Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(1):13-21. doi:10.7150/ijbs.16769 This issue Cite
Research Paper
MicroRNA-135a Regulates Apoptosis Induced by Hydrogen Peroxide in Rat Cardiomyoblast Cells
Ning Liu, Yong-Feng Shi, Hong-Ying Diao, Yang-Xue Li, Yan Cui, Xian-Jing Song, Xin Tian, Tian-Yi Li, Bin Liu ![]()
Department of Cardiology, The Second Hospital of Jilin University, Changchun, Jilin, 130041, China.
Received 2016-7-7; Accepted 2016-9-27; Published 2017-1-1
Abstract

Oxidative stress and apoptosis are the most important pathologic features of ischemic heart disease. Recent research has indicated that microRNAs (miRs) play an essential role in apoptosis. However, whether miRs might regulate B cell lymphoma-2 (Bcl-2) protein in apoptosis during ischemic heart disease is still unclear. The aim of this study, therefore, was to confirm the regulation of microRNA-135a (miR-135a) in oxidative stress injuries induced by hydrogen peroxide (H2O2) in rat cardiomyoblast cells H9c2. To this end, we analyzed the effects of H2O2 treatment on miR-135a expression in rat cardiomyocytes. Furthermore, we upregulated and inhibited miR-135a using mimics and inhibitors, respectively, and examined the effects on cell viability and apoptosis-related proteins. We observed that miR-135a was markedly up-regulated under H2O2 treatment in rat cardiomyoblast cells. Overexpression of miR-135a blocked the Bcl-2 protein and enhanced the apoptosis induced by H2O2, and miR-135a inhibition restored Bcl-2 protein expression. Interestingly, miR-135a inhibition did not attenuate H2O2-induced apoptosis with Bcl-2 knockdown. The results of the present study indicate that miR-135a regulates H2O2-induced apoptosis in H9c2 cells via targeting Bcl-2, and that miR-135a may be a novel therapeutic target for ischemic heart disease.
Keywords: MicroRNA-135a, Rat cardiomyoblast cells, Apoptosis, Bcl-2, Hydrogen peroxide.
Introduction
Ischemic heart disease is a common health problem and one of the leading causes of cardiac dysfunction in the world [1]. One potential therapeutic approach for ischemic heart disease is to reduce oxygen consumption, inhibit cardiomyocyte apoptosis, increase coronary flow, and induce revascularization [2, 3]. Ischemia and reperfusion activate cardiac myocyte apoptosis and oxidative stress, which is an important feature in the progression of ischemic heart disease [4, 5]. Hydrogen peroxide (H2O2)-induced cardiac cell apoptosis plays a key role in the pathogenesis of cardiac oxidative stress injury [6-8].
Apoptosis is a type of programmed cell death, and there are two major apoptotic signaling pathways in the cardiovascular system [9]. Both the intrinsic pathways and extrinsic pathways are associated with ischemic heart diseases [10, 11]. The intrinsic pathway is mainly regulated by the B cell lymphoma-2 (Bcl-2) superfamily, such as Bax, Bak, Bcl-2 and Bcl-XL [12]. Bcl-2 has been shown to regulate the apoptotic cascade, and it can block apoptosis by preventing cytochrome c release and mitochondrial permeabilization in ischemic heart disease [13, 14]. Recent studies have demonstrated that overexpression of Bcl‑2 in cardiac myocytes significantly reduced cardiac myocyte apoptosis and cardiac dysfunction in ischemic heart disease [15-17]. Furthermore, the ratio of antiapoptotic to proapoptotic proteins plays an important role in the regulation of apoptosis in cardiac cell injury [17, 18]. However, the detailed mechanism by which Bcl-2 is regulated during disease progression requires further investigation.
MicroRNAs (miRs) are small noncoding RNAs that play important roles in gene expression by inhibiting translation or inducing target molecules degradation [19-21]. MiRs also have roles in the functions of the healthy and diseased heart, such as cardiac hypertrophy, myocardial infarction, myocardial fibrosis, cardiac arrhythmia and heart failure [22-26]. An increasing number of reports have confirmed that miRs function as a critical apoptotic regulator in heart cells [27-29]. MiRs can function as either pro-apoptotic or anti-apoptotic factors [29-31]. Our previous studies have found that the expression of miRNA is different in rat myocardial injury models induced by psychological stress and microRNAs-135a (miR-135a) is significantly downregulated in chronic psychological stress [32]. Several studies have shown that miR-135a acts as a pro-apoptotic factor in many cancer cell lines [33-35].
