Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(12):1555-1567. doi:10.7150/ijbs.13833 This issue Cite
Research Paper
Anticancer Effects of a New SIRT Inhibitor, MHY2256, against Human Breast Cancer MCF-7 Cells via Regulation of MDM2-p53 Binding
Eun Young Park1, Youngwoo Woo1, Seong Jin Kim1, Do Hyun Kim1, Eui Kyung Lee1, Umasankar De2, Kyeong Seok Kim2, Jaewon Lee1, Jee H. Jung1, Ki-Tae Ha3, Wahn Soo Choi4, In Su Kim2, Byung Mu Lee2, Sungpil Yoon2, Hyung Ryong Moon1 ![]() , Hyung Sik Kim2
, Hyung Sik Kim2 ![]()
1. College of Pharmacy, Pusan National University, San 30, Jangjeon-dong, Geumjeung-gu, Busan, 609-735, Republic of Korea;
2. School of Pharmacy, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, Suwon 440-746, Republic of Korea;
3. Department of Korean Medical Science, School of Korean Medicine and Korean Medicine Research Center for Healthy Aging, Pusan National University, Yangsan, Gyeongsangnam-do, Republic of Korea;
4. Department of Immunology, School of Medicine, Konkuk University, Chungju 380-701, Republic of Korea.
Received 2015-9-13; Accepted 2016-9-30; Published 2016-12-6
Abstract

The sirtuins (SIRTs), a family of NAD+-dependent class III histone deacetylase, are involved in various biological processes including cell survival, division, senescence, and metabolism via activation of the stress-response pathway. Recently, inhibition of SIRTs has been considered a promising anticancer strategy, but their precise mechanisms of action are not well understood. In particular, the relevance of p53 to SIRT-induced effects has not been fully elucidated. We investigated the anticancer effects of a novel SIRT inhibitor, MHY2256, and its efficacy was compared to that of salermide in MCF-7 (wild-type p53) and SKOV-3 (null-type p53) cells. Cell viability, SIRT1 enzyme activity, cell cycle regulation, apoptosis, and autophagic cell death were measured. We compared sensitivity to cytotoxicity in MCF-7 and SKOV-3 cells. MHY2256 significantly decreased the viability of MCF-7 (IC50, 4.8 μM) and SKOV-3 (IC50, 5.6 μM) cells after a 48 h treatment period. MHY2256 showed potent inhibition (IC50, 0.27 mM) against SIRT1 enzyme activity compared with nicotinamide (IC50, >1 mM). Moreover, expression of SIRT (1, 2, or 3) protein levels was significantly reduced by MHY2256 treatment in both MCF-7 and SKOV-3 cells. Flow cytometry analysis revealed that MHY2256 significantly induced cell cycle arrest in the G1 phase, leading to an effective increase in apoptotic cell death in MCF-7 and SKOV-3 cells. A significant increase in acetylated p53, a target protein of SIRT, was observed in MCF-7 cells after MHY2256 treatment. MHY2256 up-regulated LC3-II and induced autophagic cell death in MCF-7 cells. Furthermore, MHY2256 markedly inhibited tumor growth in a tumor xenograft model of MCF-7 cells. These results suggest that a new SIRT inhibitor, MHY2256, has anticancer activity through p53 acetylation in MCF-7 human breast cancer cells.
Keywords: SIRT inhibitor, MHY2256, MDM2, p53, apoptosis, autophagy.
Introduction
Sirtuins (SIRTs) are a family of NAD+-dependent class III histone deacetylases (HDACs) [1, 2]. There are seven human SIRTs with diverse subcellular locations and functions. SIRT1, SIRT6, and SIRT7 are nuclear proteins, but SIRT1 has been shown to move between the nucleus and cytoplasm. SIRT3, SIRT4, and SIRT5 are mitochondrial proteins, and SIRT2 is predominantly located in the cytoplasm. SIRT2 and SIRT3 also shuttle between different cell components [3-6]. These proteins are involved in critical cellular processes including stress response, cell survival, metabolism, senescence, aging, and tumorigenesis via deacetylation of many substrates [7, 8].
SIRT1 is the most extensively studied a member of SIRT family and is up-regulated in several types of tumors such as prostate, skin, lung, and colon cancers as well as chemo-resistant ovarian and breast cancer cell lines [9, 10]. SIRT1 regulates histone or non-histone proteins, including DNA repair proteins, cell signaling molecules, and transcriptional regulators including nuclear factor (NF)-κB, FOXO3, p53, PPAR-γ, PGC-1α, E2F1, and pRb [11-13]. Therefore, a number of SIRT modulators induce cancer cell growth inhibition, cell cycle arrest, apoptotic cell death, and increased sensitivity in combination with chemotherapeutic agents (cisplatin and camptothecin) [14, 15]. Recent studies have observed that p53 is not only a target of SIRT1, but also SIRT2. SIRT2 also deacetylates histone H3 lysine 56 (H3K56) and α-tubulin shares non-histone substrates of FOXO1/3 and p53 with SIRT1, and regulate the cell cycle in cancer cells [16]. SIRT3 is involved in DNA repair and cell survival via deacetylation of Ku-70 [7]. SIRT6 promotes resistance to DNA damage and can suppress apoptosis and cell cycle arrest [17]. Several types of SIRT inhibitors have been discovered that exert NAD+-dependent inhibition of substrates such as nicotinamide, suramin, cambinol, EX-527, sirtinol, and salermide [18, 19]. Salermide, a compound structurally related to sirtinol, shows a potent inhibition of SIRT1/2 and induces apoptosis in cancer cells via a SIRT1-dependent manner [20]. Based on these findings, selective SIRTs inhibitors that mainly inhibit SIRT1 or SIRT2 with a low affinity against other SIRTs subfamilies have been developed [3, 18].
Tumor suppressor p53, a key transcription factor, is activated in response to diverse forms of cellular stress and facilitates induction of cell cycle arrest, apoptosis, and senescence. Functional inactivation or mutation of p53 is a common feature of human cancer [21]. Activation of p53 has been considered an attractive cancer therapeutic strategy, and is controlled through regulation of protein expression level via mouse double minute 2 homolog (MDM2). MDM2, a negative regulator of p53, is implicated in the development of tumors with wild-type p53. MDM2 inactivates p53 through degradation of the p53 protein via the ubiquitin-proteasome pathway or by directly blocking the p53 transactivation domain. Thus, coordinated interplay between histone acetytransferases (HAT) and HDACs in the regulation of p53 acetylation is believed to play a significant role in p53-mediated apoptosis. Recently, SIRT inhibitors have also been shown to block MDM2 through inhibition of ubiquitin ligase activity or MDM2-p53 binding [22]. Several MDM2 inhibitors have potential anti-tumor activity in various human cancer cells via induction of cell cycle arrest and apoptosis via p53 activation [23-25]. Moreover, a different strategy has also been reported for p53 activation through inhibition of SIRT1-catalyzed p53 deacetylation or prevention of MDM2-mediated p53 degradation. Therefore, blocking the MDM2-p53 binding by inhibiting p53 deacetylation via a SIRT1 inhibitor could serve as an alternative strategy for the development of an anti-cancer therapy.
