Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Engineered nucleases in genome...
Working mechanism of CRISPR-Cas9
Technical advance of CRISPR-Cas9...
CRISPR screening: a new epoch...
CRISPR: friendly access to...
Future aspects
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(12):1427-1436. doi:10.7150/ijbs.17421 This issue Cite
Review
CRISPR-Cas9: from Genome Editing to Cancer Research
Si Chen*, Heng Sun*, Kai Miao*, Chu-Xia Deng ![]()
Faculty of Health Sciences, University of Macau, Macau SAR, China.
*equal contribution
Received 2016-9-1; Accepted 2016-9-19; Published 2016-11-4
Abstract

Cancer development is a multistep process triggered by innate and acquired mutations, which cause the functional abnormality and determine the initiation and progression of tumorigenesis. Gene editing is a widely used engineering tool for generating mutations that enhance tumorigenesis. The recent developed clustered regularly interspaced short palindromic repeats-CRISPR-associated 9 (CRISPR-Cas9) system renews the genome editing approach into a more convenient and efficient way. By rapidly introducing genetic modifications in cell lines, organs and animals, CRISPR-Cas9 system extends the gene editing into whole genome screening, both in loss-of-function and gain-of-function manners. Meanwhile, the system accelerates the establishment of animal cancer models, promoting in vivo studies for cancer research. Furthermore, CRISPR-Cas9 system is modified into diverse innovative tools for observing the dynamic bioprocesses in cancer studies, such as image tracing for targeted DNA, regulation of transcription activation or repression. Here, we view recent technical advances in the application of CRISPR-Cas9 system in cancer genetics, large-scale cancer driver gene hunting, animal cancer modeling and functional studies.
Keywords: CRISPR, cancer, genome editing, large-scale screening, animal model.
Introduction
Cancer is a malignant disease characterized by accumulation of multiple genetic and epigenetic alterations in whole genome [1]. The mutated cancer driver genes usually dominate cancer proceeding, and determine the future of tumorigenesis [2]. However, cancer initiation processes are out of observation, and some operable and time-saving approaches are necessary to manipulate the mammalian genome, and to mimic the cancer initiation and progress [3]. Large-scale genomic screening is a possible solution to monitor the tumorigenesis caused by incident gene mutations; and CRISPR system rightly provides a tool to manipulate genes in whole genome, making them visible and traceable.
Genetically engineered mouse models have been used as powerful tools for studying cancers, although they are usually time-consuming, technically challenging and financially costing [4]. The recently developed CRISPR-Cas9 system could greatly facilitate the generation of mutant mice by simultaneously modifying several genes at one step [5]. For instance, Randall et al. (2014) established lung adenocarcinoma mice by generating modifications on three top significant genes in lung adenocarcinoma. Using Cas9-CRISPR system, they knocked out p53 and Lkb1, and knocked in KrasG12D, leading to spontaneous adenocarcinoma in the lung of mutant mice [6].
Shortly after CRISPR-Cas9 system was applied to mammalian genome editing, scientists continually modified the system to increase its efficiency, reduce the off-target effect and simplify its delivery method [7-9]. The system is also modified for new applications besides gene editing, such as imaging targeted DNA, and regulating transcription activation or repression, etc.
Engineered nucleases in genome editing
Genome editing is a strategy to modify genomic DNA and change the genetic information artificially, including gain-of-function (gene knockin, gene mutation, gene labeling and gene activation) and loss-of-function (gene knockout, and gene mutation) [3]. This technology was developed based on the discovery that DNA double strand breaks (DSBs) could stimulate endogenous DNA repair machinery, mainly through homology-directed repair (HDR) and nonhomologous end-joining (NHEJ) [10]. HDR works when the impaired sites have homologue DNA sequences in the nucleus. This mechanism protects genetic information because homologous DNA will be used as templates for the repair. If homologous DNA is absent, NHEJ works to repair the DSBs. Lacking of templates, NHEJ easily loses genetic information and introduces insertions or deletions (indels) into damaged sites [11]. When site-specially inducing DSBs, the NHEJ could be used to introduce gene alteration in cell lines or animal organs. Nowadays, three nucleases are widely used in genome editing: zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN) and CRISPR [7].
In 1990s, ZFN was applied to site-specific gene editing. ZFN consists of DNA binding domain and DNA cleavage domain. The DNA binding domain contains an array of Cys2His2 zinc fingers (ZFs); and each ZF unit catches a single atom of zinc like a finger by about 30 amino acids and recognizes 3bp of DNA [12]. The DNA cleavage domain is the FokI restriction endonuclease, which can be guided to target sites as dimers and to achieve effective genome editing [13-15] (Table 1, Figure 1). Since this editing system needs to be synthesized commercially and is difficult to use, ZFN was gradually replaced by other systems.