However, whether miR-135a might regulates Bcl-2 during the development of cardiac cell injury, thereby affecting ischemic heart disease progression is not known. Therefore, our aim was to unravel the effects of miR-135a on apoptosis, and to investigate the underlying mechanisms in H9c2 cells. In the present study, we exposed the cells to H2O2 and detected the expression of miR-135a.
Materials and methods
Cell culture
The embryonic rat cardiac myoblast H9c2 cell line was obtained from Department of Cardiology of the Second Hospital of Jilin University. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM; GIBCO, Carlsbad, USA) supplemented with 10% fetal bovine serum (Hyclone, Australia). The cells were propagated at 37°C under 5% CO2.
Cell viability assays
H9c2 cells were plated in 96-multiwell plates at 1×104 cells/well at 24 h before treatment. The cells were then treated with increased concentrations of H2O2 (0, 0.1, 0.25, 0.5, and 1 mM) for 24 h. Each treatment was repeated in five wells. H9c2 were then incubated with MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) (10 µl; 5 mg/ml in PBS; Sigma, St. Louis, MO, USA) for 4 h at 37°C under 5% CO2. Subsequently, 150 µl of dimethylsulfoxide was added to dissolve the formazan crystals of each group. After shaking for 10 min, the absorbance values were measured at a wavelength of 570 nm by a Microplate Reader (Molecular Devices, Sunnyvale, CA).
Transfection
H9c2 cells were transfected using Lipofectamine® 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Cells were plated on six-well plates and transfected at 80% confluence with the negative control (NC), miR-135a mimics or miR-135a inhibitor (GenePharma, Shanghai, CHINA).
Small interfering RNA-mediated gene silencing
H9c2 cells were seeded in 6-well plates. Small interfering RNAs targeting Bcl-2 or a scramble sequence (GenePharma, Shanghai, CHINA) were transfected into H9c2 cells using Lipofectamine® 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
RNA extraction and quantitative real-time PCR (qPCR)
Total RNA from H9c2 cells was extracted using a miRcute miRNA Isolation Kit (Tiangen Biotech, Beijing, China). To quantify the RNA expression levels, cDNA was synthesized using the miRcute miRNA First-Strand cDNA Synthesis Kit (Tiangen Biotech, Beijing, China) and qPCR was carried out using the miRcute miRNA qPCR Detection Kit (SYBR Green; Tiangen Biotech, Beijing, China). All reactions were conducted in triplicate in a LightCycler® 480 Real-Time PCR System (Roche, Basel, Switzerland). Fold changes in miR-135a expression between treatments and controls were determined using 2 -ΔΔCt method [36] and normalized to U6 RNA expression as an internal reference. The PCR forward primer for mature miR-135a was designed as 5′-GCGCCGTATGGCTTTTTATTCCTA-3′, and the PCR reverse primer for mature miR-135a was obtained from the commercial miRcute miRNA qPCR Detection Kit. Primer sequences are as follows: U6, 5′-AACGCTTCACGAATTTGCGT-3′ (sense) and 5′CTCGCTTCGGCAGCACA-3′ (antisense), Bcl-2, 5′-TGCAGAGATGTCCAGTCAGC-3′ (sense) and 5′-CATCCACAGAGCGATGTTGT-3′ (antisense), and GAPDH, 5′-AACGACCCCTTCATTGAC-3′ (sense) and 5′-TCCACGACATACTCAGCAC-3′ (antisense).
Flow cytometry analysis
After exposure to different experimental conditions, cells were trypsinized and incubated with propidium iodide (PI, 1µg/ml) and Annexin V-FITC (1 µg/ml; Invitrogen, Carlsbad, CA) for 15 min at 37°C. Samples were then analyzed for apoptosis by a FACScan flow cytometer (FCM; Bechman Coulter, CA).
Western blot analysis
H9c2 cells grown in 100-mm Petri dishes were treated with 1mM H2O2 for 24h. Total proteins were extracted from H9c2 cells using RIPA buffer, and then the cell lysates were centrifuged at 3,000×g for 30 min. The concentrations of protein were determined using a Protein Assay Kit (Bio-Rad, Hercules, CA, USA). For western blot analysis, equivalent amounts of proteins were resolved on 12% gels by SDS-polyacrylamide gel electrophoresis and then transferred onto PVDF membranes (Millipore, Bedford, MA). After blocking in 10% nonfat dry milk at room temperature for 1 h, the membranes were incubated with the relevant primary antibody overnight at 4°C. The β-actin, Bcl-2, Bax and caspase-3 antibodies (all used at 1:1000 dilution) were obtained from Cell Signaling Technology (CST, BSN, USA). On the following day, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (Thermo, Waltham, MA, USA) for 1 h at room temperature. The immunoreactive bands were visualized by the ECL detection system (Amersham/GE Healthcare, Pittsburgh, PA). The reactive bands were measured by a Tanon GIS gel imager system and the protein levels were quantified by densitometry using the Quantity One software.