HDAC inhibitors have been tested in clinical trials for breast, cervical, and ovarian cancers, which are common cancers in women. We presume that more specific targeted inhibitors for class III HDACs (SIRTs) will facilitate personalized treatment plans for these cancers. In the present study, we investigated whether MHY2256 inhibits SIRT expression and exerts antitumor activity through activation of the p53 pathway in p53-dependent cancer cells. In order to investigate the relationship between MHY2256 and p53 expression, we compared two cancer cell lines, p53 wild-type (MCF-7) and p53 null-type (SKOV-3). SIRT inhibitors up-regulate p53 in some cancers [17,18]. However, the exact role of SIRTs in p53 activation remains unclear. A novel SIRT inhibitor, MHY2256, was synthesized, and we investigated its anticancer activity against various human cancer cell lines. In addition, the anticancer potency of MHY2256 was compared with that of salermide, a selective SIRT1/2 inhibitor. To achieve evaluation of MHY2256 ability in SIRT inhibition, cell viability, cell cycle regulation, and the levels of apoptosis- and autophagy-related molecules were measured.
Materials and Methods
Drug and chemicals
5-(3,5-Di-tert-butyl-4-hydroxybenzylidene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (MHY2256) was kindly provided by Prof. Hyung Ryong Moon (College of Pharmacy, Pusan National University, Busan, South Korea). Salermide was purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell culture medium, fetal bovine serum (FBS), and other reagents were obtained from Gibco Invitrogen Corporation (Carlsbad, CA, USA). The primary antibodies for poly-ADP-ribose polymerase (PARP), Bax, Bcl-2, p53, and horseradish peroxidase-conjugated secondary antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All SIRT primary antibodies were purchased from Cell Signaling (Beverly, MA, USA). All other chemicals were purchased from Sigma-Aldrich. MHY2256 and salermide were dissolved in dimethylsulfoxide (DMSO) and stored at -20ºC until use. These agents were diluted to appropriate concentrations with culture medium containing 1% FBS. The final concentration of DMSO was less than 0.1% (vol/vol) and was also present in the corresponding controls.
Cell lines and culture conditions
The MCF-7 (wild-type p53) human breast cancer and SKOV-3 (null-type p53) ovarian cancer cells lines were purchased from the American Type Culture Collection (Manassas, VA, USA). These cancer cell lines were grown in Dulbecco's Modified Eagle's Medium (Gibco, Rockville, MD, USA) containing 10% heat-inactivated fetal bovine serum (FBS), and 1% penicillin/streptomycin (Gibco). Cells were maintained as monolayers in a humidified atmosphere containing 5% CO2 at 37ºC, and culture medium was replaced every two days.
SIRT1 activity assay
SIRT1 activity was measured with the SensoLyte® 520 fluorimetric SIRT1 activity assay kit (AnaSpec, Fremont, CA) according to the manufacturer's instructions. Nicotinamide was used as a reference compound. Briefly, SIRT1 enzymes were incubated with vehicle or various concentrations of test compound at 37oC in the presence of an SIRT1 fluorimetric substrate. The SIRT1 assay developer, which produces a fluorophore in the reaction mixture, was added, and the fluorescence was measured using VICTOR X2 (Perkin Elmer, Waltham, MA, USA) with excitation at 490 nm and emission at 520 nm. The measured activities were calculated using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Cell viability assay
Cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl- tetrazolium bromide (MTT, 5mg/ml, Sigma). The cultures were initiated in 96-well plates at a density of 2.5x10³ cells per well. After 48 h of incubation, cells were exposed to salermide or MHY2256 at various concentrations and cultured for additional 48 h. After incubation, 15 µl of MTT reagent was added to each well and incubated for 4 h at 37ºC in the dark. The supernatant was aspirated, and formazan crystals were dissolved in 100 µl of DMSO at 37ºC for 15 min with gentle agitation. The absorbance per well was measured at 540 nm using the VERSA Max Microplate Reader (Molecular Devices Corp., Sunnyvale, CA, USA). Data from three independent experiments was analyzed and normalized to the absorbance of wells containing media only (0%) and to that of untreated cells (100%). IC50 values were calculated from sigmoidal dose response curves with SigmaPlot 10.0 software.
Flow cytometry analysis
The cells were treated with various concentrations of MHY2256 for 48 h, and were harvested separately. Cells (1x106) were then washed with PBS containing 1% BSA and fixed in chilled 95% ethanol, and stained with cold propidium iodine (PI) solution (10 μg/ml PI and 100 μg/ml RNase in PBS), and incubated in the dark for 30 min at room temperature. Data acquisition and analysis were carried out using a Guava EasyCyte Plus Flow Cytometer (Merck Millipore, Billerica, MA, USA).
Annexin V/7-AAD binding assay
The Annexin V/7-AAD binding assay was performed according to the manufacturer's instruction using the MuseTM Annexin V & Dead Cell Kit (Millipore, Billerica, MA, USA). The cells were treated with MHY2256 and salermide for 48 h. The total number of cells were counted after trypsinization and washed twice with cold PBS. The cell pellet was resuspended in 1% FBS media at a density of 1x103 cells per ml and incubated with 100 μl of Muse Annexin V & dead cell reagent for 15 min at room temperature in the dark. The samples were then immediately analyzed using Guava EasyCyte Plus Flow Cytometer (Millipore, Billerica, MA, USA).
Western blot analysis
Cells were treated with drugs for 48 h, harvested via trypsinization, and washed twice with cold PBS. For total protein isolation, cells were suspended in PRO-PREP™ protein extract solution (iNtRON, Seongnam, Korea). Protein concentrations ware measured using a protein assay kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer's instructions. The cell extract with 20 μg protein was loaded on 6-15% SDS-polyacrylamide (PAGE) gel. After electrophoresis, gels were transferred to a polyvinylidene difuride (PVDF) membrane (Millipore, Billerica, MA, USA). The membrane was incubated for 1 h in TNA (10 mM Tris-Cl, pH-7.6, 100 mM NaCl, and 0.5% Tween 20) buffer containing 5% skim milk. Next, the membrane was incubated with various primary antibodies at 4°C overnight. After washing for 1 h with TNA buffer, membranes were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies (1:10000, Santa Cruz, CA, USA) for 30 min at room temperature. The blots were developed using an enhanced chemiluminescence (ECL)-plus kit (Amersham Biosciences, Amersham, Buckinghamshire, UK).
Acridine orange staining
The cells were seeded in T-25 flasks and were treated with MHY2256 for 48 h at 70% confluence. At the appropriate time points, cells were incubated with acridine orange (1 μg/ml) in serum-free medium at 37˚C for 15 min. The acridine orange was removed, and fluorescent micrographs were obtained using an inverted fluorescence microscope (Axiovert 200 fluorescence microscope; Zeiss, Thornwood, NY). The cytoplasm and nucleus of the stained cells fluoresced bright green, whereas the acidic autophagic vacuoles fluoresced bright red. Cells were treated with 200 nmol/l bafilomycin A1 for 30 min before the addition of acridine orange in order to inhibit the acidification of autophagic vacuoles. To quantify the development of acidic vesicular organelles (AVOs), cells were stained with acridine orange (1 μg/ml) for 15 min, and removed from the plate with trypsin-EDTA, and collected in phenol red-free growth medium. Green (510-530 nm) and red (650 nm) fluorescence emission from 1x104 cells illuminated with blue (488 nm) excitation light was measured with a Guava EasyCyte Plus Flow Cytometer (Merck Millipore, Billerica, MA, USA).