Comparison of ZFN, TALEN, and CRISPR-Cas9.
| Time first introduced into mammalian(year) | DNA recognition pattern | DNA modification pattern | Validation time | Relative efficiency* | Relative specificity* | Clinical development | |
|---|---|---|---|---|---|---|---|
| ZFN | 2000 | Zinc finger protein | Fok1 nuclease fused with ZFs | 8 weeks | ++ | + | Phase 1/2 |
| TALEN | 2011 | TAL protein | Fok1 nuclease fused with TALENs | 8 weeks | ++ | +++ | Phase 1 |
| CRISPR-Cas9 | 2013 | Single strand guide RNA | Cas9 nuclease | 2-4 weeks | +++ | ++++ | Preclinical |
Abbreviations: ZFN, zinc finger nuclease; TALEN, transcription activator-like effector nuclease; Cas9, CRISPR associated protein 9; TAL, transcription activator-like. The asterisk (*) markers mean relative activity and specificity of each technology compared with others.
The comparison of working mechanism among ZFN, TALEN and CRISPR. ZFN consists of a zinc finger DNA-binding domain and DNA cleavage domain of the FokI type IIS restriction endonuclease; TALEN similarly fuses FokI endonuclease with DNA-binding domain. CRISPR-Cas9 system recognizes site-specific DNA sequences through sgRNA instead of protein, and its specificity highly elevates compared with ZFN and TALEN. These systems all create double-strand breaks (DSBs) near the targeted DNA locus and initiate the DNA repair procedures.
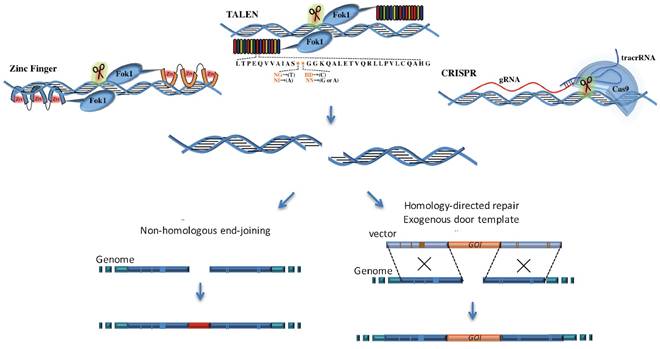
TALEN is another engineered nuclease, which shows better specificity and efficiency than ZFN. Similar to ZFN, TALEN consists of DNA-binding domain and DNA cleavage domain [16]. The non-specific DNA cleavage domain is FokI endonuclease, which has cleavage ability and works only as a dimer to cut target DNA. DNA-binding protein was first discovered in Xanthomonas bacteria, which typically consists of a tandem array of 15 to 19 modules. Each module contains 34 amino acids residues [17]; and every residue could recognize corresponding 1-4bp nucleotide sequences. By ranging the modules in different ways, the enzyme could target specific sequences with relatively high frequency [17-19]. However, the major challenge for TALEN is to clone the large modules in series, joint these modules in designed order by ligase in an efficient way. Another technique barrier is low screening efficiency for successful targeted cells [20]. Such difficulties limit its application to genome editing in recent years.
CRISPR system appears as a more powerful tool for gene editing of mammalian genome [21, 22] (Table 1 and Figure 1). As scientists continually strive to improve the system, CRISPR systems exhibit little side effects and excellent efficiency in genome editing, and extend their application to cancer research, gene therapy and functional studies [23-25]. Early in 1987, CRISPR was originally discovered as an adjacent region related to alkaline phosphatase gene in bacteria (Escherichia coli) [26], but its powerful function remained undiscovered for many decades. By observing that spacer sequence exhibits homology to extra chromosomal elements, Bolotin et al. (2006) demonstrated that CRISPR works as immune machinery against invasive genetic elements [27]. Makarova et al. (2006) reported that CRISPR-derived immunity might be conducted by RNA interference [28]. Later in 2007, cas genes were discovered to be responsible for the resistance and spacer content. Early in 2013, the CRISPR-Cas9 system was first applied to human genome editing and achieved high targeting frequencies [9, 29].
CRISPR-Cas9 system rapidly dominated the gene-editing field and showed great advantages during the past three years. First, it is extremely easy with high successful rate for making targeting constructs since it is a short guide RNA (sgRNA) based system. Researchers only need to design specific sgRNA (about 20 bp) for different targets and construct them into the Cas9 plasmid. Second, CRISPR-Cas9 shows higher specificity and efficiency compared with ZFN and TALEN [7], though the efficiency may vary among different species, methods and purposes. Lastly, by modifying the structure of CRISPR-Cas9, this system could be widely applied to different areas other than gene editing.
Working mechanism of CRISPR-Cas9
CRISPR-Cas9 nucleases were first discovered as small-RNA-based adaptive immune system, which mainly protect prokaryotes from infectious bacteria or viruses [30]. The CRISPR system can sense the invaded DNA or RNA via CRISPR loci, which is arranged of short spacer sequences and interval short repeated segments. Studies show some spacer sequences are homologues to the DNA of invaded plasmid or virus [27], leading to the hypothesis that CRISPR system works as immune machinery in prokaryotes [27, 31, 32]. Further studies supported the hypothesis, that the invaded DNA is recognized by CRISPR, and cas proteins cleave exogenous DNA, protecting the host by destructing the invader's genetic codes.
