Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
Conflicts of interest
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(10):1213-1224. doi:10.7150/ijbs.15496 This issue Cite
Research Paper
miR-628 Promotes Burn-Induced Skeletal Muscle Atrophy via Targeting IRS1
Yonghui Yu*, Xiao Li*, Lingying Liu*, Jiake Chai ![]() , Zhang Haijun, Wanli Chu, Huinan Yin, Li Ma, Hongjie Duan, Mengjing Xiao
, Zhang Haijun, Wanli Chu, Huinan Yin, Li Ma, Hongjie Duan, Mengjing Xiao
Department of Burn & Plastic Surgery, the First Affiliated Hospital of PLA General Hospital, Beijing, 100048 China.
*These authors contributed equally to this work.
Received 2016-3-10; Accepted 2016-8-8; Published 2016-9-15
Abstract

Skeletal muscle atrophy is a common clinical feature among patients with severe burns. Previous studies have shown that miRNAs play critical roles in the regulation of stress-induced skeletal muscle atrophy. Our previous study showed that burn-induced skeletal muscle atrophy is mediated by miR-628. In this study, compared with sham rats, rats subjected to burn injury exhibited skeletal muscle atrophy, as well as significantly decreased insulin receptor substrate 1 (IRS1) protein expression and significantly increased skeletal muscle cell apoptosis. An miRNA array showed that the levels of miR-628, a potential regulator of IRS1 protein translation, were also clearly elevated. Second, L6 myocyte cell apoptosis increased after induction of miR-628 expression, and IRS1 and p-Akt protein expression decreased significantly. Expression of the cell apoptosis-related proteins FoxO3a and cleaved caspase 3 also increased after induction of miR-628 expression. Finally, forced miR-628 expression in normal rats resulted in increased cell apoptosis and skeletal muscle atrophy, as well as changes in IRS1/Akt/FoxO3a signaling pathway activity consistent with the changes in protein expression described above. Inhibiting cell apoptosis with Z-VAD-FMK resulted in alleviation of burn-induced skeletal muscle atrophy. In general, our results indicate that miR-628 mediates burn-induced skeletal muscle atrophy by regulating the IRS1/Akt/FoxO3a signaling pathway.
Keywords: miR-628, Burn, IRS1, Skeletal muscle atrophy.
Introduction
The main factor underlying skeletal muscle atrophy is excessive protein degradation. It is one of the most common clinical features of patients with cancer-related cachexia [1, 2], sepsis [3], severe burns [4], and chronic kidney disease [5]. Burn-induced skeletal muscle atrophy is a complicated process that results in prolonged hospital days and delayed wound healing [6]. Previous studies have shown that it may last at least 9-12 months after complete wound healing [7]. Thus, skeletal muscle atrophy plays a critical role in the recoveries and prognoses of burn patients.
Ubiquitin proteasome-mediated protein degradation plays an important role in muscle skeletal atrophy [8]. Damaged and misfolded proteins are bound by ubiquitin. With the assistance of E2 and E3 ligases, proteasomes capture poly-ubiquitinated proteins and trigger their degradation. Muscle RING finger 1 (MuRF1) and muscle atrophy F-box (MAFbx) are muscle-specific E3 ligases [9]. Clinical and animal model studies have shown that MuRF1 and MAFbx expression levels are significantly increased during skeletal muscle atrophy [10]. MG132, a proteasome inhibitor, suppresses MuRF1 and MAFbx expression and significantly alleviates skeletal muscle atrophy [11].
Skeletal muscle cell apoptosis is also observed during skeletal muscle atrophy, and increased skeletal muscle cell apoptosis contributes to muscle atrophy [12, 13]. Inflammatory factors, such as tumor necrosis factor α (TNF-α), are pivotal inducers of skeletal muscle cell apoptosis. TNF-α induces C2C12 myotube apoptosis and atrophy [14] and also inactivates the PI-3K/Akt signaling pathway and induces L6 myotube atrophy [15]. Previous studies have shown that PI-3K/Akt activation is impaired after burn injury and that insulin receptor substrate 1 (IRS1) is an upstream regulator of the PI-3K/Akt signaling pathway [16]. IRS1 is also a critical mediator of cell apoptosis. IRS1 deficiency promotes cell apoptosis [17], and IRS1 overexpression contributes to cell survival [18]. Therefore, the IRS1/PI-3K/Akt signaling pathway plays a pivotal role in regulating cell apoptosis and skeletal muscle atrophy.
The transcription factor forkhead box protein O (FoxO) is a downstream effector of Akt, which regulates cell apoptosis and skeletal muscle atrophy. FoxO is phosphorylated by Akt in the cell cytoplasm and degraded, whereas dephosphorylated FoxO translocates into the nucleus and triggers apoptosis-related gene expression [19]. Akt inactivation results in FoxO transactivation and eventually skeletal muscle atrophy [20]. FoxO3a, a member of the FoxO family, plays a critical role in regulating skeletal muscle atrophy. Dexamethasone (Dex) activates the AMPK/FoxO3 signaling pathway and induces skeletal muscle atrophy [21]. The protein deacetylase SIRT1 helps suppress muscle atrophy and promotes muscle growth by blocking FoxO3 activation [22]. Aberrant FoxO activation plays a pivotal role in skeletal muscle atrophy.
miRNAs, small noncoding RNAs, are important regulators of skeletal muscle development. miR-128a inhibits myoblast cell proliferation by repressing IRS1 protein expression, and reducing miR-128a levels induces myofiber hypertrophy [23]. miR-182 attenuates atrophy-related gene expression by targeting FoxO3, thereby alleviating skeletal muscle atrophy [24]. The muscle-specific miRNAs miR-1 and miR-206 are also critical regulators of skeletal muscle atrophy. In Dex-induced muscle atrophy, miR-1 overexpression suppresses HSP70 protein expression and reduces Akt phosphorylation, which enhances FoxO3 activation [25]. miR-206 promotes skeletal muscle satellite cell proliferation and differentiation by inhibiting Pax 7 expression [26], and miR-206 plays a critical role in halting amyotrophic lateral sclerosis progression [27]. These findings indicate that miRNAs are potential therapeutic targets in the treatment of skeletal muscle atrophy.
Previous reports have shown that miR-628 is upregulated in an NF-κB-dependent manner upon lipopolysaccharide (LPS) induction. miR-628 also plays a role in mediating Toll-like receptor signaling via feedback inhibition [28]. In this study, we found that miR-628 levels were significantly increased in rat tibialis anterior muscles after burn injury, which we attributed to FoxO3a activation secondary to IRS1 suppression, as forced miR-628 expression suppressed IRS1 protein expression, activated FoxO3a and promoted skeletal muscle cell apoptosis and muscle atrophy.