Statistical analysis
All of the data are representative of three independent experiments performed as triplicate determinations. Statistical analyses of the data were evaluated using one-way ANOVA. Values of P < 0.05 were considered statistically differences.
Results
Expression of miR-135a is decreased in H9c2 cells treated with H2O2
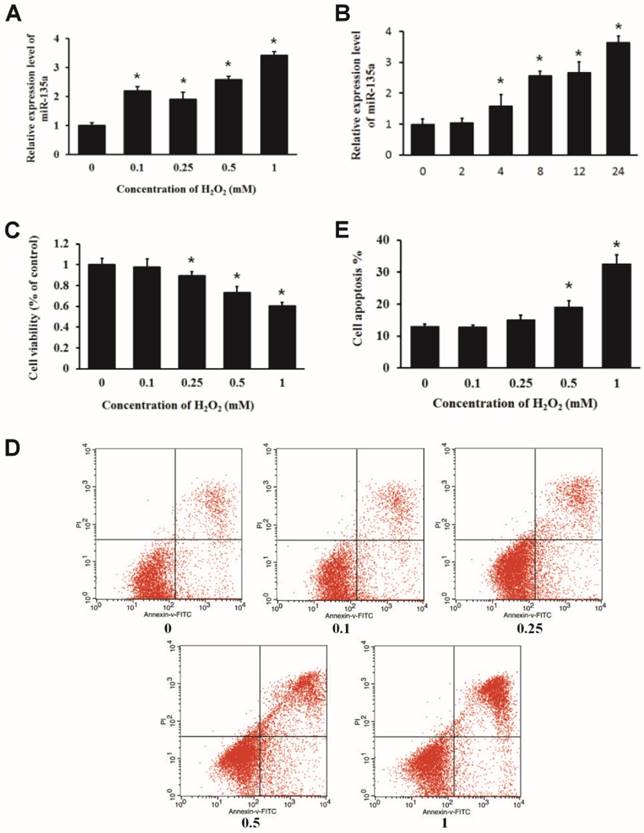
We treated H9c2 cells with increasing doses of H2O2 for 24 h or 1 mM H2O2 for 2, 4, 8, 12 and 24 h, and examined the relative expression of miR-135a by qPCR. We found that H2O2 treatment upregulated the expression of miR-135a in H9c2 cells (Fig. 1A and B). MTT assays indicated that H2O2 inhibited the cell viability in a dose-dependent manner (Fig. 1C). H9c2 cells were subjected to PI and Annexin V-FITC staining. Flow cytometry analysis was used to quantify apoptotic cell populations. H2O2 treatment resulted in an increase in the apoptotic cell population (Fig. 1D and E). These findings demonstrate that H2O2 induced apoptosis of embryonic rat cardiac myoblasts H9c2 cells and increased the expression of miR-135a.
Expression of miR-135a is increased in H9c2 cells treated with H2O2. The relative expression of miR-135a was measured by quantitative real-time PCR (qPCR). (A) H9c2 cells were treated with 0, 0.1, 0.25, 0.5 or 1 mM of H2O2 for 24 h, or with (B) 1 mM of H2O2 for 0, 2, 4, 8, 12, or 24 h. (C) Cells were treated with varying doses of H2O2 for 24 h. Cell viability was determined by the MTT assay. (D) H9c2 cells were stained with propidium iodide (PI) and Annexin V-FITC. The apoptosis positive stained cells were counted by flow cytometry. (E) Quantitation of the apoptosis rate. All data are presented as means ± SD (n = 3). *P < 0.05 vs. control.