According to the structure variation of cas genes and their organization styles, the system mainly contains three types of prokaryotic CRISPR immune systems [33]. Due to its high efficiency and simplicity, the widely used CRISPR system in mammalian derives from type II CRISPR system in S. pyogenes. The system composes of a guide RNA component and a Cas9 nuclease as an enzyme component [28]. The guide RNA component is a single-guide RNA (sgRNA) fused with two key RNA units (CRISPR RNA, also named crRNA; and trans-activating crRNA, also called tracrRNA) [9]. The activation of Cas9 nuclease depends on sgRNA recognition of target DNA sequence. sgRNA strictly recognizes the protospacer adjacent motif (PAM) at the end of target DNA sequence, then unwinds the double strand DNA. PAM is a special marker for Cas9 to distinguish the target DNA from its own genome, lack of PAM will lead to failure of unwinding the target. PAM constitutes of an NAG or an NGG [34] nucleotides for Streptococcus pyogenes-initiated Cas9. Actually, different Cas9 orthologous correspond to different PAM sequences [35]. When sgRNA hybridizes to the complementary targeted nucleotides, the Cas9 unwinds double strand DNA immediately upstream of the target sequences and start the cleavage [35]. Unlike searching for target sequences by protein-DNA interaction, CRISPR-Cas9 system mediates specificity through RNA-DNA hybridization, which is much easier and more efficient [36]. By cleaving on target sequence, CRISPR-Cas9 system causes DSBs, triggering NHEJ or HDR, leading to indels in target DNA. Enormous studies reported efficient gene knockout and knockin in somatic cells or organisms by CRISPR-Cas9 system in the past years [37].
Technical advance of CRISPR-Cas9 system
Since its establishment, the CRISPR-Cas9 has been widely utilized in genome editing. Furthermore, with further innovation of this system, lots of interesting and useful modifications expand the application of CRISPR-Cas9 to many other research fields recently, which mainly rely on the binding of the nuclease Cas9 to DNA target via a programmable manner (Figure 2).
Application of CRISPR-Cas9 system in new fields. In usual cases, the cleavage activity is removed while sgRNA remains as a tool for genomic gene/RNA location. By fusing the Cas9 protein with functional domains, the system could be applied to tracking genomic loci/mRNA, transcriptional activation/ repression and base modification.
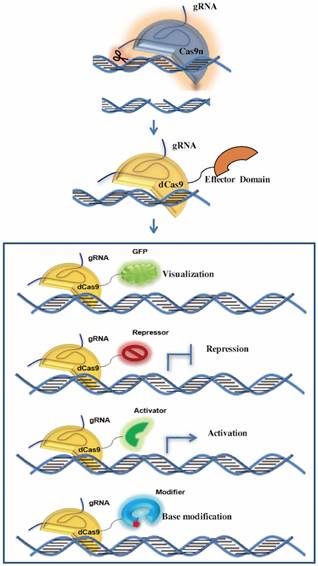
In many cases, Cas9 remains as targeting tools without cleavage ability. Cas9 cleaves the complementary and non-complementary strands of the target DNA by using its two conserved nuclease domains, RuvC and HNH respectively. The endonuclease activity of HNH domains can be abolished via an H840A site mutation while the D10A mutation can eliminate the function of the RuvC domain [28]. In addition, each or both of these two catalytically inactive mutations does not affect the binding of Cas9 to its target through a programmable manner, which facilitates the application of CRISPR-Cas9 in many other ways as well as generating DNA DSBs for genome editing, such as transcription regulation, genomic loci and mRNA imaging, genomic base editing and so on.
Cas9 nickase (Cas9n) induces single-strand nicks
Cas9n consists of one inactivating mutation (D10A or H840A) in the endonuclease cleavage domains while keeps the other cleavage domain functional. Therefore, instead of generating DSBs by the wild type Cas9, Cas9n is capable of inducing single-strand nicks, which is prone to be repaired by the high-fidelity base excision repair pathway. Hence, by combining Cas9n with a pair of appropriate offset sgRNAs that are complementary to opposite strands of the target site, it is possible to generate a staggered DSB for activating the DNA repair process. Since the double nicking strategy needs Cas9n to generate two nicks in the on-target double strand DNA with the help of two adjacent gRNAs, and rarely induce double strand breaks on the off-target sites, it dramatically reduces the off-target occurrence compared to the wild-type Cas9. Thus, the specificity of the CRISPR-Cas9 system can be further improved without sacrificing its on-target cleavage efficiency [38]. The double-nicking method offers a great solution for genome editing of cultured cells or organisms with only minute off-target mutations, especially for the studies need strict control of the unwanted mutations.
Dead Cas9 (dCas9) regulates transcriptional level
The catalytically dead Cas9 mutant (dCas9) is generated by double mutations to block enzymatic activities of both the RuvC and HNH domains in the Cas9 nuclease [39]. Once targeted to the promoter of a given gene, dCas9 itself is able to perturb the gene expression without modifying the DNA sequence through blocking the transcription initiation and elongation by the dCas9-sgRNA complex. Further, more robust gene expression repression could be achieved by fusing dCas9 to transcriptional repressors. Fusing four copies of the transcription activator VP16 (named VP64) or a single copy of p65 activation domain to dCas9 could also efficiently enhance the target gene expression [40]. To expand the application of CRISPR-dCas9 mediated transcription regulation, multiple sgRNAs could be delivered to silence or activate multiple genes simultaneously.