Materials and methods
Plasmids and reagents
Mature rat miR-628 (AUGCUGACAUAUUUACGAGAGG) was cloned into a pcDNA6.2-GW/EmGFP-miR vector (GenePharma Co., Ltd., Shanghai, China) using a BLOCK-iT Pol II miR RNAi Expression Vector Kit (Life Technologies, Grand Island, NY, USA). The rat IRS1 mRNA 3'-UTR sequence (1069 base pairs) was cloned into a pGL3 luciferase assay vector (IRS1 3'-UTR luciferase reporter). Antibodies against IRS1, Akt, p-Akt S473, FoxO3a, p-FoxO3a S253, MuRF-1, MAFbx and cleaved caspase 3 were purchased from Cell Signaling Technologies (Beverly, MA, USA). β-Actin antibodies were purchased from Sigma-Aldrich (St Louis, MO, USA). The pan-caspase inhibitor Z-VAD-FMK was purchased from Abcam (Cambridge, MA, USA).
Animal model
This study was conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and was approved by the Animal Care and Use Committee of the First Affiliated Hospital of PLA General Hospital. And the rats were pair-fed in all the animal experiments. A thirty percent total body surface area (TBSA) and full-thickness burn model was established, and anesthetics and analgesics were used as described in our previous reports [29, 30]. After burn injury, 1 ml of PBS or Z-VAD-FMK (3.5 mg/kg·body weight) was immediately injected intraperitoneally.
A pcDNA6.2-GW/EmGFP-miR-628 vector was used to induce miR-628 expression. Normal SD rats were randomly divided into two groups, and 1 ml of miR-628 expression vector (250 μg/kg·body weight) or vehicle (250 μg/kg·body weight) was injected via the tail vein.
Cell culture and transfection
L6 myoblasts and 293T cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum, 1% penicillin/streptomycin, and 2 mM glutamine (Life Technologies, Grand Island, NY, USA) and incubated at 37℃ with 5% CO2. Cells were seeded into 6-well plates after reaching 70~80% confluence. miR-628 or vehicle was transiently transfected into L6 myoblasts, and an IRS1 3'-UTR luciferase reporter was cotransfected into 293T cells with miR-628 or vehicle using Lipofectamine 2000 reagent (Life Technologies, Grand Island, NY, USA), in accordance with the manufacturer's instructions. Transfectants were collected for further analysis at the indicated times.
Luciferase assay
293T cells were seeded into 6-well plates after reaching 70~80% confluence, and an IRS1 3'-UTR luciferase reporter was cotransfected into the cells with miR-628 or vehicle. After 24 h, transfectants were collected, and cell pellets were lysed using luciferase substrate (Promega, San Luis Obispo, CA, USA). Luciferase levels were measured using a Microplate Luminometer LB 96V, according to the manufacturer's instructions (Promega, San Luis Obispo, CA, USA).
Flow cytometry assay
After 48 h of transfection, L6 myoblasts were trypsinized and collected by centrifugation at 1500 r.p.m. for 5 min. The cell pellets were washed twice with ice-cold phosphate-buffered saline and then fixed in ice-cold 70% ethanol at -20℃ overnight. The cells were washed with phosphate-buffered saline, and cell apoptosis was analyzed via flow cytometry (Beckman, Indianapolis, IN, USA) after the cells were stained for 15 min with propidium iodide (PI) buffer (0.1% Triton X-100, 0.2 mg/ml RNase A, 0.05 mg/ml propidium iodide).
Real-time polymerase chain reaction (PCR)
Total RNA was extracted from tibialis anterior muscle samples or L6 myoblasts using Trizol reagent (Life Technologies, Grand Island, NY, USA), according to the manufacturer's instructions. A SuperScript III First-Strand Synthesis system (Life Technologies, Grand Island, NY, USA) was employed for first-strand cDNA synthesis with oligdT(20) primers. Specific primers (Sangon Biotech, Shanghai, China) were used for PCR amplification. The sequences of the primers used in this study were as follows: rat IRS1 (forward: 5'-AAGCAGCACCCTCAAGACTG-3' and reverse: 5'-AGGTCTTTCATTCTGCCTGTGA-3') and rat GAPDH (forward: 5'-ATGGAGAAGGCTGGGGCTC-3' and reverse: 5'-CCTTCCACGATGCCAAAGTT-3'). Data were analyzed using the 2-△△T method.
Real-time PCR for miR-628
Tibialis anterior muscle samples or L6 myoblast transfectants were collected, and total RNA was extracted using an miRNeasy Mini Kit (Qiagen, Valencia, CA, USA). A miScript II RT kit (Qiagen, Valencia, CA, USA) was employed for first-strand cDNA synthesis, and miR-628 expression was subsequently detected using an miScript SYBR Green PCR system (Qiagen, Valencia, CA, USA) with a ViiA 7TM Real-time PCR System (Applied Biosystems, Foster City, CA, USA). The following primers were used in this study: miR-628 (forward: 5'-GGGGGATGCTGACATATTTAC-3' and reverse: 5'-CAGTGCGTGTCGTGGAGT-3') and U6 (forward: 5'-GCTTCGGCAGCACATATACTAAAAT-3' and reverse: 5'-CGCTTCACGAATTTGCGTGTCAT-3'). Initial activation was performed at 95℃ for 10 min, followed by 40 cycles of denaturation at 95℃ for 10 sec and annealing and extension at 60℃ for 60 sec. Data were analyzed using the 2-△△T method.
Frozen-section analysis
Rat tibialis anterior muscle tissues injected with miR-628 or vehicle were collected at the indicated times and immediately embedded in optimum cutting temperature compound. Subsequently, approximately 5-μm-thick sections were cut at -20℃. These sections were transferred to microscope slides and observed via fluorescence microscopy. Images were taken using an inverted fluorescent microscope (Leica, Wetzlar, Germany).
Western blotting
Total proteins were extracted from tissue or cell samples using RIPA buffer (MACGENE Biotech, Beijing, China), and a BCA protein assay kit was used for protein quantification. Sixty micrograms of total protein from each sample was separated by SDS-PAGE, followed by 5% dry milk blocking and labeling with specific primary antibodies and HRP-conjugated secondary antibodies. Bands were subsequently detected using ECL western blotting substrate, and quantitative analysis was carried out using AlphaEaseFC 4.0 software.
TUNEL analysis
After being collected at the indicated times, the tibialis anterior muscle samples were fixed with 4% paraformaldehyde. After dehydration, the tissues were embedded in paraffin. Tissue sections were subsequently subjected to TUNEL staining using an In Situ Cell Death Detection Kit (Roche Applied Science, Indianapolis, IN, USA). Relative apoptosis ratios were calculated using Image Pro Plus 6.0 software.
Statistical analysis
Student's t-test was used to determine statistical significance. Data are expressed as the mean ± SD. Differences were considered significant at p≤0.05.
