preconditioning in neonatal brain. Ann Neurol. 2004;56:611-623
26. Lin WY, Chang YC, Lee HT. et al. CREB activation in the rapid, intermediate, and delayed ischemic preconditioning against hypoxic-ischemia in neonatal rat. J Neurochem. 2009;108:847-859
27. Bonni A, Brunet A, West AE. et al. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358-1362
28. Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11-14
29. Mantamadiotis T, Lemberger T, Bleckmann SC. et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47-54
30. Tanaka K. Alteration of second messengers during acute cerebral ischemia - adenylate cyclase, cyclic AMP-dependent protein kinase, and cyclic AMP response element binding protein. ProgNeurobiol. 2001;65:173-207
31. Hara T, Hamada J, Yano S. et al. CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region. J Neurochem. 2003;86:805-814
32. Fang D, Li Z, Zhong-ming Q. et al. Expression of bystin in reactive astrocytes induced by ischemia/reperfusion and chemical hypoxia in vitro. BiochimBiophysActa. 2008;1782:658-663
33. Li Z, Ya K, Xiao-Mei W. et al. Ginkgolides protect PC12 cells against hypoxia-induced injury by p42/p44 MAPK pathway-dependent upregulation of HIF-1alpha expression and HIF-1DNA-binding activity. J Cell Biochem. 2008;103:564-575
34. He W, Qian Zhong M, Zhu L. et al. Ginkgolides mimic the effects of hypoxic preconditioning to protect C6 cells against ischemic injury by up-regulation of hypoxia-inducible factor-1 alpha and erythropoietin. Int J Biochem Cell Biol. 2008;40:651-662
35. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-408
36. Du F, Zhu L, Qian ZM. et al. Hyperthermic preconditioning protects astrocytes from ischemia/reperfusion injury by up-regulation of HIF-1 alpha expression and binding activity. BiochimBiophysActa. 2010;1802:1048-1053
37. Cartharius K, Frech K, Grote K. et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933-2942
38. Dimova EY, Jakubowska MM. et al. CREB binding to the hypoxia-induciblefactor-1 responsive elements in the plasminogen activator inhibitor-1 promoter mediates the glucagon effect. ThrombHaemost. 2007;98:296-303
39. Gonzalez G.A, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675-680
40. Yamamoto KK, Gonzalez G A, Biggs WH. et al. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature. 1988;334:494-498
41. Matthews R, Guthrie C, Wailes L. et al. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107-6116
42. Sun P, Enslen H, Myung P. et al. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527-2539
43. Ginty D, Bonni A, Greenberg M. Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell. 1994;77:713-725
44. Tan Y, Rouse J, Zhang A. et al. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629-4642
45. Du K, Montminy M. CREB Is a Regulatory Target for the Protein Kinase Akt/PKB. J Biol Chem. 1998;273:32377-32379
46. Li XY, Zhan XR, Liu XM. et al. CREB is a regulatory target for the protein kinase Akt/PKB in the differentiation of pancreatic ductal cells into islet β-cells mediated by hepatocyte growth factor. BiochemBiophys Res Commun. 2011;404:711-716
47. Jacinto E, Lorberg A. TOR regulation of AGC kinases in yeast and mammals. Biochem J. 2008;410:19-37
48. Carloni S, Girelli S, Scopa C. et al. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy. 2010;6:366-377
49. Li X, Han X, Llano J. et al. Mammalian target of rapamycin inhibition in macrophages of asymptomatic HIV+ persons reverses the decrease in TLR-4-mediated TNF-alpha release through prolongation of MAPK pathway activation. J Immunol. 2011;187:6052-6058
50. Kim JE, Kang JS, Lee WJ. Vitamin C Induces Apoptosis in Human Colon Cancer Cell Line, HCT-8 Via the Modulation of Calcium Influx in Endoplasmic Reticulum and the Dissociation of Bad from 14-3-3beta. Immune Network. 2011;12:189-195
51. Donnelly SC, Haslett C, Reid PT. et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 1997;3:320-323
52. Baugh JA, Donnelly SC. Macrophage migration inhibitory factor: a neuroendocrine modulator of chronic inflammation. J. Endocrinol. 2003;179:15-23
53. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791-800
54. Baugh JA, Gantier M, Li L. et al. Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. BiochemBiophys Res Commun. 2006;347:895-903
Author contact
![]() Corresponding authors: Zhong Ming Qian (E-mail: zhongming.qiancom) Laboratory of Neuropharmacology, Fudan University School of Pharmacy,826 Zhang Heng Road, Shanghai 201203, PRC. Ya Ke: yakeedu.hk; Li Zhu: zhulili65com.
Corresponding authors: Zhong Ming Qian (E-mail: zhongming.qiancom) Laboratory of Neuropharmacology, Fudan University School of Pharmacy,826 Zhang Heng Road, Shanghai 201203, PRC. Ya Ke: yakeedu.hk; Li Zhu: zhulili65com.