MiR-135a is functionally involved in apoptosis induced by H2O2 in H9c2 cells
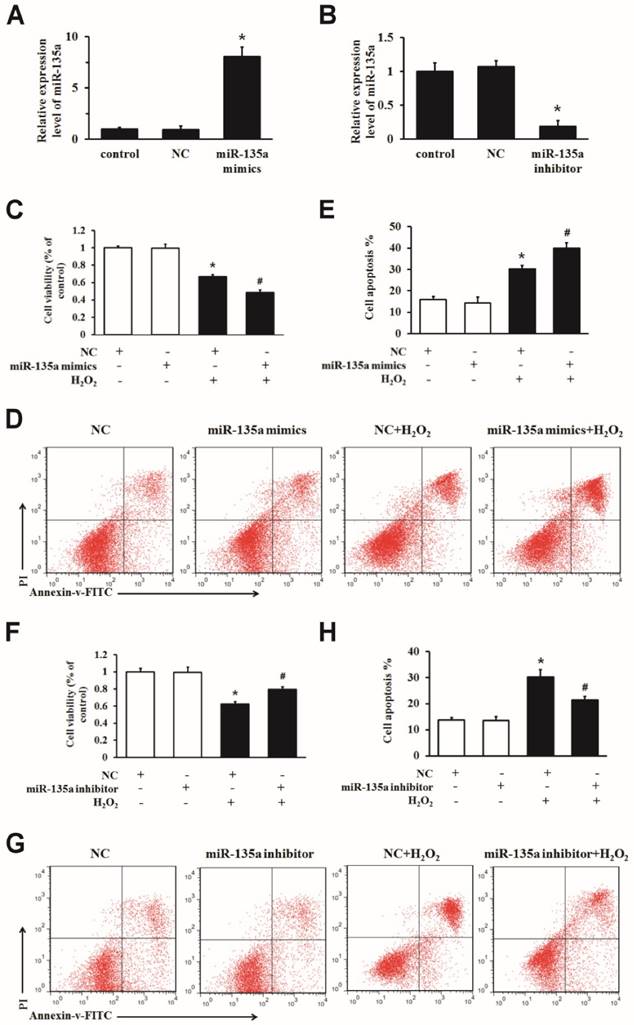
To determine whether miR-135a regulates apoptosis, we transfected miR-135a mimics, inhibitor or NC into H9c2 cells. The expression of miR-135a was markedly changed after transfection (Fig. 2A and B). We increased the expression of miR-135a in H9c2 cells exposed to H2O2 for 24 h, and then examined the growth inhibition and apoptosis ratio. Transfection of the miR-135a mimics decreased the cell viability and increased apoptosis after H2O2 treatment compared to the NC + H2O2 group (Fig. 2C, D and E). As shown in Fig. 2F, G and H, inhibition of miR-135a expression increased the cell viability and decreased apoptosis after H2O2 treatment compared to the NC + H2O2 group. Together, these findings demonstrate that miR-135a promotes H2O2-induced apoptosis in H9c2 cells, and that inhibition of miR-135a attenuates apoptosis.
miR-135a is functionally involved in apoptosis induced by H2O2 in H9c2 cells. qPCR was used to determine the expression of miR-135a in H9c2 cells. Cells were transfected with negative control (NC) or miR-135a mimics (A), or with NC or miR-135a inhibitor (B). Cells were subsequently treated with 1 mM H2O2 for 24 h and cell viability was assessed using the MTT assay (C and F). The cells were stained with PI and Annexin V-FITC. The positive stained cells were counted by flow cytometry (D and G). Quantitation of the apoptosis rate (E and H). All data are presented as means ± SD (n = 3). *P < 0.05 vs. NC. #P < 0.05 vs. NC + H2O2.
The effect of miR-135a mimics on apoptotic proteins in H9c2 cells
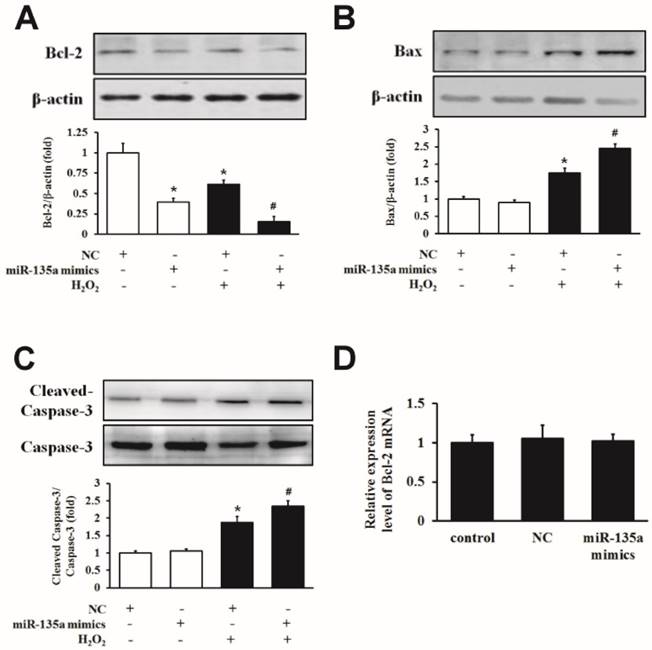
To examine the mechanism underlying the induction of apoptosis in more detail, we examined the expression of Bcl-2, Bax and caspase-3 proteins. Western blot analysis showed that H9c2 cells transfected with miR-135a mimics showed a decrease in Bcl-2 protein expression compared to the NC group. Importantly, the level of the Bcl-2 protein was markedly decreased in the group of miR-135a mimics + H2O2 group compared to the NC + H2O2 group (Fig. 3A). Furthermore, the level of apoptotic proteins Bax and cleaved caspase-3 were increased after miR-135a mimics transfection following H2O2 treatment compared to the NC + H2O2 group (Fig. 3B and C). However, there was no change in the expression of Bcl-2 mRNA in either group (Fig. 3D). These results suggest that miR-135a overexpression can affect apoptosis-associated proteins under H2O2 exposure.