Results
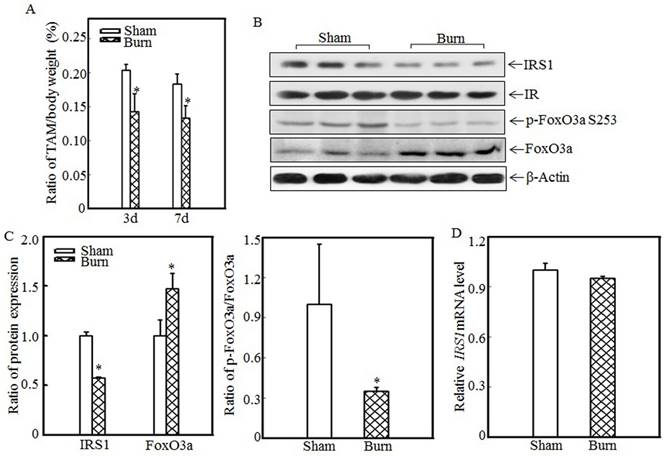
Burn injury induced skeletal muscle atrophy and suppressed IRS1 expression
Skeletal muscle atrophy is a common clinical feature among burn patients. To elucidate the molecular mechanisms underlying burn-induced skeletal muscle atrophy, we established a 30% TBSA burn model, as described in the “Material and methods” section. Tibialis anterior muscle samples were collected from sham and burn rats. Absolute data regarding body weights and muscle mass are shown in Table S1 as supplementary data. Tibialis anterior weight/body weight ratios were calculated. As shown in Fig. 1A, these ratios were significantly decreased in burn rats compared with sham rats at the indicated times, indicating that skeletal muscle atrophy was successfully introduced in this animal model. Previous reports showed that downregulating IRS1 expression activates FoxO and promotes muscle atrophy [31]; therefore, we detected IRS1 protein expression in the tibialis anterior muscles of sham and burn rats. Compared with the sham rats, the burn rats exhibited significantly decreased IRS1 protein expression (Fig. 1B). Our previous study also demonstrated that Akt phosphorylation was decreased after burn injury [29], as increased levels of FoxO3a activation were also detected. After burn injury, FoxO3a phosphorylation at Ser 253, which promotes FoxO3a protein degradation, was significantly decreased (Fig. 1B), and total FoxO3a protein expression was significantly increased (Fig. 1B), indicating that burn injury results in FoxO3a activation, which may promote skeletal muscle atrophy. Subsequent quantification analysis indicated that IRS1 protein expression was decreased by approximately 40% and FoxO3a level was significantly increased after burn injury in burn rats compared with sham rats (Fig. 1C, left panel) and that the p-FoxO3a/FoxO3a ratio was also significantly decreased (Fig. 1C, right panel). However, IRS1 mRNA expression levels were comparable in the tibialis anterior muscles of sham and burn rats (Fig. 1D), indicating that burn injury-induced impairments in IRS1 protein expression were introduced not at the transcription level but at the post-transcription level.
Burn-injury induced skeletal muscle atrophy and repressed IRS1 expression. The 30% TBSA burn injury model was established as described in the “Materials and methods” section. At 3 d or 7 d after injury, tibialis anterior muscle samples were collected from sham and burn rats (n=5 in each group), and tibialis anterior muscle weights and body weights were measured. Then, the tibialis anterior muscle weight/body weight ratio was calculated. The ratio was significantly decreased after burn injury, and the asterisk (*) indicates a significant difference (p<0.05) (A). IRS1, FoxO3a and p-FoxO3a at S253 expression in the tibialis anterior muscles of sham and burn rats at 3 d after injury was detected via western blotting. The protein expression levels of IRS1 and p-FoxO3a at S253 were decreased, and the total protein expression level of FoxO3a was increased in the burn group (B). Quantitative analysis of protein expression in sham and burn rats was performed. Burn injury significantly reduced IRS1 protein expression and the p-FoxO3a/FoxO3a ratio. The asterisk (*) indicates a significant difference (p<0.05) (C). The levels of IRS1 mRNA expression in the tibialis anterior muscles of sham and burn rats at 3 d after injury were also analyzed using real-time PCR. The levels of IRS1 mRNA expression in sham and burn rats were comparable (D).
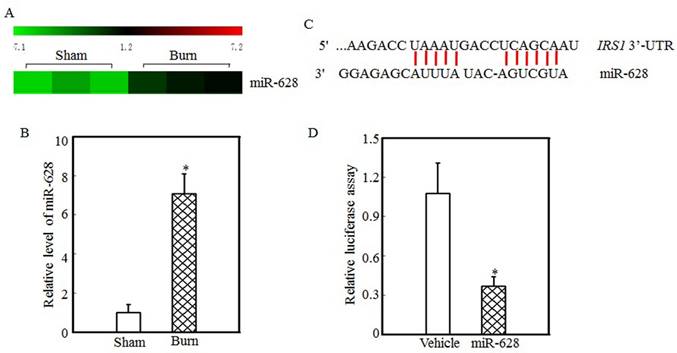
miR-628 might be associated with IRS1 protein expression regulation
Protein translation and degradation are two methods of post-transcription regulation, and miRNAs play a critical role in regulating protein translation. Our previous report indicated that the expression levels of many miRNAs were altered in the tibialis anterior muscles of burn rats and that the expression of miR-628 was significantly increased after burn injury [32]. miR-628 expression levels in the tibialis anterior muscles of sham and burn rats were determined using an miRNA array and are shown in Fig. 2A. These results indicate that burn injury induced miR-628 expression. RT-PCR was used to measure miR-628 expression levels in the tibialis anterior muscle and showed that the levels of miR-628 expression were 6-fold higher in burn rats than in sham rats (Fig. 2B). Subsequent experiments focused on whether miR-628 regulates IRS1 protein translation. First, TargetScan software analysis indicated that miR-628 binds to the 3'-UTR of IRS1 (Fig. 2C), indicating that miR-628 regulates IRS1 protein translation. Second, a luciferase reporter containing the IRS1 3'-UTR was constructed and co-transfected into 293T cells with vehicle or miR-628. Twenty-four hours later, the cells were collected, and luciferase levels were detected. As shown in Fig. 2D, miR-628 expression significantly decreased luciferase activity, which indicated that miR-628 directly binds to the 3'UTR of IRS1 mRNA and suppresses its translation.
IRS1 protein expression was regulated by miR-628 after burn injury. miRNA array analysis of the tibialis anterior muscles of sham and burn rats at 3 d after injury was performed using a miRCURY™ LNA Array (v.18.0) system, as previously described [29]. The expression of miR-628 in sham and burn rats is shown, and miR-628 expression was increased after burn injury (A). Real-time PCR was used to measure miR-628 expression in the tibialis anterior muscles of sham and burn rats. The level of miR-628 expression in the burn group was approximately 7 times higher than that in the sham group, and the asterisk (*) indicates a significant difference (p<0.05) (B). The potential miRNA binding site in the IRS1 mRNA 3'-UTR was analyzed using TargetScan software (C). The full length of the rat IRS1 mRNA 3'-UTR sequence was amplified and cloned into a pGL3 luciferase reporter vector (IRS1 3'-UTR luciferase reporter), and the IRS1 3'-UTR luciferase reporter was cotransfected with miR-628 or vehicle into 293T cells. After 24 h of transfection, the cells were collected, and luciferase activity was measured. Luciferase levels were dramatically reduced after miR-628 transfection, and the asterisk (*) indicates a significant difference (p<0.05) (D).