Tumor xenograft model
Six-week-old female nude mice (BALB-c nu/nu) weighing approximately 20 g were housed in filtered-air laminar-flow cabinets at a controlled temperature (22 ± 2°C) with a 12 h light/dark cycle. The institutional animal care committee of Sungkyunkwan University approved the experimental procedure. MCF-7 cells (5×105 cells/0.1 ml) in serum-free medium containing 50% Matrigel were injected subcutaneously. Prior to drug administration, the mice were randomized to four groups (n = 6): Group 1 was the control group; Group 2 was treated with doxorubicin (4 mg/kg); Group 3 was treated with salermide (30 mg/kg); and Group 4 was treated with MHY2256 (5 mg/kg). Doxorubicin (once/week) and MHY2256 (twice/week) were injected intraperitoneally (i.p.) for 4 weeks. Tumor volume (V) was calculated using the following standard formula: V (mm3) = 0.52(ab2), where a is the length and b is the width of tumor. Body weights were recorded before dosing and at termination.
Statistical analysis
The data were expressed as the mean ± SEM of at least three independent experiments. Statistical analysis was performed using one-way analysis of the variance (ANOVA) followed by Bonferroni's multiple comparison tests. *p <0.05; **p<0.01 were considered statistically significant.
Results
p53 proteins are differentially expressed in human cancer cells
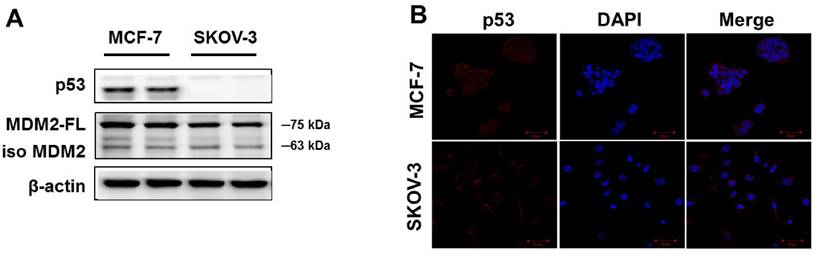
To confirm p53 expression levels in the human cancer cell lines, two types of human cancer cells were cultured without drugs and Western blot analysis was performed. As shown in Figure 1A, high p53 expression was observed in MCF-7 cells. The SKOV-3 did not show any p53 expression. Similar results were observed using immunofluorescence analysis (Figure 1B). The p53 protein was mainly located in the cytoplasm of MCF-7 cells, whereas SKOV-3 cells showed p53 accumulations in the nucleus. However, the expression level of MDM2 did not differ in any of the cancer cell lines.
Basal expression levels of p53 and MDM2 proteins in human cancer cell lines. (A) The cells were cultured in the absence of any drug treatment and were analyzed using Western blot analysis with antibodies to p53, MDM2, and β-actin (as a loading control). (B) Immunofluorescence for p53 proteins levels and fluorescence detection of p53 using rhodamine red-tagged secondary antibody was observed using confocal microscopy (Magnification x400).
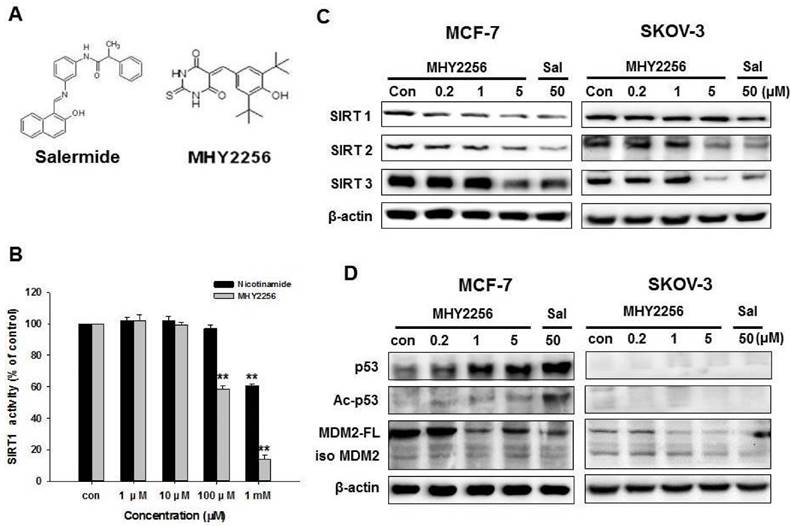
MHY2256 inhibits SIRT1 enzyme activity and SIRT protein expression in human cancer cells
The chemical structure of MHY2256 and salermide is presented in Fig. 2A. For screening, we selected MHY2256, which has high cytotoxicity and low IC50. The ranges for 0.1 μM MHY2256 were selected based on our screening data with high cytotoxicity. After we analyzed the IC50 for MHY2256 in MCF-7 cells, we selected three concentrations (0.2, 1, and 5 μM) for further analysis; 50 μM salermide was also selected based on previous studies. The effect of MHY2256 on SIRT1 enzyme activity was measured using the SensoLyte® 520 SIRT1 assay kit. Nicotinamide was used as a reference SIRT1 inhibitor. As shown in Fig. 2B, MHY2256 significantly inhibited SIRT1 enzyme activity in a concentration-dependent manner. The concentration of MHY2256 that was required for 50% inhibition (IC50 value) of SIRT1 enzyme activity was 0.27 mM, which was lower than that of nicotinamide (IC50 > 1 mM). Next, the effects of MHY2256 on SIRT protein level were examined via Western blot analysis using specific antibodies against SIRTs. Salermide, a SIRT inhibitor, was used as a positive control. SIRT1 is down-regulated only in MCF-7 cells after MHY2256 treatment, not in SKOV-3, suggesting that existence of p53 or other factor needs to reduce SIRT1 levels by MHY2256, and that this is missing in SKOV-3 cancer cells (Figure 2C). To further investigate the effects of SIRT inhibition, acetylated p53, the SIRT1/2 target protein, were measured. MHY2256 markedly increased acetylated p53 levels in MCF-7 cells, but not SKOV-3 cells. As expected, the expression level of total p53 was up-regulated in MCF-7 cells after MHY2256 treatment. Moreover, MHY2256 markedly reduced expression of MDM2, an important negative regulator of p53, in both cancer cell lines (Figure 2D).
Effect of MHY2256 on sirtuin (SIRT) and p53 expression in MCF-7 and SKOV-3 cells. (A) Chemical structure of MHY2256 used in the present study. (B) Effects of MHY2256 and nicotinamide on SIRT1 activity. SIRT1 enzyme activity was measured using the SensoLyte® 520 FRET SIRT1 assay kit. The results are presented as the percentage of activity relative to the control in each group. Statistical analysis was performed using one-way analysis of the variance (ANOVA) followed by Bonferroni's multiple comparison tests. **p <0.01 indicates significant differences between control and treatment groups. (C) Expression levels of SIRT1-3 in MHY2256- and salermide-treated cells were detected using Western blot analysis. (D) Effects of MHY2256 on p53, acetylated p53 (Ac-p53), and MDM2 expression. MCF-7 and SKOV-3 cells were treated with MHY2256 and salermide for 48 h, and then Western blot analysis was performed.