The effect of miR-135a mimics on apoptotic proteins in H9c2 cells. Cells were treated with 1 mM H2O2 for 24 h after transfection with NC or miR-135a mimics. Western blot analysis for the expressions of Bcl-2 (A), Bax (B) and cleaved caspase-3 (C). Data are presented as means ± SD (n = 3). *P < 0.05 vs. NC. #P < 0.05 vs. NC + H2O2. (D) qPCR was used to analyze the expression of Bcl-2 after transfection with miR-135a mimics. Data are presented as means ± SD (n = 3).
Inhibition of miR-135a attenuates the apoptotic proteins induced by H2O2
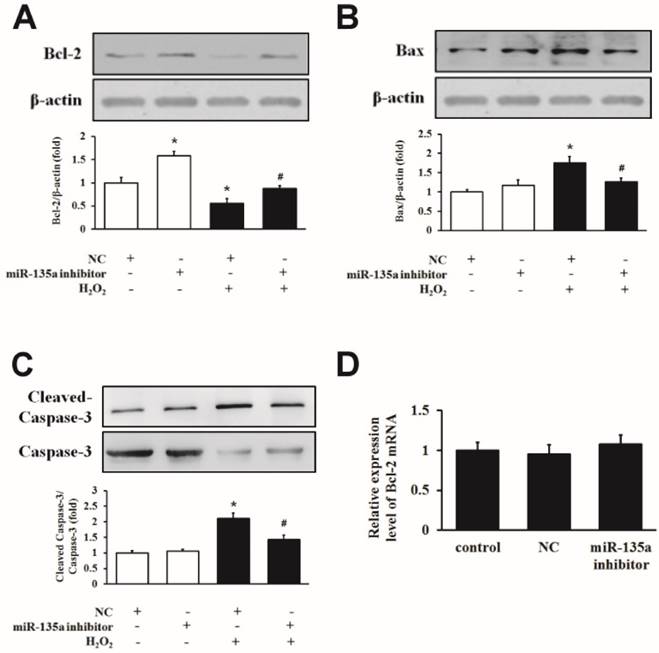
We inhibited miR-135a expression by using an inhibitor in H9c2 cells exposed to H2O2. As shown in Fig. 4A, the inhibition of miR-135a upregulated the level of Bcl-2 protein compared to the NC group, and the expression of the Bcl-2 protein was increased in the miR-135a inhibitor + H2O2 group compared to the NC + H2O2 group. The levels of Bax and cleaved caspase-3 protein were decreased in the miR-135a inhibitor + H2O2 group compared to the NC + H2O2 group (Fig. 4B and C). However, there is no change of the expression of Bcl-2 mRNA in either group (Fig. 4D). This data indicate that inhibition of miR-135a directly increases the expression of Bcl-2 protein and decreases the pro-apoptosis proteins caused by H2O2.
Inhibition of miR-135a attenuates the induction of apoptotic proteins by H2O2. H9c2 cells were treated with 1 mM H2O2 for 24 h after transfection with NC or miR-135a inhibitor. Western blot was used to determine the expression of Bcl-2 (A), Bax (B) and cleaved caspase-3 (C). Data are presented as means ± SD (n = 3). *P < 0.05 vs. NC. #P < 0.05 vs. NC + H2O2. (D) Expression of Bcl-2 mRNA after treatment with miR-135a inhibitor. Data are presented as means ± SD (n = 3).