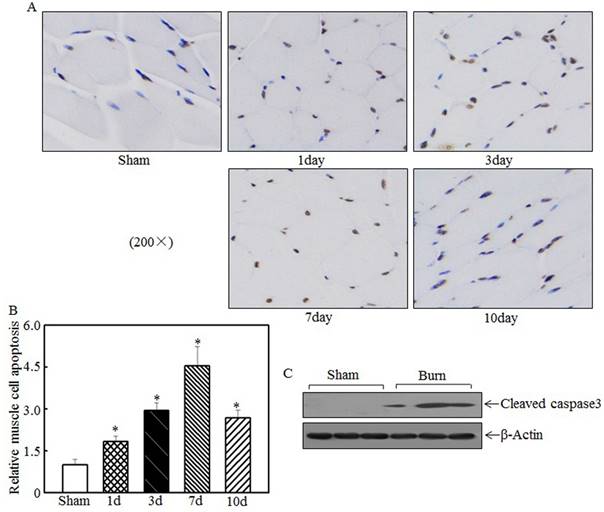
Skeletal muscle cell apoptosis caused atrophy
Skeletal muscle atrophy was partially attributed to incensed skeletal muscle cell apoptosis [15, 33]. We detected tibialis anterior muscle cell apoptosis at the indicated times after injury using TUNEL staining. Compared with the sham rats, the burn rats exhibited significantly enhanced skeletal muscle cell apoptosis (Fig. 3A). Quantitative analysis indicated that 1 d after burn injury, skeletal muscle cell apoptosis was significantly increased in the burn rats compared with the sham rats. At 3 d and 7 d after injury, cell apoptosis was increased by approximately 3- to 4.5-fold in the burn rats compared with the sham rats (Fig. 3B). Subsequent experiments analyzed the expression of cleaved caspase 3, a pro-apoptotic protein, at 7 d after injury. As shown in Fig. 3C, cleaved caspase 3 levels were significantly elevated after burn injury. These results indicate that cell apoptosis is associated with skeletal muscle atrophy.
Burn injury promoted skeletal muscle cell apoptosis. Tibialis anterior muscle samples from sham or burn rats were collected at 1 d, 3 d, 7 d, and 14 d after injury. TUNEL staining was employed to detect cell apoptosis at the indicated times, and images were taken using an inverted microscope (200×). Muscle cell apoptosis was observed in the samples from day 1 to day 10 post-injury, especially on day 7 (A). Quantitative analysis of skeletal cell apoptosis was performed, and significantly increased levels of cell apoptosis were noted after burn injury. The asterisk (*) indicates a significant difference compared with the sham group (B). Increased caspase 3 fragmentation was noted in tibialis anterior muscle samples from sham and burn rats via western blotting at 7 d after injury (C).
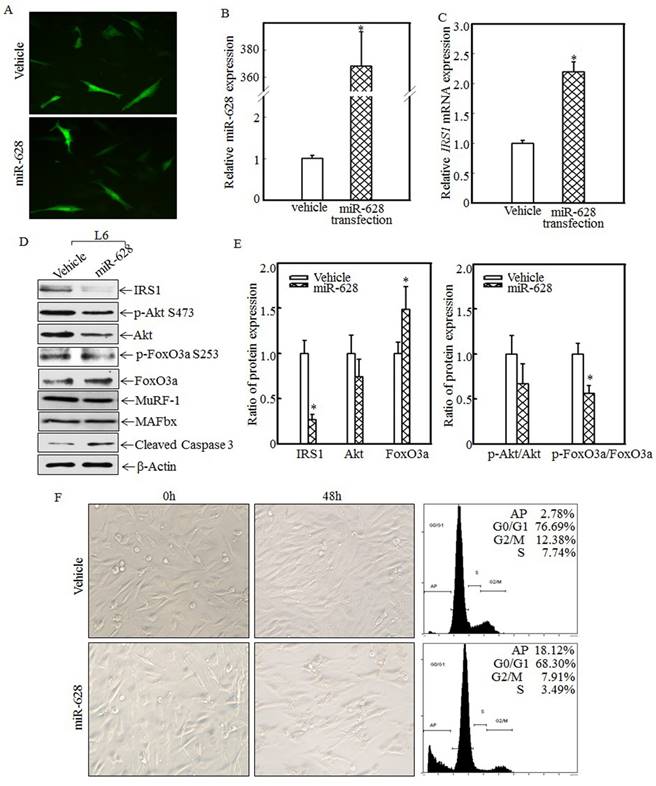
Forced miR-628 expression promoted L6 myoblast apoptosis
To study the relationship between miR-628 expression and skeletal muscle cell apoptosis, L6 myoblasts were transfected with vehicle or miR-628. After 48 h of transfection, green fluorescence was detected using an inverted fluorescence microscope. As shown in Fig. 4A, green fluorescence was observed in both groups, indicating that vehicle and miR-628 were successfully transfected into L6 myoblasts. Real-time PCR was used to quantify miR-628 and IRS1 mRNA levels after transfection. Compared with the vehicle group, the miR-628-transfected group exhibited significantly increased miR-628 expression (Fig. 4B) and significantly increased IRS1 mRNA expression (Fig. 4C). The enhanced expression of IRS1 mRNA may have been due to miR-628-mediated inhibition of IRS1 protein translation. The expression of IRS1 and its downstream proteins was also analyzed by western blotting. After miR-628 transfection, IRS1 protein expression was significantly suppressed, and Akt phosphorylation at Ser473 was also significantly decreased (Fig. 4D). FoxO3a phosphorylation at Ser 253 was also impaired, resulting in increased total FoxO3a levels (Fig. 4D). FoxO3a activation induced cell apoptosis, and cleaved caspase 3 levels were significantly increased after miR-628 transfection; However, the E3 ligase proteins MuRF-1 and MAFbx had no significant difference (Fig. 4D). Quantitative data also demonstrated that IRS1 protein expression (Fig. 4E, left panel) and the p-FoxO3a/FoxO3a ratio (Fig. 4E, right panel) were significantly decreased. FoxO3a protein level in L6 cell transfected with miR-628 was dramatically increased (Fig. 4E, left panel). These results suggest that miR-628 promotes L6 myoblast apoptosis by regulating the IRS1/Akt/FoxO3a signaling pathway. Cell morphology was subsequently observed, and images were taken after 48 h of transfection. Forced miR-628 expression resulted in significantly increased L6 myoblast cell proliferation and subsequent cell shrinkage and death (Fig. 4F, left panel). Quantitative analysis of cells in both groups after 48 h of transfection showed (Fig. 4F, right panel) that the rate of apoptosis in L6 myoblasts transfected with miR-628 was 18.12%, which was significantly higher than the rate of apoptosis in cells transfected with vehicle (2.78%), indicating that miR-628 transfection induces L6 myoblast apoptosis.