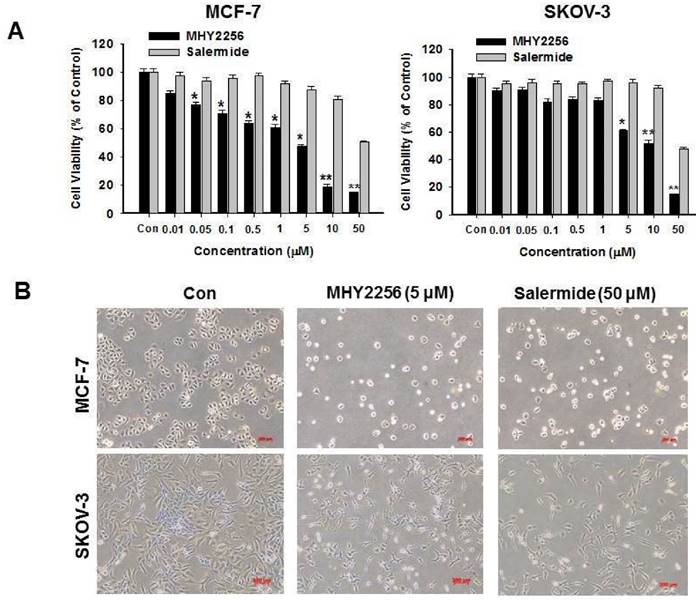
MHY2256 suppresses human cancer cell viability
MCF-7 or SKOV-3 human cancer cells were treated with different MHY2256 doses for 48 h. The cytotoxicity of MHY2256 was assessed using the MTT assay. MHY2256 significantly reduced the viability of human cancer cells in a concentration-dependent manner (Figure 3A). MHY2256 had better inhibitory effects against MCF-7 (IC50 = 4.8 μM) and SKOV-3 (IC50 = 5.6 μM) cells after 48 h of treatment compared with salermide (IC50 values 36.5-50.9 μM). Based on these results, all further experiments were performed after a 48h treatment of MHY2256. MHY2256 also exhibited the typical morphological changes in these cells (Figure 3B); the cells changed to a rounded shape or shrunk after MHY2256 treatment. We found that the SKOV-3 IC50 was much higher than that of MCF-7 cells (Figure 3A). SIRT1 protein levels were only decreased by MHY2256 in MCF-7 cells (Figure 2C), suggesting that MHY2256 targets SIRT1 to increase high cytotoxicity.
Effects of MHY2256 and salermide on cell viability. (A) The cells were treated with MHY2256 and salermide at various concentrations (0.1-50 μM) for 48 h. Viable cells were detected using MTT assay, and viability was determined as the ratio between treated cells and untreated controls. Data are presented as the mean ± SEM of three independent experiments. Statistical analysis was performed using one-way analysis of the variance (ANOVA) followed by Bonferroni's multiple comparison test. *p<0.05, **p <0.01 indicate significant differences between control and treatment groups. (B) Morphological changes in human cancer cells after MHY2256 and salermide treatment. MCF-7 and SKOV-3 cells were incubated for 48 h with the indicated drug concentrations and the cell morphology was examined microscopically (Magnification x100).
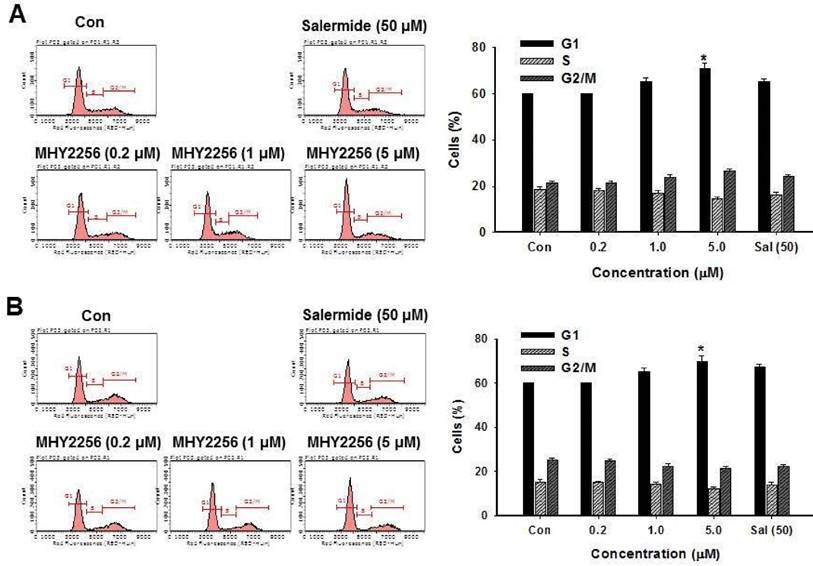
MHY2256 inhibits cell cycle distribution
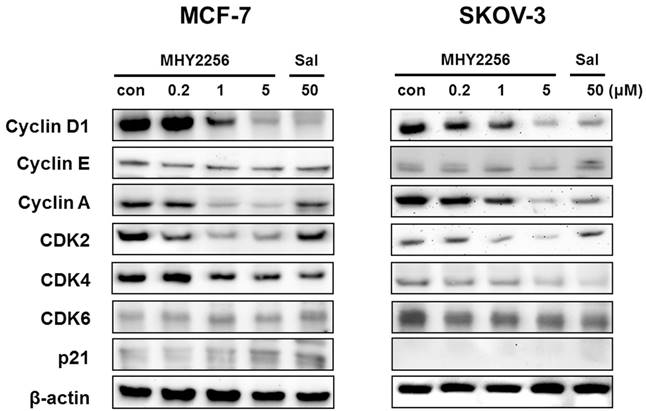
SIRT1 inhibitors block the proliferation of cancer cells via cell cycle arrest at a specific phase depending on exposure conditions [13, 14]. The effects of MHY2256 on cell cycle distribution were examined via flow cytometry. Cells were treated with the indicated concentrations of MHY2256 (0.2, 1, or 5 μM) and salermide (50 μM) for 48 h. MHY2256 markedly induced cell cycle arrest at the G1 phase in MCF-7 (Figure 4A) and SKOV-3 (Figure 4B) cells. The MHY2256-mediated cell cycle distribution was similar to that of salermide. The effects of MHY2256 on the expression levels of cell cycle-related proteins were confirmed using Western blot analysis. MHY2256 significantly decreased the expression of cyclin D1, CDK2 and CDK4, which indicates that these molecules are closely associated with the G1 cell cycle check point (Figure 5). In addition, MHY2256 significantly increased the expression of p21 in a concentration-dependent manner in MCF-7 and SKOV-3 cells. The level of cyclin A was also decreased in human cancer cells after MHY2256 treatment. In FACS analysis, we observed that G1 arrest by MHY2256 was slightly increased in both cancer cells. Further analysis over short periods of MHY2256 treatment might be needed to observe detailed cell cycle distribution changes.
Effects of MHY2256 or salermide on cell cycle regulation. MCF-7 (A) and SKOV-3 (B) cells were treated with the indicated concentrations for 48 h. Cells stained with propidium iodide (PI) were subjected to flow cytometric analysis to determine the cell distributions in each phase of the cell cycle. Statistical analysis was performed using one-way analysis of the variance (ANOVA) followed by Bonferroni's multiple comparison tests. *p<0.05 indicate significant differences between control and treatment groups.