Moreover, a recently established signal amplification system, termed SunTag, could also be applied to further enhance the silence or activation of gene expression by dCas9. The Sun Tag system harnesses the high affinity and specificity of the binding between antibody and antigen to construct a protein-tagging platform to achieve the signaling amplification. Tanenbaum et al (2014) on one hand fused the antigen peptide with the dCas9, and on the other hand fused the correspondent single chain antibody with multiple copies of transcription activator VP64. Thus, based on the exact guidance of gRNA for dCas9 and the special binding between antigen and antibody, these transcription regulators could be recruited to the promoter of the target gene for enhanced transcription regulation by utilizing the dCas9-Sun Tag system [41].
Apart from constitutive activation or suppression of target genes, spatiotemporally specific transcription regulation could be realized by combining tunable genetics tools with CRISPR-Cas9 system, such as expressing dCas9 under the inducible TetO promoter [42] or utilizing the optogenetics technology to control CRISPR-Cas9 activation with blue light [43, 44]. Compared with CRISPR-Cas9 mediated genome editing, dCas9 provides a highly versatile platform to achieve flexible, tunable, reversible and efficient gene expression regulation, which serves as a useful resource for gene function and mechanism studies in vitro and in vivo.
CRISPR imaging of genomic loci and mRNA
Based on specific nucleic acid targeting, the CRISPR-dCas9 platform can also be repurposed to probe the genome. Currently, at least three strategies have been developed for genomic loci imaging by harnessing CRISPR-dCas9 system. The first and simplest one directly uses dCas9-EGFP fusions to image and track genomic loci in live cells [45]. The limitation of this system is the low brightness for imaging because only one molecular fused EGFP could be used here, though it could be slightly improved by simultaneously delivering several sgRNAs targeting a closely adjacent locus. The second CRISPR imaging method is devised on the base of dCas9-SunTag system mentioned above. While dCas9 nucleotides-specifically targeting the genomic DNA by sgRNA, the fused Sun-Tag recruits GFP or other fluorescent proteins to light the genomic loci. This dCas9-SunTag system can recruit a number of GFPs, so the imaging lightness is much stronger than the simple dCas9-EGFP fusion [41]. Recently, a third CRISPR imaging tool was developed to achieve dual-color visualization of target genomic loci by cleverly combining the programmable dCas9 platform with the one-to-one viral RNA and RNA-binding protein association system [46]. Mechanistically, sgRNAs and viral RNAs scaffolds are expressed as chimeric transcripts, which recruit both the co-expressed dCas9 and viral RNA-binding proteins that can be fluorescently tagged to specific genomic sites.
Moreover, such dual-color imaging method could be modified to achieve simultaneous imaging of up to six chromosomal loci in individual live cells by fusing two or three viral RNAs together with one sgRNA and so recruiting more than one viral RNA-binding protein to the same locus [47]. Altogether, these CRISPR imaging tools will certainly facilitate the studies of chromosomes conformation and dynamics as well as epigenetic regulatory mechanisms.
Programmable RNA tracking by Cas9 (RCas9) in live cells is another recently developed CRISPR imaging application [48]. O'Connell (2014) demonstrated that Cas9 could also bind RNAs with the existence of the PAM as part of an oligonucleotide (PAMmer) that hybridizes to the target RNA [49]. Because the Cas9 target-searching mechanism relies on PAM sequences [50], a PAM including PAMmer hybridized with mRNA (while genomic DNA just contains non-PAM sequence) exclusively targets RNA but not DNA. With the marker of the PAMmer, co-expressed dCas9-GFP fusions could recognize endogenous, unmodified mRNAs without affecting abundance of mRNA or amount of translated protein, which serves as a very useful tool for tracing RNA in a programmable manner. In addition, the RCas9 could apply to studies of RNA-protein interactions by site-specifically targeting aimed proteins to RNAs.
Base editor
Recently, Alexis Komor et al (2016) created a genomic base editor by engineering fusions of a cytidine deaminase enzyme with CRISPR-dCas9 to fulfill single base editing (C→T or G→A) without double-stranded DNA cleavage [51]. While the sgRNA guided the nuclease dead Cas9 to the genomic DNA with a target C and opened the DNA double helix, the fused deaminase enzyme converted the single strand target cytosine (C) to uracil (U). Then, the resulting U: G heteroduplex can be corrected to an A: T base pair through DNA replication or DNA repair. The base editor converts cytidines within a window of approximately five nucleotides with high efficiency and very low indels forming or off-target ratio, which extend the extent and efficiency of genome editing and provides a great potential for correcting a variety of point mutations that may be responsible for human diseases.
CRISPR screening: a new epoch for driver gene discovery in cancer research
Cancer genome carries a diversity of genetic aberrations, which are accumulated by innate and acquired mutations, and trigged by a succession of clonal expansions [52]. A major challenge in clarifying cancer initiation and progression is to identify the genes driving tumor evolution [2]. Most clinical cancer samples include a chaotic mixture of mutations that have appeared during disease progression. Initiation and subsequently occurring mutations act coordinately to produce aberrant physical and biologic processes, leading to complex mix of characteristics. However, it is the initial mutations that dominate cancer initiation and determine its progression and eventually metastasis [53, 54].