miR-628 overexpression induced L6 myoblasts apoptosis. L6 myoblasts were seeded into 6-well plates. After the cells reached 70~80% confluence, miR-628 and vehicle were transfected into L6 myoblasts using Lipofectamine 2000. Green fluorescence was observed at 48 h after transfection using an inverted fluorescence microscope, and images were taken (200×) (A). Then, the cells were collected, and miR-628 (B) and IRS1 mRNA (C) expression was detected using real-time PCR. Both miR-628 and IRS1 mRNA expression was upregulated in L6 cells transfected with miR-628 (n=3 in each group). The asterisk (*) indicates a significant difference (p<0.05). After 48 h of transfection, the protein expression levels of IRS1, Akt, p-Akt at S473, FoxO3a and p-FoxO3a at S253 were analyzed via western blotting. Akt phosphorylation at S473 was decreased upon miR-628 overexpression, resulting in FoxO3a phosphorylation at S253. Phosphorylated FoxO3a at S253 was inactivated and degraded by the proteasome. Decreased levels of phosphorylated FoxO3a at S253 resulted in increased total FoxO3a protein and cleaved caspase 3 levels (D). Quantitative analysis of IRS1 protein levels and the p-FoxO3a/FoxO3a ratio was performed and showed that IRS1 protein expression and the p-FoxO3a/FoxO3a ratio were reduced after ectopic miR-628 expression. The asterisk (*) indicates a significant difference (p<0.05) (E). After transfection, cell morphology was observed, and images were taken using an inverted microscope at the indicated times (200×). miR-628 overexpression resulted in slight morphological alterations and cell growth inhibition, implying that forced miR-628 expression induces cell apoptosis. Flow cytometry was used to measure the L6 cell apoptotic ratio after 48 h of transfection and showed that ectopic miR-628 expression significantly increased L6 cell apoptosis (F).
miR-628 overexpression in rats induced cell apoptosis and skeletal muscle atrophy
The effects of miR-628 overexpression on cell apoptosis and skeletal muscle atrophy were also analyzed. A rat model of miR-628 overexpression was established by injecting plasmids expressing miR-628 into the tail vein. At 3 d and 7 d after injection, tibialis anterior muscle samples were collected, and green fluorescence was detected. Green fluorescence was observed in both the vehicle group and the miR-628 group at the indicated times after injection (Fig. 5A). Real-time PCR was used to quantify miR-628 expression after injection. As shown in Fig. 5B, miR-628 expression was significantly elevated at the indicated times. These results indicate that miR-628 was successfully expressed in the tibialis anterior muscle after tail vein injection. Cell apoptosis in the tibialis anterior muscle was subsequently measured using TUNEL staining. Imaging (Fig. 5C) and quantitative analysis (Fig. 5D) indicated that cell apoptosis was dramatically increased at both 3 d and 7 d after miR-628 injection. Subsequent analysis of the levels of IRS1 and its downstream proteins showed that IRS1 expression was significantly decreased in the miR-628-treated group compared with that in the vehicle group (Fig. 5E). Additionally, the levels of Akt phosphorylation at Ser 473 and FoxO3a phosphorylation at Ser 253 were significantly decreased, and the level of total FoxO3a protein expression was increased, resulting in increased cell apoptosis (Fig. 5E). The expression of cleaved caspase 3, a pro-apoptotic protein, was also enhanced after miR-628 injection. There was no difference about the protein levels of MuRF-1 and MAFbx (Fig. 5E). Quantitative analysis indicated that ectopic miR-628 expression resulted in significantly decreased IRS1 protein expression and increased FoxO3a protein level (Fig. 5F, left panel), and a significantly decreased p-FoxO3a/FoxO3a ratio (Fig. 5F, right panel).
Forced miR-628 expression in rats accelerated cell apoptosis and skeletal muscle atrophy. Plasmids containing vehicle or miR-628 were injected into rats via the tail vein. At 3 d or 7 d after injection, tibialis anterior muscle samples were collected. Frozen-section analysis was used to observe GFP expression in the tibialis anterior muscle at the indicated times via inverted fluorescence microscopy, and images were taken (200×) (A). Quantitative analysis of miR-628 levels in the tibialis anterior muscle was performed using real-time PCR (n=3 in each group). miR-628 levels were significantly increased in rats injected with plasmids expressing miR-628 compared with rats injected with vehicle. The asterisk (*) indicates a significant difference (p<0.05) (B). TUNEL staining was employed to analyze cell apoptosis in tibialis anterior muscle samples from different groups. Images (C) were taken using an inverted microscope (200×), and the relative cell apoptosis ratio was measured (D). These results indicated that miR-628 overexpression induced muscle cell apoptosis. The asterisk (*) indicates a significant difference (p<0.05). The protein levels of IRS1, Akt, p-Akt at S473, FoxO3a and p-FoxO3a at S253 in the tibialis anterior muscle were detected at 3 d after injection using western blotting. The protein expression levels of IRS1, p-Akt at S473 and p-FoxO3a at S253 were reduced, and the protein expression levels of FoxO3a and cleaved caspase 3 were increased after miR-628 overexpression (E). Quantitative analysis of IRS1 protein levels and the p-FoxO3a/FoxO3a ratio was performed. The asterisk (*) indicates a significant difference (p<0.05) (F). The weights of whole body and that of tibialis anterior muscle were measured, and the tibialis anterior muscle weight/body weight ratio was calculated (n=5 in each group). The ratio was significantly decreased in the miR-628-overexpression group compared with the vehicle group. The asterisk (*) indicates a significant difference (p<0.05) (G).
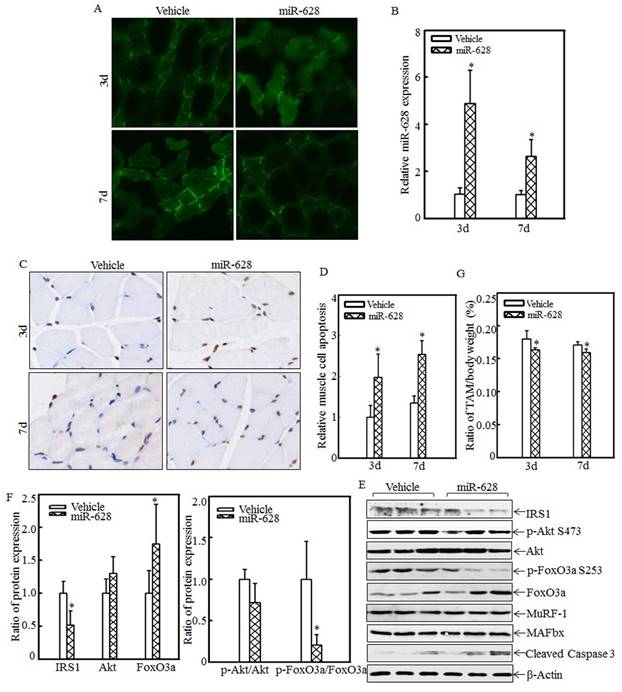
These results indicated that miR-628 expression activates the IRS1/Akt/FoxO3a signaling pathway and enhances cell apoptosis. Finally, absolute data regarding body weight and muscle mass are shown in Table S2 as supplementary data, and the tibialis anterior muscle weight/body weight ratio was calculated. Compared with the vehicle group, the miR-628-treated group exhibited a significantly decreased ratio at the indicated times (Fig. 5G). These results indicate that miR-628 overexpression promotes skeletal muscle cell apoptosis and muscle atrophy.