Effects of MHY2256 on expression levels of cell cycle regulatory proteins. MCF-7 and SKOV-3 cells were treated with MHY2256 (0, 0.2, 1, and 5 μM) or salermide (50 μM) for 48 h, and then protein levels were detected using Western blot analysis. The cells were homogenized, and the proteins were isolated. Aliquots of proteins were immunoblotted with specific primary antibodies against p21, cyclin D1, cyclin E, cyclin A, CDK4, CDK6 and CDK2. Equal loading and transfer were verified by re-probing the membranes with β-actin antibody.
MHY2256 induces apoptosis in human cancer cells
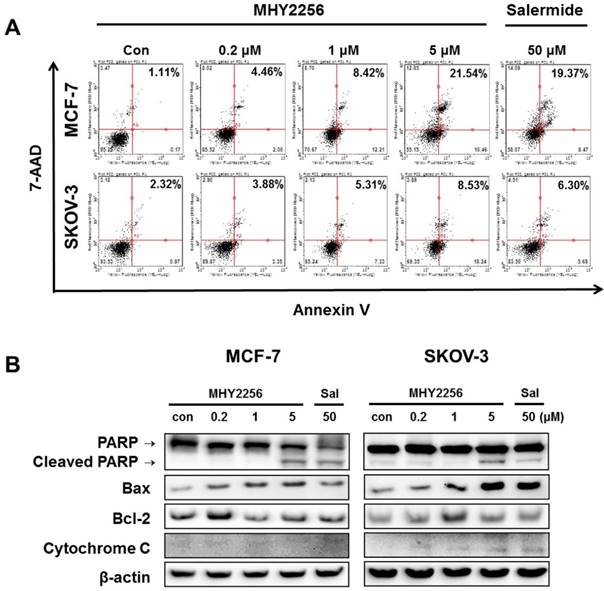
To assess the extent of apoptosis induced in human cancer cells after MHY2256 treatment, Annexin V/7-AAD double staining was performed. The percentage of late-stage apoptotic cells was increased in a concentration-dependent manner after treatment with MHY2256 (Figure 6A). In addition, both 1 μM and 5 μM concentrations of MHY2256 increased early apoptosis in MCF-7 and SKOV-3 cells (Figure 6A), whereas late apoptosis at those concentrations was increased only in MCF-7 cells, suggesting that p53 expression contributes to late apoptosis in the presence of MHY2256. To elucidate the apoptotic pathway underlying the cytotoxic effect of MHY2256, the expression of apoptosis-relative proteins were measured using Western blot analysis. MHY2256 significantly increased the levels of cleaved PARP and cytochrome c release. In addition, bax expression was markedly increased, while bcl-2 levels were decreased in MCF-7 cells, but not SKOV-3 cells (Figure 6B). Considering both cell viability analysis and annexin V staining, MHY2256 exhibits strong cytotoxicity in p53 wild-type MCF-7, but not in p53-null SKOV-3 cancer cells. We conclude that induction of apoptosis by MHY2256 is highly dependent on p53 expression levels.
Effect of MHY2256 on apoptosis. (A) Assay for apoptosis of MHY2256 and salermide treatment in MCF-7 (a) and SKOV-3 (b) cells. Cells were treated with MHY2256 or salermide for 48 h at the indicated concentrations. MHY2256-induced apoptosis was examined using annexin V/7-AAD double staining. Representative flow cytometry scatter plots indicate the percentage of cells in the early and late phases of apoptosis after drug treatment. (B) Effect of MHY2256 on expression of proteins related to apoptotic cell death. Cells were treated with MHY2256 or salermide for 48 h at the indicated concentrations, and Western blot analysis was performed to determine apoptosis-related protein levels. The protein levels were normalized by comparison to β-actin levels.
MHY2256 induces autophagic cell death
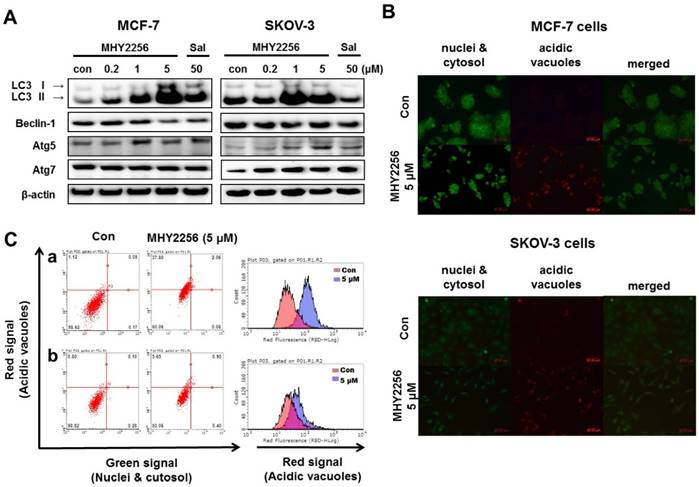
Our previous study indicated that a SIRT inhibitor, sirtinol, significantly increased autophagic cell death in MCF-7 cells [19]. Recent studies suggested that LC3 is the first protein recruited to the autophagosome membrane. The conversion of the soluble form LC3-I to the autophagic vesicle-associated form LC3-II is considered a specific marker of autophagosomes promotion [26, 27]. To assess MHY2256-induced autophgic cell death, Western blot analysis and acridine orange staining were performed. As shown in Fig. 7A, MHY2256 markedly increased the level of LC3-II in both cell lines and autophagy-related gene 5 (ATG5) and ATG7 increased in SKOV-3 cells at low MHY2256 (0.2 and 1 μM). We found that salermide did not significantly increase autophagy-related protein levels (Figure 7A), suggesting that MHY2256 could be a novel SIRT inhibitor with mechanisms of action that very from those of well-known SIRT inhibitor salermide. The induction of autophagy was confirmed by acridine orange staining. Acridine orange is a lysosomotropic agent that accumulates in acidic organelles in a pH-dependent manner. At neutral pHs, acridine orange is a hydrophobic green fluorescent molecule. In acidic compartments, acridine orange becomes protonated and trapped within the organelle and forms aggregates that emit red fluorescence [28]. After acridine orange staining, control cells primarily displayed green fluorescence with slight red fluorescence, which indicated a lack of acidic vesicular organelles (AVOs). However, MHY2256-treated cells showed a significant increase in red fluorescence AVOs at 48 h (Figure 7B). Flow cytometry analysis after acridine orange staining also indicated an increase in red fluorescence intensity with drug treatment. Histogram profiles show the mean fluorescence intensity of control and drug-treated cells (Figure 7C). Furthermore, levels of Bax were markedly increased by MHY2256 in p53-null SKOV-3 cells (Figure 6B), suggesting that SKOV-3 induces an increase in autophagy with lower cytotoxicity. We propose that increased autophagy in SKOV-3 contributes to the lower observed levels of MHY2256 cytotoxicity, since late apoptosis in SKOV-3 was not significantly increased by MHY2256 (Figure 6A) when compared to p53 wild-type MCF-7 cells.
Effects of MHY2256 on the expression of autophagy-related proteins in MCF-7 and SKOV-3 cells. (A) Cells were treated with MHY2256 and salermide for 48 h at the indicated concentrations, and Western blot analysis was performed with LC3-I/II, Beclin-1, Atg5, and Atg7. Protein levels were normalized by comparison to β-actin levels. (B) Immunofluorescence microscopy of acridine orange-stained MCF-7 and SKOV-3 cells treated for 48 h at the indicated drug concentration (magnification, x200). (C) Histogram profiles of control and drug-treated MCF-7 (a) and SKOV-3 (b) cells analyzed with flow cytometry.