Large-scale genomic screening is a powerful technology capable of detecting the genes whose mutation initiates various cancers [52]. Whole genomic gene knockdown (by siRNA or shRNA) and overexpression (expression of open reading frames) strategies are widely used to screen for such genes in vitro, in vivo, or both [55-57]. However, the poor knockdown efficiency of siRNA and shRNA strategies is problematic, making gene silent difficult and library screening labor intensive.
Loss-of-function screening in vitro
The CRISPR-Cas9 system is frequently utilized in loss-of-function mutation studies [58], which makes knockout strategy applicable to large-scale screening successfully. Using CRISPR-Cas9 system, several outstanding studies have discovered novel tumor suppressors in human somatic cell lines [9, 29, 58-62] (Table 2). In 2014, Zhang lab established the lentiviral system of a genome-scale CRISPR-Cas9 knockout (GeCKO) library [60]. There are 64751 unique guide sequences cloned into the lentivirus plasmids, which target to 18080 human genes for gene knockout. Using this system, they screened the candidate genes responding to vemurafenib, a therapeutic RAF inhibitor in a melanoma model [60]. The pioneering work provides scientists worldwide with cheap and accessible library plasmids in Addgene. In the same issue in Science, Wang et al. (2014) constructed a similar pooled, loss-of-function lentiviral sgRNA library containing 73000 sgRNAs to screen two human cell lines, and discovered genes in DNA mismatch repair pathway involved in drug resistance to the nucleotide analog 6-thioguanine [29]. This area is so active that four months later, another team reported its new lentiviral CRISPR libraries for human being with more efficient screening markers. Differing from establishing whole genome libraries [60], they only picked 291 interesting genes to construct a small library based on previous studies, the smaller pool screening has more clear and specific functional direction [63]. Their system contains two plasmids, plasmid A expressing Cas9 and organic cation transporter 1 (OCT1) protein, whose overexpression was reported to enhance U6 promoter activity to elevate the expression level of sgRNAs [64]. Plasmid B expresses sgRNA with GFP, GFP serves as functional screening tag. Cells with GFP signals indicate sgRNA successfully integrated and expressed, which may be the most convenience of this system.
Application of Large-scale genetic screening by CRISPR-Cas9 library.
| Year | Approach | Alteration | Targeted species | Subjective | Delivery | Discovery | Reference |
|---|---|---|---|---|---|---|---|
| 2014 | In vitro | Loss-of-function | Human being | melanoma cell line | CRISPR-Cas9 knockout (GeCKO) library by 1 vector | a. LentiCRISPR library b. Genes for vemurafenib resistance | [60] |
| 2014 | In vitro | Loss-of-function | Human being | KBM7, CML cell line | pooled sgRNA library by 1 vector | a. 4 Lenti CRISPR library divided by functions b. Genes response to 6-thioguanine. | [29] |
| 2014 | In vitro | Loss-of-function | Human being | HeLa cell line | sgRNA library by 2 vectors | Lenti CRISPR library in two vector system with OCT1 and GFP expression | [63] |
| 2014 | In vitro | Loss-of-function | Mouse | ESCs | pooled sgRNA library by 1 vector | a. Lenti CRISPR library in 1 vector system b. 31 genes for clostridium septicum alpha-toxin or 6-thioguanine resistance | [59] |
| 2014 | In vitro | Gain-of-function | Human Being | 293FT cells | Pooled sgRNA library by 1 vector | a. Activator library b. Genes for a BRAF inhibitor resistance | [70] |
| 2014 | In vitro | Activation or repression | Human being | K562 cells | Pooled sgRNA library by 2 vectors | a. large-scale CRISPRa and CRISPRi libraries b. Essential genes, regulators of differentiation and tumor suppressors. | [67] |
| 2015 | In vivo | Loss-of-function | Mouse | Immunocompromised mice | GeCKO library | Genes for tumor growth and metastasis | [53] |
| 2015 | In vitro | Loss of function | Human being | KBM7, K562, Raji and Jiyoye | Pooled sgRNA library by 2 vectors | a. CRISPR pooled library optimized for cleavage activity, b. Genes required for proliferation and survival. | [71] |
| 2015 | In Vitro | Loss-of-function | Human being | DLD1, HCT116, RPE1, HeLa, GBM, A375 | Pooled sgRNA library by 2 vectors | a. Lenti CRISPR library of all human protein-coding genes without genomic off-target sites b. Oncogenic drivers specific to different cancer cell lines | [61] |
| 2016 | In vitro | Loss-of-function | Human being Mouse | A375 cell line | Pooled sgRNA library by 1/2 vectors | Reduced off-target lenti CRISPR library | [72] |
| 2016 | In vivo | Loss-of-function | Mouse | Pancreas in KARSmu, BRAC2-/- mouse | Pooled sgRNA library on px330 | a. DNA library of CRISPR b. Electronic transfection | [70] |
| 2016 | In vitro | Enhancer | Human | BJ-RASG12V | Pooled sgRNA library by 2 vectors | a. Libraries for p53 and ERα -bound enhancers b. Functional enhancer elements in mediating p53 and ERα gene regulation. | [69] |
Loss-of-function screening in vivo
Soon after large-scale screen in vitro, Zhang's team pioneered an in vivo loss-of-function Cas9 screening for tumor development and metastasis [53]. They used a lentivirus system (mGeCKOa) for sgRNA library delivery. The mGeCKOa is a pooled loss-of-function library containing 67405 sgRNAs targeting 20611 genes, 1175 microRNA precursors and 1000 control sgRNAs (termed non-targeting sgRNAs) [65]. In the study, they mainly focused on tumorigenesis and metastasis in lung cancer. They infected mouse lung cancer cell lines with mGeCKOa, followed by transplanting the infected cells into immunocompromised mice. Six weeks later, they took out orthotopic and metastasizing tumors sequenced for enriched sgRNAs. The enriched sgRNAs were a small portion of whole genome according to the result, indicating that specific loss-of-function mutations trigger tumor initiation and metastasis. This work provides a robust in vivo model to screen the cancer driver genes. Moreover, this model could be designed to clarify other onco-genotypes, genetic diseases, and metastasis in target organs [66]. Beside subcutaneous transplantation in this study, other cell delivery methods (orthotopic transplantation or intravenous injection) may provide localized information mimicking physical and biological situations.