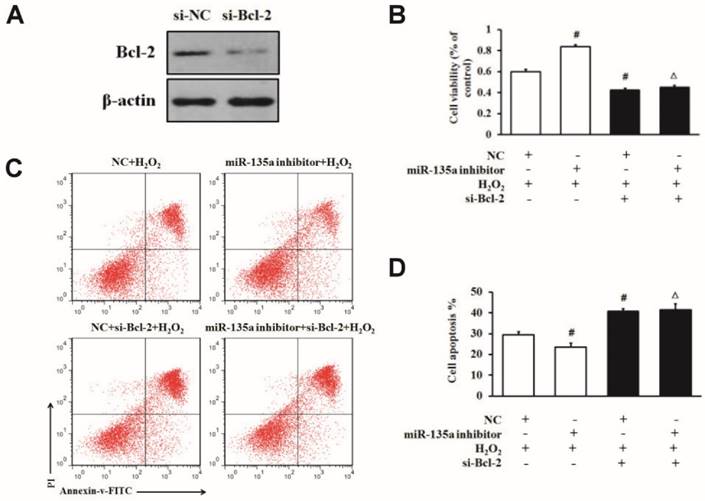
MiR-135a regulates apoptosis induced by H2O2 via targeting Bcl-2 protein
To further confirm whether Bcl-2 protein is involved in this apoptosis induction, we specifically knocked down Bcl-2 in H9c2 cells by using siRNA. We found that the level of Bcl-2 protein was lower in the si-Bcl-2 group than in the si-NC group (Fig. 5A). To determine whether Bcl-2 is directly involved in the apoptosis pathway of miR-135a in H9c2 cells exposed to H2O2, we used Bcl-2 siRNA after the downregulation of miR-135a by inhibitor in H9c2 cells. As shown in Fig. 5B, C and D, the transfection of si-Bcl-2 decreased the cell viability and increased cell apoptosis after H2O2 treatment compared to the NC + H2O2 group. Moreover, co-transfection with miR-135a and si-Bcl-2 decreased the cell viability and increased apoptosis after H2O2 treatment in comparison with the miR-135a inhibitor + H2O2 group. In the NC + H2O2 group (with normal Bcl-2 expression), miR-135a inhibition protected cells from apoptosis under H2O2 exposure. Interestingly, in the group treated with Bcl-2 siRNA, miR-135a inhibition did not protect cells from apoptosis induced by H2O2.
miR-135a regulates H2O2-induced apoptosis via Bcl-2 modulation. (A) The expression of Bcl-2 protein was detected by western blot in cells transfected with si-Bcl-2. (B) H9c2 cells were transfected with the miR-135a inhibitor, si-Bcl-2 or both, followed by treatment with 1 mM H2O2 for 24 h. Cell viability was determined by the MTT assay. (C) Cells were stained with PI and Annexin V-FITC. Positively stained cells were counted by flow cytometry. (D) Quantitation of the apoptosis rate. All data are presented as means ± SD (n = 3). #P < 0.05 vs. NC + H2O2. ∆P < 0.05 vs. miR-135a inhibitor + H2O2.
Discussion
Clinical observations have suggested that myocardial infarction is one of the leading causes of death in the world [37, 38], and apoptosis may be involved in the process of ischemic cardiomyoblast cells [30]. Apoptosis is a major feature of ischemic heart disease [39, 40], and Bcl-2 protects cells from apoptotic stimuli [41-43]. H2O2 has been widely used to simulate conditions of oxidative stress, and H2O2 is considered to play a key role in ischemic cardiomyoblast cells by stimulating apoptosis [44, 45]. We treated rat cardiomyoblast cells H9c2 with H2O2 and found significant apoptosis accompanied by caspase-3 activation. Previous studies have indicated that Bcl-2, an anti-apoptotic protein, plays an important role by controlling the permeabilization of the mitochondrial outer membrane [46, 47]. We evaluated the expression of Bcl-2 protein by western blot and found that the level of Bcl-2 was downregulated in cardiomyocytes treated with H2O2. These results suggested that Bcl-2 is involved in apoptosis under H2O2 treated conditions.
Recent data have suggested that miRs regulate apoptosis in ischemic heart disease [48]. In our study, we found that miR-135a plays a significant role in regulating apoptosis in rat cardiomyoblast cells. The expression of miR-135a was strongly upregulated in H9c2 cells after H2O2 treatment in a dose-dependent manner. To determine the potential role of miR-135a in regulating apoptosis induced by H2O2, the expression of miR-135a was modulated by miR-135a mimics and inhibitor. Upregulation of miR-135a by miR-135a mimics in H9c2 cells exacerbated apoptosis induced by H2O2. In contrast, inhibition of miR-135a expression attenuated cardiomyocyte apoptosis. These results suggested that miR-135a participates in the regulation of H2O2-mediated apoptosis and may be a new target for the treatment of cardiac cell injury.
To determine whether Bcl-2 is involved in miR-135a-mediated H2O2-induced cardiomyocytes apoptosis, we evaluated the expression of Bcl-2 proteins after transfected with miR-135a mimics and inhibitor. We found that the level of Bcl-2 protein was downregulated in cardiomyocytes treated with miR-135a mimics, and the suppression of miR-135a directly enhanced the expression of Bcl-2 protein. Interestingly, we did not detect any changes in Bcl-2 mRNA expression after treatment with the miR-135a mimics and inhibitor. Furthermore, we knocked down the expression of Bcl-2 in H9c2 cells with si-Bcl-2. We found that inhibition of miR-135a did not increase the cell viability or attenuate apoptosis in cells with Bcl-2 knockdown. These results demonstrated that the Bcl-2 protein might be a target of miR-135a in cardiomyocytes, and miR-135a regulates apoptosis partly through the regulation of Bcl-2 protein. In summary, our data investigate for the first time that miR-135a regulated H2O2-induced apoptosis via the modulation of Bcl-2.