MHY2256 inhibits MCF-7 cell tumors in a xenograft model
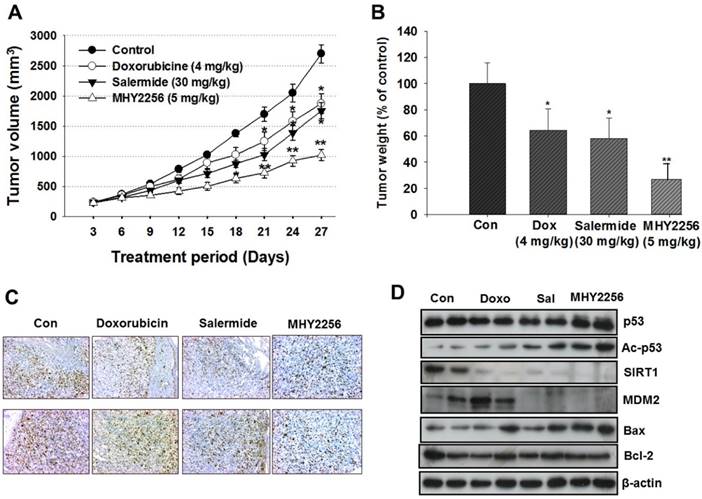
To evaluate the in vivo effects of MHY2256, nude mice were inoculated with MCF-7 cells and treated with doxorubicin (4 mg/kg), salermide (30 mg/kg), or MHY2256 (5 mg/kg) for 4 weeks. Doxorubicin and salermide time-dependently inhibit tumor growth compared to the control group. Moreover, MHY2256 (5 mg/kg) treatment significantly reduced tumor volume by 60% relative to the control group (Figure 8A), and tumor weights were reduced by approximately 70% (Figure 8B). No significant adverse effects related to MHY2256 treatment were observed. Ki67 is a representative marker for cancer cell proliferation. Immunohistochemistry analysis showed high expression of Ki67-positive cells in the tumor tissues of the control group, while the expression intensity of Ki67 was markedly lower in tumor tissues of MHY2256-treated mice (Figure 8C). Consistent with the inhibitory action of MHY2256 on SIRT1 expression in vitro, SIRT1 was markedly reduced and MDM2 protein concentration was also significantly reduced in tumor tissues from the MHY2256-treated group compared to the control group. Acetylated p53 proteins were markedly elevated in MHY2256-treated tumors (Figure 8D). Taken together, these data suggest that the inhibitory effects of MHY2256 on tumor cell proliferation and down-regulation of MDM2 expression in vitro correlated with in vivo anti-cancer effects.
Effects of MHY2256, salermide, and doxorubicin on the growth of MCF-7 tumors in nude mice. Mice with established tumors were randomized into four groups. Vehicle control, doxorubicin (4 mg/kg/week, i.p.), salermide (30 mg/kg/week, i.p.) or MHY2256 (5 mg/kg, twice/week, i.p.) were administered to the tumor-bearing mice. (A) The mean tumor volumes for each treatment group are indicated. (B) Each bar represents the inhibition rate (% of control) of mean tumor weight. The results are presented as the mean ± SEM per group. Statistical analysis was performed using one-way analysis of the variance (ANOVA) followed by Bonferroni's multiple comparison test. *p<0.05, **p <0.01 indicate significant differences between control and treatment groups. (C) The tumors were fixed in 10% formalin and embedded in paraffin. Immunohistochemical staining for Ki-67 were measured in tumors. Magnification x200. Scale bar = 50 μm. The representative images were recorded under a 40x objective lens. (D) Expression levels of p53, acetylated p53, MDM2, Bax, and Bcl-2 were measured in tumor tissues using Western blot analysis. Western blots are representative of three independent experiments.
Discussion
In the present study, we demonstrated the potent antitumor activity and apoptosis induced by MHY2256 in MCF-7 human breast cancer cells with wild-type p53 and autophagic cell death in p53-null SKOV-3 cells. Previous studies also reported inhibition of cell growth in various types of cancer by a few different SIRT inhibitors [27, 28]. Most researchers have described the antitumor effects of SIRT inhibitors in cancer cells, which are closely attributed to the activation of p53 [31, 32]. In contrast, several studies have shown the antitumor activity of SIRT inhibitors in various human cancer cells through p53-independent pathways [33, 34]. The data in the present study indicate that activation of the apoptosis-related signal pathway via up-regulation of p53 plays a pivotal role in MHY2256-induced cell death, as mentioned in another report on SIRT inhibitors [20]. SIRT inhibitors can also be potentially useful as therapeutic agents because over-expression of SIRTs has been demonstrated in many human cancer cells [35-37].
To investigate the possible role of SIRTs on human cancer cell growth, we analyzed the sensitivity of MHY2256-mediated cytotoxicity in human cancer cell lines including wild-type p53, mutated p53, or null p53. MHY2256 inhibited cell proliferation in all cancer cell lines, with an IC50 ranging between 1.3 and 5.5 μM at 48 h as determined using the MTT assay. The cytotoxicity of MHY2256 against these cancer cells was more potent than that of salermide, a well-known SIRT1/2 inhibitor. Furthermore, MHY2256 significantly decreased SIRT1, SIRT2, and SIRT3 expression levels in human cancer cells, and SIRT1 enzyme activity was markedly inhibited in an in vitro assay, indicating greater effective potency than nicotinamide. Recently, several studies have described the apoptotic effects of SIRT inhibitors and have shown that these effects depend on the type of cancerous cells [38, 39]. Although the molecular role of SIRT inhibitors against different cancer cells with or without p53 has been suggested, little is known about the specific mechanisms of action. Therefore, we examined the apoptotic potency of MHY2256 in MCF-7 and SKOV-3 cells, since these cell lines have a substantial SIRT1 expression level, different p53 statuses, and are common malignancies in women. Mutation or functional inactivation of p53 has been reported in more than 50% of all cancer types and has been recognized as a major event in carcinogenesis [21]. In particular, p53, a target for acetylation by SIRT1 and SIRT2, can exert anti-proliferative effects through cell cycle arrest, apoptosis, senescence, and various types of stressors [31]. As expected, MHY2256 increased acetylated p53 in MCF-7 cells. Consequently, MHY2256 induced cell cycle arrest and apoptosis in cancer cells by increasing p53 transcription. Previous studies demonstrated that SIRT1 inhibitors induced p53 stability or inhibited p53 expression in p53 mutant type cells [40, 41]. As we demonstrated, expression of wild-type p53 was significantly increased in MCF-7 cells afterMHY2256 treatment. Therefore, inhibition of SIRT1 activity by MHY2256 leads to elevated p53 acetylation and transactivation, resulting in enhanced apoptosis.
The most important function of activated p53 is induction of cell cycle arrest, apoptosis, and DNA repair [42, 43]. Regulation of the cell cycle by SIRT is essential in the cytotoxicity to cancer cells. However, mechanistic studies are needed to investigate the differential regulation of p53 activity on cell cycle arrest caused by SIRT1 inhibitors. To investigate whether SIRT inhibition is associated with cell cycle control, cell cycle check points were measured using Western blot analysis since cyclins/CDKs play an important role in controlling cell cycle regulation. The present study demonstrated that MHY2256 significantly increased G1 in MCF-7 cells via decreased expression of cyclin D1, CDK2, and CDK4. Previous studies indicated that the CDK inhibitor p21 was up-regulated via p53-dependent and p53-independent mechanisms of HDAC inhibitors [42-45]. Similarly, our data indicate that MHY2256 significantly increase the expression of p21 in MCF-7cells but not SKOV-3 cells.