Gain-of-function and other types of screenings
Besides loss-of-function screening by CRISPR-Cas9 system, several studies provide genomic screening for gain-of-function, such as transcriptional activation or activation of enhancers [67-69]. Gozde et al. (2016) identified enhancer elements by large-scale screening via CRISPR-Cas9 system. In order to find functional enhancers and demonstrate their role in mediating p53 and ERα, they targeted Cas9 to transcriptional factor binding sites of p53 in enhancer region via specific sgRNAs [69]. This outstanding study expanded the scope of CRISPR-Cas9 to map the characteristics of noncoding genome.
With some modifications, the Cas9 screening could be applied to studies in different ways. A very recent study modified the delivery method by electronic transfection, it provides a new and rapid method for mutant generation, especially in adult organs [70] (Table 2). They used electroporation-based vector delivery approach to transfect plasmid mixture into adult mouse pancreas, the mixed reagent includes plasmids of expressing Cas9 protein and sgRNAs. The sgRNAs target multiplexed genes which might initiate pancreatic tumor growth and metastasis. Their methods provided a possible way to simultaneously knock out several vital genes without virus, to analyze combinatorial gene-network within some typical cancers. In their work, the pancreas was mobilized after laparotomy, making it convenient for intraparenchymal injection. The CRISPR-Cas9 vectors were prepared together with GFP vector to indicate the successful injection. This approach could be applied to large-scale analysis of gene function, gene interaction in cancer and other genetic diseases as well as chromosome structural variations.
CRISPR: friendly access to animal cancer model
Besides large-scaled screening, CRISPR also accelerates the establishment of cancer models [73]. CRISPR provides a more accessible and efficient tool to introduce genetic alterations into mouse embryonic stem (ES) cells or into zygote [74]. Some successful cases were also reported in somatic organs of adult mice models by directly introducing naked DNA or virus-packaged DNA. Hanyi et al. (2013) successfully generated mice carrying mutations in multiple genes by CRISPR-Cas9, setting up a good example to introduce several genetic alterations via one step modification [5] (Table 3). By cotransfecting plasmids for expressing Cas9 and sgRNAs into mouse ES cells, they generated mutations simultaneously for five genes (Sry, Uty, Tet1, Tet2, Tet3) at one step with about 10% knockout frequency. Then they successfully generated mutated mice with Tet1 and Tet2 deficiency, the efficiency is up to about 80%. Besides DNA, they also coinjected mRNA of Cas9 and sgRNAs into pronuclear stage one-cell mouse oocytes; this strategy is safe with low toxicity. Even when concentration of Cas9 mRNA was up to 200ng/µl, blastocysts developed normally. They also tried to provide donors for Cas9 cleavage and generated mice carrying specific point mutations in two target genes with relatively high frequency.
Typical cancer mouse models established by CRISPR-Cas9 system.
| Year | Alteration | Objective | Delivery | Modified genes | Main technical creation | Reference |
|---|---|---|---|---|---|---|
| 2015 | Knock out | mESCs, mouse zygotes | mRNA of Cas9 and sgRNA | Sry, Uty, Tet1, Tet2, Tet3 | Knock out 5 genes at one step | [5] |
| 2015 | Knock in | Mouse zygotes | mRNA of Cas9 and sgRNA | Nanog, Oct4, Sox2, Mecp2 | Knock in 3kp DNA sequences at one step | [74] |
| 2014 | Knock out | Lung, brain, blood, etc. | Lentivirus, adeno-associated virus (AAV) or naked DNA | K-ras, p53, LKB1 | Establish stably Cas9 expressed mouse | [6] |
| 2015 | Knock out | Liver | Naked DNA | p53, Pten | Establish cancer model in adult wild-type mouse | [76] |
Several months later, the same team provided strategies to generate mice with larger DNA fragment insertion via CRISPR-Cas9, knocking the reporter and conditional alleles into mice models [74]. They designed sgRNA targeting stop codon of the interesting genes, and a homologous oligo with extra insertion fused to the last codon. Using similar approaches, they also generated mice carrying various modifications, including fluorescent reporter or specific tag in the Nanog, Oct4, and Sox2 genes, conditional mutation in Mecp2 gene, and a 3kb transgene cassette (IRES-eGFP-loxP-Neo-loxP) into Oct4 gene.