In summary, our findings demonstrate that the expression of miR-135a in rat cardiomyoblast cells is upregulated under H2O2 treatment. MiR-135a participates in apoptosis induced by H2O2 in H9c2 cells by downregulating Bcl-2. Overexpression of miR-135a blocked the Bcl-2 protein and increased the apoptosis induced by H2O2. Together, our results suggest that miR-135a might play a significant role in apoptosis of rat myocardial cells, and miR-135a may lead to a novel therapeutic target for ischemic heart disease.
Abbreviations
IHD: ischemic heart disease; H2O2: hydrogen peroxide; Bcl-2: B cell lymphoma-2; miR: microRNA; miR-135a: microRNA-135a; DMEM: Dulbecco's modified Eagle's medium; MTT: 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; qPCR: quantitative real-time PCR; NC: negative control; siRNA: small interfering RNA; PI: propidium iodide; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; PVDF: polyvinylidene difluoride; ECL: enhanced chemiluminescence.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NO.81570250), Norman Bethune Program of Jilin University (NO.2015412), the Finance Department of Jilin Province (NO.2014520) and the National Clinical Key Specialty Project.
Competing Interest
The authors have declared that no competing interest exists.
References
1. Raj P, Zieroth S, Netticadan T. An overview of the efficacy of resveratrol in the management of ischemic heart disease. Ann N Y Acad Sci. 2015;1348:55-67
2. Lavu M, Gundewar S, Lefer DJ. Gene therapy for ischemic heart disease. J Mol Cell Cardiol. 2011;50:742-50
3. Hashmi S, Al-Salam S. Acute myocardial infarction and myocardial ischemia-reperfusion injury: a comparison. Int J Clin Exp Pathol. 2015;8:8786-96
4. Clerk A, Kemp TJ, Zoumpoulidou G. et al. Cardiac myocyte gene expression profiling during H2O2-induced apoptosis. Physiol Genomics. 2007;29:118-27
5. Qu S, Zhu H, Wei X. et al. Oxidative stress-mediated up-regulation of myocardial ischemic preconditioning up-regulated protein 1 gene expression in H9c2 cardiomyocytes is regulated by cyclic AMP-response element binding protein. Free Radic Biol Med. 2010;49:580-6
6. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241-8
7. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181-90
8. Sverdlov AL, Elezaby A, Qin F. et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease. J Am Heart Assoc. 2016:5
9. Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671-4
10. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305-8
11. Wurstle ML, Zink E, Prehn JH. et al. From computational modelling of the intrinsic apoptosis pathway to a systems-based analysis of chemotherapy resistance: achievements, perspectives and challenges in systems medicine. Cell Death Dis. 2014;5:e1258
12. Shamas-Din A, Kale J, Leber B. et al. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714
13. Rolland SG, Conradt B. New role of the BCL2 family of proteins in the regulation of mitochondrial dynamics. Curr Opin Cell Biol. 2010;22:852-8
14. Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157-64
15. Wang Y, Zhang H, Chai F. et al. The effects of escitalopram on myocardial apoptosis and the expression of Bax and Bcl-2 during myocardial ischemia/reperfusion in a model of rats with depression. BMC Psychiatry. 2014;14:349
16. Xu T, Li D, Jiang D. Targeting cell signaling and apoptotic pathways by luteolin: cardioprotective role in rat cardiomyocytes following ischemia/reperfusion. Nutrients. 2012;4:2008-19
17. Chen Z, Chua CC, Ho YS. et al. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313-20
18. Kadenbach B, Ramzan R, Moosdorf R. et al. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion. 2011;11:700-6
19. Bhaskaran M, Mohan M. MicroRNAs: history, biogenesis, and their evolving role in animal development and disease. Vet Pathol. 2014;51:759-74
20. Gurtan AM, Sharp PA. The role of miRNAs in regulating gene expression networks. J Mol Biol. 2013;425:3582-600
21. Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441-50
22. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
23. Ikeda S, Pu WT. Expression and function of microRNAs in heart disease. Curr Drug Targets. 2010;11:913-25
24. Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18:510-25
25. Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419-29