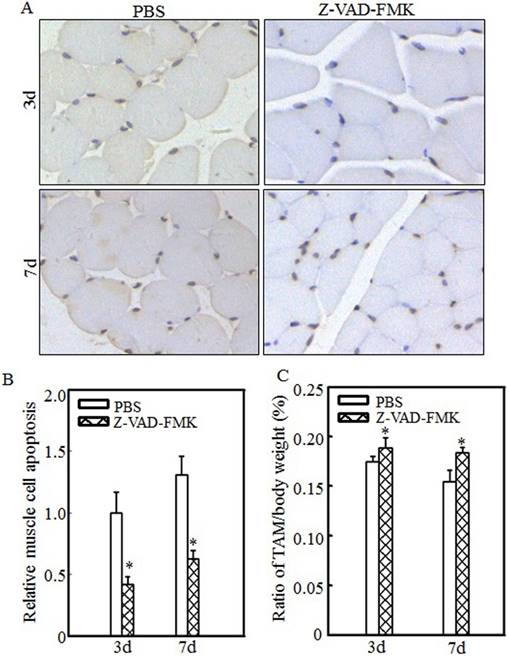
Inhibiting cell apoptosis via Z-VAD-FMK administration alleviated burn-induced muscle atrophy
To determine the relationship between cell apoptosis and burn-induced muscle atrophy, Z-VAD-FMK, a pan-caspase inhibitor, was used to suppress burn-induced cell apoptosis. After burn injury, rats were injected intraperitoneally with PBS or Z-VAD-FMK. Tibialis anterior muscle samples were collected at 3 d or 7 d after injury. Imaging (Fig. 6A) and quantitative analysis (Fig. 6B) showed that cell apoptosis was significantly inhibited after Z-VAD-FMK administration at the indicated times. Absolute data regarding body weight and muscle mass are shown in Table S3 as supplementary data, and tibialis anterior muscle weight/body weight ratios were calculated. As shown in Fig. 6C, these ratios were significantly increased in the Z-VAD-FMK-treated group compared with the PBS-treated group, indicating that Z-VAD-FMK administration effectively suppressed burn-induced muscle atrophy. These findings indicated that cell apoptosis promotes skeletal muscle atrophy and that inhibiting cell apoptosis via Z-VAD-FMK administration successfully attenuates skeletal muscle atrophy after burn injury.
Z-VAD-FMK administration abrogated burn-induced cell apoptosis and skeletal muscle atrophy. After injury, burn rats were intraperitoneally injected with PBS or Z-VAD-FMK. At 3 d and 7 d after injection, tibialis anterior muscle samples were collected (n=5 in each group). Tibialis anterior muscle cell apoptosis was analyzed using TUNEL staining at the indicated times, and images (A) were taken using an inverted microscope (200×). Quantitative analysis of cell apoptosis was carried out (B), and the asterisk (*) indicates a significant difference (p<0.05) compared with the PBS group. These data indicated that cell apoptosis was inhibited after Z-VAD-FMK injection. Tibialis anterior muscle weights and body weights were measured, and the tibialis anterior muscle weight/body weight ratio was calculated. The ratio was significantly increased after Z-VAD-FMK injection. The asterisk (*) indicates a significant difference (p<0.05) (C).
Discussion
Myogenesis plays a critical role in maintaining muscle mass, and myogenesis inactivation was observed during muscle atrophy [34]. The expression of myostatin, the negative regulator of myogenesis, was elevated, while the expression of miR-208b was significantly decreased in humans with spinal cord injury-induced muscle atrophy [35]. miR-208b overexpression decreased myostatin levels [35]. MyoD and myogenin, myogenic regulatory factors, promote myogenic differentiation. miR-26a upregulated myoD expression and promoted myogenic C2C12 myoblast differentiation [36]. During muscle regeneration, miR-26a was upregulated, and blocking miR-26a expression in the tibialis anterior muscle prevented muscle regeneration [37]. miR-186 reduced myogenic differentiation by targeting myogenin [38]. The expression levels of the muscle specific miRNAs miR-1, miR-133a/b and miR-206 were elevated in the sera of myotonic dystrophy patients with progressive muscle wasting [39]. Circulating miRNAs can be used as biomarkers of muscle differentiation and atrophy [40]. Thus, miRNAs play a pivotal role in regulating muscle atrophy. Our previous study showed that there were 4 upregulated miRNAs including miR-628, miR-107, miR-201, and miR-222 in tibialis anterior muscle after burn injury [32]. And IRS1, the critical muscle atrophy-related protein, was the potential target of miR-628. We subsequently focused on studying the possible function of miR-628 overexpression on regulating burn-induced muscle wasting. The results demonstrated that miR-628 was overexpressed in the tibialis anterior muscle after burn injury, resulting in decreased IRS1 protein translation.
IRS1, the insulin receptor substrate, is an important mediator of the insulin signaling pathway, and its expression was decreased after burn injury [16]. Our results also showed that IRS1 protein levels were decreased after burn injury and that miR-628 overexpression in both L6 myoblasts and rats significantly decreased IRS1 expression. Previous reports showed that IRS1 was associated with muscle atrophy regulation. Burn injury can induce IRS1 protein degradation via SOCS3 and ubiquitin-mediated pathway [41]. Endotoxin, which is widely exist in burn patient, serves an important role in promoting iNOS generation, mediating tyrosine nitration or decreasing tyrosine phosphorylation of IRS-1[42, 43]. C1-Ten is a protein tyrosine phosphatase of IRS1. C1-Ten dephosphorylates IRS1 at Y612, resulting in accelerated IRS1 degradation and skeletal muscle atrophy [31]. Dexamethasone and glucocorticoids are well-known inducers of muscle atrophy. After dexamethasone treatment, caveolin-1 expression in C2C12 myotubes decreased, resulting in increased IRS1 expression and muscle atrophy [44]. In a mouse model, endogenous glucocorticoids prevented IRS1-related PI3K activity and caused muscle atrophy [45]. Thus, IRS1/PI3K signaling pathway inactivation plays an important role in muscle atrophy.
FoxO is a downstream effector of the PI3K/Akt pathway. Akt activation results in FoxO phosphorylation and inhibition of its transcriptional activity [19]. FoxO family members play critical roles in muscle atrophy. In a rat model, LPS infusion downregulated Akt protein levels and cytosolic FoxO1 and FoxO3 phosphorylation, resulting in increased muscle protein degradation [46]. However, fish oil increased Akt and FoxO1 phosphorylation and improved muscle protein mass [47]. In this study, we showed that FoxO3a protein levels were elevated in the tibialis anterior muscle after burn injury and that forced miR-628 expression also elevated FoxO3a protein levels in both rats and L6 myoblasts, suggesting that enhanced FoxO3a activation induces skeletal muscle atrophy after burn injury.