Another study established a mouse model with Cre-dependent Cas9 expression, a new system for convenient gene editing in adult organs via different delivery methods [6]. Stable expression of Cas9 simplifies the introduction procedure for CRISPR system. Through lentivirus, adeno-associated virus (AAV) or naked DNA delivery of guide RNA in different organs, CRISPR-Cas9 system can easily alter genetic information in specific organs of adult mice model, such as in the lung, brain, blood, etc. Also, this method shows great elevation on sgRNA delivery and integration. For an example, after Cas9 introduction into lung by Cre recombinase, they generated lung adenocarcinoma model mice by modifying the genes of KRAS, p53 and LKB1, which are the most frequently mutated genes in lung adenocarcinoma, with the efficiency of 33% for KRAS, 46% for p53 and 17% for LKB1, respectively [75].
Roman et al (2014) described a method to set up cancer model directly in adult wild-type mouse via CRISPR system, which shortens the time consuming [76]. The authors hydrodynamicly injected DNA plasmids expressing Cas9 and sgRNA to mouse liver directly, the sgRNA target to tumor suppressors p53 and Pten [77, 78]. In their study, Pten mutation alone led to enhanced Akt phosphorylation and lipid aggradation in hepatocytes. Double targeted Pten and p53 induced liver tumor growth, consistent with the phenomenon in traditional knockout method (knocking out these two genes by Cre-LoxP system) [79-81]. Their data provides access to set up cancer model in adult mouse liver by CRISPR-Cas9 system, avoiding engineering design of Cre-Loxp system. Though the positive frequency is relatively low to cause Pten and p53 indels (4.0 ± 0.1% for Pten and 6.4 ± 0.1% for p53), respectively, this method is sufficient to generate liver tumors.
Future aspects
With the fast progression of the CRISPR-based engineering system in cancer biology. we prospect this technology will change general picture in cancer research, provide new approaches for personalized therapy, or contribute to gene therapy, immunotherapeutic applications, genetic disorder treatment, and etc.
The efficiency and simplicity of CRISPR-Cas9 system may accelerate application to gene therapy in the future. Current works show CRISPR-Cas9 permanently correct gene mutations in the liver of an adult mouse model, partly cured hereditary tyrosinemia and rescued the body weight loss phenotype [82]. It has also been shown that monkey and mouse could be genetically modified by injecting Cas9 mRNA and sgRNAs into adult organ or zygote [83, 84].
Some new systems may provide alternative possibilities in gene editing. A very recent report showed Natronobacterium gregoryi Argonaute (NgAgo) as an engineer nuclease with comparable efficiency and specificity for genome editing to Cas9 [85]. This system seems to have a supper application potential than CRISPR-Cas9 since it does not require PAM, whereas the PAM is vital for Cas9 to unwind the target sequence. However, NgAgo is currently under serious challenge, as many scientists claimed that they failed to see any endonuclease activities under various experimental conditions. The fact that laboratories worldwide could not repeat it, at least currently, casting a doubt that NgAgo could serve as an alternative gene-editing strategy to the popular CRISPR-Cas9 system [86].
As it currently stands, CRISPR-Cas9 system may sometimes suffer an off-target problem by generating DSBs at non-specific sites [87]. Considering that the gnome modifications introduced by CRISPR-Cas9 are permanent, it could be a serious limitation, if the problem is not solved, for the use of CRISPR-Cas9 as therapy. To overcome this, a number of new techniques have been developed for better designing Cas9 nuclease and sgRNA, finding target sequences, avoiding off-targets, as well as for monitoring off-target and on-target effects [88, 89].
With endeavors of researchers in the whole world, the genome-wide specificities should be further improved; and the CRISPR-Cas9 system should be becoming a more preferable technology that can be applied in all fields of life sciences. We believe it will be a big evolution in gene therapy for cancer and genetic diseases.