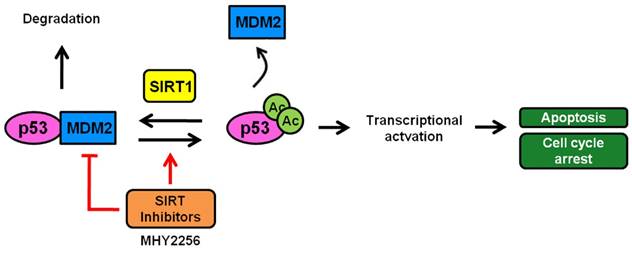
As previously reported, SIRT1 deacetylates p53 at acetylated lysine 382, which is also the target residue for ubiquitination by MDM2, resulting in the facilitation of MDM2-dependent ubiquitination or degradation of p53 [46]. p53 activity is modulated by regulation of its protein expression levels, a process mediated primarily by the ubiquitin ligase MDM2 [22]. To investigate the mechanism of p53 activation by MHY2256, the expression level of MDM2 was measured using Western blot analysis. Interestingly, MHY2256 significantly decreased the expression of MDM2 protein in MCF-7 cells. Therefore, our data indicate that MHY2256 induces p53 activation through inhibition of MDM2 and has bifunctional effects on p53 regulation activity via the inhibition of SIRT1 and MDM2 expression (Figure 9). Furthermore, acetylated p53 controls the transcriptional activity of p53 and inhibits p53 ubiquitin-mediated degradation by MDM2. A previous study demonstrated that acetylated p53 inhibited MDM2-p53 interaction and promoted the activation of proteins involved in proapoptotic, DNA repair, and cell cycle regulatory functions [47]. Results about increased p53 activity with acetylation further support the hypothesis that inhibition of SIRT1 is required for cell death and p53 acetylation in cancer cells with wild-type p53. Our results are in agreement with a study showing that SIRT inhibitors increased p53 acetylation as targets for SIRT1 and SIRT2, which is important for cell death [47]. Our results support this view and show that SIRT1 inhibition can induce cytotoxicity in MCF-7 cells, and that this cytotoxicity is enhanced in the presence of functional p53. Thus, therapeutic SIRT1 inhibition can be beneficial for the treatment of selected cancer types.
A scheme depicting the proposed role of MHY2256 as a p53 activator via reduction of MDM2 expression and SIRT inhibitors. MHY2256, a new SIRT inhibitor, has bifunctional effects on SIRT and MDM2. MHY2256 acetylates p53, and acetylated p53 allows for the disruption of MDM2-p53 interaction and promotes the activation of cell cycle-related genes and proapoptotic genes. Furthermore, MHY2256 increased p53 by reducing MDM2 expression levels.
To investigate the possible association between SIRT1 expression and autophagy, we determined the expression of autophagy-associated proteins LC3-II and beclin-1 in different types of p53-dependent cells. As shown in Figure 7, LC3-II expression was increased in p53-null SKOV-3 cells, suggesting an association between SIRT1 and autophagy. As expected, we demonstrated that the anticancer effects of MHY2256 were closely related to their ability to modulate autophagic cell death. Autophagy is another important cell death process and is a dynamic process characterized by degradation of cytoplasmic components such as mitochondria through complicated intracellular reorganization with lysosomes [48]. Induction of autophagy has been demonstrated in some cancer cells treated with various anticancer agents such as DNA-damaging agents, microtubule targeting agents, and HDAC inhibitors [49, 50]. Recently, sirtinol, a potent SIRT1 inhibitor, was shown to induce autophagic cell death in MCF-7 cells [19]. Likewise, MHY2256 markedly increased LC3-II levels and autophagy-related proteins such as Atg5 and Atg7 in MCF-7 and SKOV-3 cells. Autophagic cell death induced by MHY2256 was confirmed again via acridine orange staining. MHY2256 treatment significantly increased the acidic AVOs in the cytoplasm in MCF-7 and SKOV-3 cells. Therefore, p53 had no effect on the physical interaction between beclin-1 and Bcl-2, indicating that MHY2256-induced autophagic cell death may depend on another p53-related pathway. However, we found that Bax levels were markedly increased by MHY2256 in p53-null SKOV-3 cells, and we thus assumed that Bax might be involved in the increase of autophagy protein levels in the presence of MHY2256. Based on apoptosis and autophagy in p53 wild-type MCF-7 and p53-null SKOV-3 cells, we conclude that increased autophagy induction by MHY2256 contributes to increased apoptosis only in the presence of p53.
The anti-tumorigenic effects of MHY2256 were also demonstrated using an in vivo xenograft model, in which MCF-7 cells tumor was significantly retarded by MHY2256 injection. Significant decreases in MDM2 levels were observed in tumor tissues treated with doxorubicin, salermide, or MHY2256 compared with the control. These results suggest that MHY2256-mediated MDM2 inhibition is closely related to autophagic cell death in MCF-7 cells.
In summary, a new SIRT inhibitor, MHY2256, induced anti-proliferative effects in human cancer cells via cell cycle arrest, apoptosis, and autophagic cell death. MHY2256 also induced p53 activation by reducing MDM2 expression. Taken together, our data suggest that inhibition of SIRT1 by MHY2256 causes increased activation of p53 by decreasing MDM2 expression. MHY2256 thus provides a good opportunity for drug development in relation to p53 targeting. Therefore, MHY2256 may be used as a novel SIRT-targeted agent against human breast cancers, especially in patients with wild-type p53 expression.
Acknowledgements
This study was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (NRF-2016R1A2B2011071; NRF-2016R1A4A1011189).
Competing Interests
The authors have no competing interests to declare.