FoxO3a translocates into the nucleus and triggers apoptosis-related gene expression. Previous reports indicate that reducing Akt1 activation promotes vascular smooth muscle cell apoptosis through FoxO3a activation [48]. Our results also showed that burn injury impaired Akt activation and increased total FoxO3a protein levels, resulting in skeletal muscle cell apoptosis. Z-VAD-FMK, a pan-caspase inhibitor, significantly abrogated goniothalamin-induced coronary artery smooth muscle cell apoptosis [49]. Z-VAD-FMK administration reduced skeletal proteolysis and tumor cell apoptosis [50]. In this study, Z-VAD-FMK treatment suppressed skeletal muscle cell apoptosis in rats after injury. These results indicate that Z-VAD-FMK administration significantly decreases burn-induced skeletal muscle cell apoptosis and restores muscle mass in rats. Therefore, abrogating cell apoptosis plays an important role in attenuating skeletal proteolysis and muscle mass loss.
Conclusion
Our results indicate that miR-628 overexpression after burn injury enhances FoxO3a activation by repressing IRS1 protein expression, promotes skeletal cell apoptosis and ultimately induces skeletal muscle atrophy. Our study has provided us with new insights regarding the role of miR-628 in regulating burn-induced skeletal muscle atrophy and has shown that this molecule may be a therapeutic target in burn patients.
Abbreviations
IRS1: insulin receptor substrate 1, FoxO: forkhead box protein O, MuRF1: muscle RING finger 1, MAFbx: muscle atrophy F-box, Pax: paired box, 3'-UTR: 3' untranslated region, TBSA: total body surface area, TNF-α: tumor necrosis factor α, PI-3K: phosphatidylinositol-3 kinase, SIRT1: silent mating type information regulation 2 homolog 1, HSP70: heat shock protein 70, TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was partially supported by grants from the National Natural Science Foundation of China (NSFC81120108014, NSFC81471873, NSFC81501694 and NSFC81571894).
Author contributions
Y.H.Y. and J.K.C. conceived and designed the study. Y.H.Y., X.L. and L.Y.L. performed the experiments and collected the data. Y.H.Y., H.J.Z., W.L.C., L.M., H.N.Y., H.J.D. and M.J.X. analyzed the data. Y.H.Y. drafted the manuscript. J.K.C. approved the version to be submitted.
Conflicts of interest
The authors declare no conflicts of interest.
References
1. Chen MC, Chen YL, Lee CF, Hung CH, Chou TC. Supplementation of Magnolol Attenuates Skeletal Muscle Atrophy in Bladder Cancer-Bearing Mice Undergoing Chemotherapy via Suppression of FoxO3 Activation and Induction of IGF-1. PloS one. 2015;10:e0143594
2. Zhang LL, Wang XJ, Zhou GQ, Tang LL, Lin AH, Ma J. et al. Dose-volume relationships for moderate or severe neck muscle atrophy after intensity-modulated radiotherapy in patients with nasopharyngeal carcinoma. Scientific reports. 2015;5:18415
3. Callahan LA, Supinski GS. Sepsis-induced myopathy. Critical care medicine. 2009;37:S354-67
4. Song J, Saeman MR, De Libero J, Wolf SE. Skeletal Muscle Loss is Associated with TNF Mediated Insufficient Skeletal Myogenic Activation After Burn. Shock. 2015;44:479-86
5. Thomas SS, Dong Y, Zhang L, Mitch WE. Signal regulatory protein-alpha interacts with the insulin receptor contributing to muscle wasting in chronic kidney disease. Kidney international. 2013;84:308-16
6. Hart DW, Wolf SE, Chinkes DL, Gore DC, Mlcak RP, Beauford RB. et al. Determinants of skeletal muscle catabolism after severe burn. Annals of surgery. 2000;232:455-65
7. Pereira C, Murphy K, Jeschke M, Herndon DN. Post burn muscle wasting and the effects of treatments. The international journal of biochemistry & cell biology. 2005;37:1948-61
8. Murton AJ, Constantin D, Greenhaff PL. The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochimica et biophysica acta. 2008;1782:730-43
9. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E. et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell metabolism. 2007;6:376-85
10. Yuan L, Han J, Meng Q, Xi Q, Zhuang Q, Jiang Y. et al. Muscle-specific E3 ubiquitin ligases are involved in muscle atrophy of cancer cachexia: an in vitro and in vivo study. Oncology reports. 2015;33:2261-8
11. Caron AZ, Haroun S, Leblanc E, Trensz F, Guindi C, Amrani A. et al. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC musculoskeletal disorders. 2011;12:185
12. Bargiela A, Cerro-Herreros E, Fernandez-Costa JM, Vilchez JJ, Llamusi B, Artero R. Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Disease models & mechanisms. 2015;8:679-90
13. Chung L, Ng YC. Age-related alterations in expression of apoptosis regulatory proteins and heat shock proteins in rat skeletal muscle. Biochimica et biophysica acta. 2006;1762:103-9
14. Gallo D, Gesmundo I, Trovato L, Pera G, Gargantini E, Minetto MA. et al. GH-Releasing Hormone Promotes Survival and Prevents TNF-alpha-Induced Apoptosis and Atrophy in C2C12 Myotubes. Endocrinology. 2015;156:3239-52
15. Sishi BJ, Engelbrecht AM. Tumor necrosis factor alpha (TNF-alpha) inactivates the PI3-kinase/PKB pathway and induces atrophy and apoptosis in L6 myotubes. Cytokine. 2011;54:173-84
16. Yu Y, Chai J. The function of miRNAs and their potential as therapeutic targets in burn-induced insulin resistance (review). International journal of molecular medicine. 2015;35:305-10
17. Ramocki NM, Wilkins HR, Magness ST, Simmons JG, Scull BP, Lee GH. et al. Insulin receptor substrate-1 deficiency promotes apoptosis in the putative intestinal crypt stem cell region, limits Apcmin/+ tumors, and regulates Sox9. Endocrinology. 2008;149:261-7
18. Porter HA, Perry A, Kingsley C, Tran NL, Keegan AD. IRS1 is highly expressed in localized breast tumors and regulates the sensitivity of breast cancer cells to chemotherapy, while IRS2 is highly expressed in invasive breast tumors. Cancer letters. 2013;338:239-48
19. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochimica et biophysica acta. 2011;1813:1978-86
20. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A. et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399-412
21. Liu J, Peng Y, Wang X, Fan Y, Qin C, Shi L. et al. Mitochondrial dysfunction launches dexamethasone-induced skeletal muscle atrophy via AMPK/FOXO3 signaling. Molecular pharmaceutics. 2015