Acknowledgements
We thank members of the Deng laboratory for critical reading and discussion. This work was supported, in part, by the Chair Professor Grant (CPG) and Startup Research Grant (SRG) of University of Macau granted to Chu-Xia Deng.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
2. Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17-37
3. Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15:387-95
4. Kannan R, Ventura A. The CRISPR revolution and its impact on cancer research. Swiss Med Wkly. 2015;145:w14230
5. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910-8
6. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR. et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440-55
7. Porteus M. Genome Editing: A New Approach to Human Therapeutics. Annu Rev Pharmacol Toxicol. 2016;56:163-90
8. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N. et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819-23
9. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE. et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823-6
10. Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H. et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. Embo J. 1998;17:5497-508
11. Maeder ML, Gersbach CA. Genome-editing Technologies for Gene and Cell Therapy. Mol Ther. 2016;24:430-46
12. Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188:773-82
13. Kim YG, Chandrasegaran S. Chimeric restriction endonuclease. Proc Natl Acad Sci U S A. 1994;91:883-7
14. Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156-60
15. He J, Li Q, Fang S, Guo Y, Liu T, Ye J. et al. PKD1 mono-allelic knockout is sufficient to trigger renal cystogenesis in a mini-pig model. Int J Biol Sci. 2015;11:361-9
16. Jiang W, Marraffini LA. CRISPR-Cas: New Tools for Genetic Manipulations from Bacterial Immunity Systems. Annu Rev Microbiol. 2015;69:209-28
17. Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S. et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509-12
18. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49-55
19. Masuda T, Xu X, Dimitriadis EK, Lahusen T, Deng CX. "DNA Binding Region" of BRCA1 Affects Genetic Stability through modulating the Intra-S-Phase Checkpoint. Int J Biol Sci. 2016;12:133-43
20. Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C. et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63
21. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709-12
22. Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174-82
23. Barrangou R, May AP. Unraveling the potential of CRISPR-Cas9 for gene therapy. Expert Opin Biol Ther. 2015;15:311-4
24. Barrangou R, Dudley EG. CRISPR-Based Typing and Next-Generation Tracking Technologies. Annu Rev Food Sci Technol. 2016;7:395-411
25. Li H, Sheng C, Liu H, Liu G, Du X, Du J. et al. An Effective Molecular Target Site in Hepatitis B Virus S Gene for Cas9 Cleavage and Mutational Inactivation. Int J Biol Sci. 2016;12:1104-13
26. Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429-33
27. Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551-61
28. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-21
29. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80-4
30. Barrangou R, Marraffini LA. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell. 2014;54:234-44
31. Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7
32. Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M. et al. Rapid "open-source" engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294-301
33. Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P. et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467-77
34. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827-32
35. Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ. et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110:15644-9
36. Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116-21
37. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096
38. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE. et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380-9
39. Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB. et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32:670-6
40. Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442-51
41. Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635-46
42. Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP. et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell. 2016;18:541-53
43. Nihongaki Y, Yamamoto S, Kawano F, Suzuki H, Sato M. CRISPR-Cas9-based photoactivatable transcription system. Chem Biol. 2015;22:169-74
44. Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015;11:198-200
45. Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW. et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479-91
46. Fu Y, Rocha PP, Luo VM, Raviram R, Deng Y, Mazzoni EO. et al. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat Commun. 2016;7:11707
47. Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D. et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol. 2016;34:528-30
48. Nelles DA, Fang MY, O'Connell MR, Xu JL, Markmiller SJ, Doudna JA. et al. Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell. 2016;165:488-96
49. O'Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014;516:263-6
50. Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62-7
51. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420-4
52. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
53. Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X. et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246-60
54. van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW. et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999-2009
55. Schiano C, Casamassimi A, Rienzo M, de Nigris F, Sommese L, Napoli C. Involvement of Mediator complex in malignancy. Biochim Biophys Acta. 2014;1845:66-83
56. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X. et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171-84
57. Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D. et al. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science. 2014;343:309-13
58. Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B. et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell. 2008;135:852-64
59. Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267-73
60. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84-7
61. Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G. et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515-26
62. Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ. et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096-101
63. Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y. et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487-91
64. Lin BR, Natarajan V. Negative regulation of human U6 snRNA promoter by p38 kinase through Oct-1. Gene. 2012;497:200-7
65. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783-4
66. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603-7
67. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH. et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647-61
68. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583-8
69. Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K. et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol. 2016;34:192-8
70. Maresch R, Mueller S, Veltkamp C, Ollinger R, Friedrich M, Heid I. et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat Commun. 2016;7:10770
71. Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J. et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015;12:673-83
72. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184-91
73. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC. et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423-7
74. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370-9
75. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543-50
76. Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS. et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514:380-4
77. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283-96
78. Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA. et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468:572-5
79. Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J. et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774-83
80. Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ. et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci U S A. 2004;101:2082-7
81. Katz SF, Lechel A, Obenauf AC, Begus-Nahrmann Y, Kraus JM, Hoffmann EM. et al. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology. 2012;142:1229-39 e3
82. Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M. et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32:551-3
83. Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M. et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31:681-3
84. Li W, Teng F, Li T, Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol. 2013;31:684-6
85. Gao F, Shen XZ, Jiang F, Wu Y, Han C. DNA-guided genome editing using the Natronobacterium gregoryi Argonaute. Nat Biotechnol. 2016;34:768-73
86. Cyranoski D. Replications, ridicule and a recluse: the controversy over NgAgo gene-editing intensifies. Nature. 2016;536:136-7
87. Martin F, Sanchez-Hernandez S, Gutierrez-Guerrero A, Pinedo-Gomez J, Benabdellah K. Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview. Int J Mol Sci. 2016:17
88. Rastogi A, Murik O, Bowler C, Tirichine L. PhytoCRISP-Ex: a web-based and stand-alone application to find specific target sequences for CRISPR/CAS editing. BMC Bioinformatics. 2016;17:261
89. Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB. et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148
Author contact
![]() Corresponding author: Chu-Xia Deng, cxdengmo.
Corresponding author: Chu-Xia Deng, cxdengmo.