References
1. Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26(37):5489-504
2. Roth M, Chen WY. (2013) Sorting out functions of sirtuins in cancer. Oncogene. 2014;33(13):1609-20
3. Cen Y. Sirtuins inhibitors: The approach to affinity and selectivity. Biochim. Biophys. Acta. 2010;1804(8):1635-44
4. Milner J. Cellular regulation of SIRT1. Curr. Pharm. Des. 2009;15(1):39-44
5. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149-59
6. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell. 2005;16(10):4623-35
7. Olmos Y, Brosens JJ, Lam EW. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updat. 2011;14(1):35-44
8. Villalba JM, Alcaín FJ. Sirtuin activators and inhibitors. Biofactors. 2012;38(5):349-59
9. Chu F, Chou PM, Zheng X, Mirkin BL, Rebbaa A. Control of multidrug resistance gene mdr1 and cancer resistance to chemotherapy by the longevity gene sirt1. Cancer Res. 2005;65(22):10183-87
10. Lim CS. Human SIRT1: a potential biomarker for tumorigenesis? Cell. Biol. Int. 2007;31(6):636-7
11. Yi J, Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta. 2010;1804(8):1684-9
12. Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, Nemoto S, Finkel T, Gu W, Cress WD, Chen J. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006;8(9):1025-31
13. Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69(5):1702-05
14. Kojima K, Ohhashi R, Fujita Y, Hamada N, Akao Y, Nozawa Y, Deguchi T, Ito M. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem. Biophys. Res. Commun. 2008;373(3):423-8
15. Kim HB, Kim MJ, Lee SH, Lee JW, Bae JH, Kim DW, Dao TT, Oh WK, Kang CD, Kim SH. Amurensin G, a novel SIRT1 inhibitor, sensitizes TRAIL-resistant human leukemic K562 cells to TRAIL-induced apoptosis. Biochem. Pharmacol. 2012;84(3):402-10
16. Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW. SIRT Inhibitors Induce Cell Death and p53 Acetylation through Targeting Both SIRT1 and SIRT2. Mol. Cancer Ther. 2010;9(4):844-55
17. Jia G, Su L, Singhal S, Liu X. Emerging roles of SIRT6 on telomere maintenance, DNA repair, metabolism and mammalian aging. Mol. Cell Biochem. 2012;364(1-2):345-50
18. Villalba JM, Alcain FJ. Sirtuin activators and inhibitors. Biofactors. 2012;38(5):349-59
19. Wang J, Kim TH, Ahn MY, Lee J, Jung JH, Choi WS, Lee BM, Yoon KS, Yoon S, Kim HS. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012;41(3):1101-9
20. Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, Varela-Rey M, Rotili D, Nebbioso A, Ropero S, Montoya G, Oyarzabal J, Velasco S, Serrano M, Witt M, Villar-Garea A, Imhof A, Mato JM, Esteller M, Fraga MF. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28(6):781-91
21. Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J. Pathol. 2012;223(2):116-26
22. Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007;13(1):23-31
23. Soares J, Pereira NA, Monteiro A, Leão M, Bessa C, Dos Santos DJ, Raimundo L, Queiroz G, Bisio A, Inga A, Pereira C, Santos MM, Saraiva L. Oxazoloisoindolinones with in vitro antitumor activity selectively activate a p53-pathway through potential inhibition of the p53-MDM2 interaction. Eur. J. Pharm. Sci. 2014;66C:138-47
24. Leão M, Pereira C, Bisio A, Ciribilli Y, Paiva AM, Machado N, Palmeira A, Fernandes MX, Sousa E, Pinto M, Inga A, Saraiva L. Discovery of a new small-molecule inhibitor of p53-MDM2 interaction using a yeast-based approach. Biochem. Pharmacol. 2013;85(9):1234-45
25. Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009;49:223-41
26. Lai SC, Devenish RJ. LC3-associated phagocytosis (LAP): Connections with host autophagy. Cells. 2012;1(3):396-408
27. Wang L, Chen M, Yang J, Zhang Z. LC3 fluorescent puncta in autophagosomes or in protein aggregates can be distinguished by FRAP analysis in living cells. Autophagy. 2013;9(5):756-69
28. Chiarelli R, Agnello M, Roccheri MC. Sea urchin embryos as a model system for studying autophagy induced by cadmium stress. Autophagy. 2011;7(9):1028-34
29. Hoffmann G, Breitenbücher F, Schuler M, Ehrenhofer-Murray AE. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 2014;289(8):5208-16
30. Amengual JE, Clark-Garvey S, Kalac M, Scotto L, Marchi E, Neylon E, Johannet P, Wei Y, Zain J, O'Connor OA. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood. 2013;122(12):2104-13
31. Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, Dong H, Wei J, Song J, Zhang DD, Fang D. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell. 2012;46(4):484-94
32. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107(2):137-48
33. Sonnemann J, Marx C, Becker S, Wittig S, Palani CD, Krämer OH, Beck JF. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br. J. Cancer. 2014;110(3):656-67
34. Nosho K, Shima K, Irahara N, Kure S, Firestein R, Baba Y, Toyoda S, Chen L, Hazra A, Giovannucci EL, Fuchs CS, Ogino S. SIRT1 histone deacetylase expression is associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod. Pathol. 2009;22(7):922-32
35. Stünkel W, Peh BK, Tan YC, Nayagam VM, Wang X, Salto-Tellez M, Ni B, Entzeroth M, Wood J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007;2(11):1360-8
36. Lim CS. SIRT1: tumor promoter or tumor suppressor? Med. Hypotheses. 2006;67(2):341-4
37. Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67(14):6612-8
38. Kong S, Kim SJ, Sandal B, Lee SM, Gao B, Zhang DD, Fang D. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J. Biol. Chem. 2011;286(19):16967-75
39. Vergnes B, Vanhille L, Ouaissi A, Sereno D. Stage-specific antileishmanial activity of an inhibitor of SIR2 histone deacetylase. Acta. Trop. 2005;94(2):107-15
40. Ahn MY, Jung JH, Na YJ, Kim HS. A natural histone deacetylase inhibitor, Psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol. Oncol. 2008;108(1):27-33
41. Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, Huber LJ. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell Biol. 2006;26(1):28-38
42. Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, Sun J, Yu Y, Zhou W, heng Q, Wu M, Otterson GA, Zhu WG. Acetylation of p53 at lysine 73/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21 (Waf1/Cip1). Mol. Cell Biol. 2006;26(7):2782-90
43. Yi YW, Kang HJ, Kim HJ, Kong Y, Brown ML, Bae I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget. 2013;4(7):984-94
44. Liu PY, Chan JY, Lin HC. Modulation of the cyclin-dependent kinase inhibitor p21 (WAF1/Cip1) gene by Zac1through the antagonistic regulators p53 and histone deacetylase 1in HeLa Cells. Mol. Cancer Res. 2008;6(7):1204-14
45. Patra N, De U, Kim TH, Lee YJ, Ahn MY, Kim ND, Yoon JH, Choi WS, Moon HR, Lee BM, Kim HS. A novel histone deacetylase (HDAC) inhibitor MHY219 induces apoptosis via up-regulation of androgen receptor expression in human prostate cancer cells. Biomed Pharmacother. 2013;67(5):407-15
46. Hirano Y, Ronai Z. A new function for p53 ubiquitination. Cell. 2006;127(4):675-7
47. Kamel C, Abrol M, Jardine K, He X, Mcburney MW. Sirt1 fails to affect p53-mediated biological functions. Aging Cell. 2006;5(1):81-8
48. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J. Pathol. 2010;221(1):3-12
49. Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2004;101(52):18030-5
50. Notte A, Leclere L, Michiels C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharmacol. 2011;82(5):427-34
Author contact
![]() Corresponding authors: Prof. Hyung Sik Kim, School of Pharmacy, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, Suwon, Gyeonggi-do 440-746, Republic of Korea; E-mail: hkimsedu; Tel.: +82-31-290-7789; Fax: +82-31-292-8800. Prof. Hyung Ryong Moon, College of Pharmacy, Pusan National University. San 30, Jangjeon-dong, Geumjeung-gu, Busan 609-735, Republic of Korea; Email: mhr108ac.kr; Tel.: +82-51-510-2815; Fax: +82-51-513-6754.
Corresponding authors: Prof. Hyung Sik Kim, School of Pharmacy, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, Suwon, Gyeonggi-do 440-746, Republic of Korea; E-mail: hkimsedu; Tel.: +82-31-290-7789; Fax: +82-31-292-8800. Prof. Hyung Ryong Moon, College of Pharmacy, Pusan National University. San 30, Jangjeon-dong, Geumjeung-gu, Busan 609-735, Republic of Korea; Email: mhr108ac.kr; Tel.: +82-51-510-2815; Fax: +82-51-513-6754.