22. Lee D, Goldberg AL. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. The Journal of biological chemistry. 2013;288:30515-26
23. Motohashi N, Alexander MS, Shimizu-Motohashi Y, Myers JA, Kawahara G, Kunkel LM. Regulation of IRS1/Akt insulin signaling by microRNA-128a during myogenesis. Journal of cell science. 2013;126:2678-91
24. Hudson MB, Rahnert JA, Zheng B, Woodworth-Hobbs ME, Franch HA, Price SR. miR-182 attenuates atrophy-related gene expression by targeting FoxO3 in skeletal muscle. American journal of physiology Cell physiology. 2014;307:C314-9
25. Kukreti H, Amuthavalli K, Harikumar A, Sathiyamoorthy S, Feng PZ, Anantharaj R. et al. Muscle-specific microRNA1 (miR1) targets heat shock protein 70 (HSP70) during dexamethasone-mediated atrophy. The Journal of biological chemistry. 2013;288:6663-78
26. Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X. et al. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. The Journal of cell biology. 2010;190:867-79
27. Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL. et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549-54
28. Jun H, Ying H, Daiwen C, Bing Y, Xiangbing M, Ping Z. et al. miR-628, a microRNA that is induced by Toll-like receptor stimulation, regulates porcine innate immune responses. Scientific reports. 2015;5:12226
29. Yu Y, Chai J, Zhang H, Chu W, Liu L, Ma L. et al. miR-194 Promotes burn-induced hyperglycemia via attenuating IGF-IR expression. Shock. 2014;42:578-84
30. Feng Y, Chai J, Chu W, Ma L, Zhang P, Duan H. Combination of ketamine and xylazine exacerbates cardiac dysfunction in severely scalded rats during the shock stage. Experimental and therapeutic medicine. 2013;6:641-8
31. Koh A, Lee MN, Yang YR, Jeong H, Ghim J, Noh J. et al. C1-Ten is a protein tyrosine phosphatase of insulin receptor substrate 1 (IRS-1), regulating IRS-1 stability and muscle atrophy. Mol Cell Biol. 2013;33:1608-20
32. Haijun Z, Yonghui Y, Jiake C, Hongjie D. Detection of the MicroRNA expression profile in skeletal muscles of burn trauma at the early stage in rats. Ulusal travma ve acil cerrahi dergisi = Turkish journal of trauma & emergency surgery: TJTES. 2015;21:241-7
33. Dupont-Versteegden EE. Apoptosis in skeletal muscle and its relevance to atrophy. World journal of gastroenterology. 2006;12:7463-6
34. Shi H, Verma M, Zhang L, Dong C, Flavell RA, Bennett AM. Improved regenerative myogenesis and muscular dystrophy in mice lacking Mkp5. The Journal of clinical investigation. 2013;123:2064-77
35. Boon H, Sjogren RJ, Massart J, Egan B, Kostovski E, Iversen PO. et al. MicroRNA-208b progressively declines after spinal cord injury in humans and is inversely related to myostatin expression. Physiological reports. 2015:3
36. Wong CF, Tellam RL. MicroRNA-26a targets the histone methyltransferase Enhancer of Zeste homolog 2 during myogenesis. The Journal of biological chemistry. 2008;283:9836-43
37. Dey BK, Gagan J, Yan Z, Dutta A. miR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes & development. 2012;26:2180-91
38. Antoniou A, Mastroyiannopoulos NP, Uney JB, Phylactou LA. miR-186 inhibits muscle cell differentiation through myogenin regulation. The Journal of biological chemistry. 2014;289:3923-35
39. Koutsoulidou A, Kyriakides TC, Papadimas GK, Christou Y, Kararizou E, Papanicolaou EZ. et al. Elevated Muscle-Specific miRNAs in Serum of Myotonic Dystrophy Patients Relate to Muscle Disease Progress. PloS one. 2015;10:e0125341
40. Tasca E, Pegoraro V, Merico A, Angelini C. Circulating microRNAs as biomarkers of muscle differentiation and atrophy in ALS. Clinical neuropathology. 2016;35:22-30
41. Lu XM, Tompkins R, Fischman A. Burn injury-induced IRS-1 degradation in mouse skeletal muscle. International journal of burns and trauma. 2013;3:37-48
42. Pilon G, Charbonneau A, White PJ, Dallaire P, Perreault M, Kapur S. et al. Endotoxin mediated-iNOS induction causes insulin resistance via ONOO(-) induced tyrosine nitration of IRS-1 in skeletal muscle. PloS one. 2010;5:e15912
43. Sugita M, Sugita H, Kim M, Mao J, Yasuda Y, Habiro M. et al. Inducible nitric oxide synthase deficiency ameliorates skeletal muscle insulin resistance but does not alter unexpected lower blood glucose levels after burn injury in C57BL/6 mice. Metabolism: clinical and experimental. 2012;61:127-36
44. Son YH, Lee SJ, Lee KB, Lee JH, Jeong EM, Chung SG. et al. Dexamethasone downregulates caveolin-1 causing muscle atrophy via inhibited insulin signaling. The Journal of endocrinology. 2015;225:27-37
45. Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. The Journal of clinical investigation. 2009;119:3059-69
46. Crossland H, Constantin-Teodosiu D, Gardiner SM, Constantin D, Greenhaff PL. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. The Journal of physiology. 2008;586:5589-600
47. Liu Y, Chen F, Odle J, Lin X, Zhu H, Shi H. et al. Fish oil increases muscle protein mass and modulates Akt/FOXO, TLR4, and NOD signaling in weanling piglets after lipopolysaccharide challenge. The Journal of nutrition. 2013;143:1331-9
48. Tucka J, Yu H, Gray K, Figg N, Maguire J, Lam B. et al. Akt1 regulates vascular smooth muscle cell apoptosis through FoxO3a and Apaf1 and protects against arterial remodeling and atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:2421-8
49. Chan KM, Rajab NF, Siegel D, Din LB, Ross D, Inayat-Hussain SH. Goniothalamin induces coronary artery smooth muscle cells apoptosis: the p53-dependent caspase-2 activation pathway. Toxicological sciences: an official journal of the Society of Toxicology. 2010;116:533-48
50. Wang Q, Li C, Peng X, Kang Q, Deng D, Zhang L. et al. Combined treatment of carfilzomib and z-VAD-fmk inhibits skeletal proteolysis and apoptosis and ameliorates cancer cachexia. Medical oncology. 2015;32:100
Author contact
![]() Corresponding author: Chai Jiake, Ph.D. Department of Burn & Plastic Surgery, the First Affiliated Hospital of PLA General Hospital, 51 Fucheng Road, Haidian District, Beijing, 100048 China. Tel: +86-10-66867972 Fax: +86-10-68989181 Email: cjk304com.
Corresponding author: Chai Jiake, Ph.D. Department of Burn & Plastic Surgery, the First Affiliated Hospital of PLA General Hospital, 51 Fucheng Road, Haidian District, Beijing, 100048 China. Tel: +86-10-66867972 Fax: +86-10-68989181 Email: cjk304com.