26. Meder B, Keller A, Vogel B. et al. MicroRNA signatures in total peripheral blood as novel biomarkers for acute myocardial infarction. Basic Res Cardiol. 2011;106:13-23
27. Li P. MicroRNAs in cardiac apoptosis. J Cardiovasc Transl Res. 2010;3:219-24
28. Eisenhardt SU, Weiss JB, Smolka C. et al. MicroRNA-155 aggravates ischemia-reperfusion injury by modulation of inflammatory cell recruitment and the respiratory oxidative burst. Basic Res Cardiol. 2015;110:32
29. Hu Q, Luo W, Huang L. et al. Apoptosis-related microRNA changes in the right atrium induced by remote ischemic perconditioning during valve replacement surgery. Sci Rep. 2016;6:18959
30. Liu Q, Du GQ, Zhu ZT. et al. Identification of apoptosis-related microRNAs and their target genes in myocardial infarction post-transplantation with skeletal myoblasts. J Transl Med. 2015;13:270
31. Glass C, Singla DK. MicroRNA-1 transfected embryonic stem cells enhance cardiac myocyte differentiation and inhibit apoptosis by modulating the PTEN/Akt pathway in the infarcted heart. Am J Physiol Heart Circ Physiol. 2011;301:H2038-49
32. Cui Y, Bai Y, Wang XD. et al. Differential expression of miRNA in rat myocardial tissues under psychological and physical stress. Exp Ther Med. 2014;7:901-6
33. Zhao J, Li X, Zou M. et al. miR-135a inhibition protects A549 cells from LPS-induced apoptosis by targeting Bcl-2. Biochem Biophys Res Commun. 2014;452:951-7
34. Shin EA, Sohn EJ, Won G. et al. Upregulation of microRNA135a-3p and death receptor 5 plays a critical role in Tanshinone I sensitized prostate cancer cells to TRAIL induced apoptosis. Oncotarget. 2014;5:5624-36
35. Dang Z, Xu WH, Lu P. et al. MicroRNA-135a inhibits cell proliferation by targeting Bmi1 in pancreatic ductal adenocarcinoma. Int J Biol Sci. 2014;10:733-45
36. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
37. Writing Group M, Mozaffarian D, Benjamin EJ. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38-60
38. Joki Y, Ohashi K, Yuasa D. et al. Neuron-derived neurotrophic factor ameliorates adverse cardiac remodeling after experimental myocardial infarction. Circ Heart Fail. 2015;8:342-51
39. Albrecht M, Zitta K, Bein B. et al. Remote ischemic preconditioning regulates HIF-1alpha levels, apoptosis and inflammation in heart tissue of cardiosurgical patients: a pilot experimental study. Basic Res Cardiol. 2013;108:314
40. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674-99
41. Besbes S, Mirshahi M, Pocard M. et al. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget. 2015;6:12862-71
42. Liu N, Xu Y, Sun JT. et al. The BH3 mimetic S1 induces endoplasmic reticulum stress-associated apoptosis in cisplatin-resistant human ovarian cancer cells although it activates autophagy. Oncol Rep. 2013;30:2677-84
43. Nagel SA, Keuper M, Zagotta I. et al. Up-regulation of Bcl-2 during adipogenesis mediates apoptosis resistance in human adipocytes. Mol Cell Endocrinol. 2014;382:368-76
44. Haidarali S, Patil CR, Ojha S. et al. Targeting apoptotic pathways in myocardial infarction: attenuated by phytochemicals. Cardiovasc Hematol Agents Med Chem. 2014;12:72-85
45. Shi YF, Liu N, Li YX. et al. Insulin protects H9c2 rat cardiomyoblast cells against hydrogen peroxide-induced injury through upregulation of microRNA-210. Free Radic Res. 2015;49:1147-55
46. Li MX, Dewson G. Mitochondria and apoptosis: emerging concepts. F1000Prime Rep. 2015;7:42
47. Inserte J, Cardona M, Poncelas-Nozal M. et al. Studies on the role of apoptosis after transient myocardial ischemia: genetic deletion of the executioner caspases-3 and -7 does not limit infarct size and ventricular remodeling. Basic Res Cardiol. 2016;111:18
48. Song MA, Paradis AN, Gay MS. et al. Differential expression of microRNAs in ischemic heart disease. Drug Discov Today. 2015;20:223-35
Author contact
![]() Corresponding author: Bin Liu, Department of Cardiology, The Second Hospital of Jilin University. Address: 218 Ziqiang Street, Changchun, Jilin, 130041, China. Tel./Fax: +86-431-88796739. E-mail address: liubin3333sina.com.
Corresponding author: Bin Liu, Department of Cardiology, The Second Hospital of Jilin University. Address: 218 Ziqiang Street, Changchun, Jilin, 130041, China. Tel./Fax: +86-431-88796739. E-mail address: liubin3333sina.com.