Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(3):327-338. doi:10.7150/ijbs.17751 This issue Cite
Research Paper
Lyophilized Powder of Catalpol and Puerarin Protected Cerebral Vessels from Ischemia by Its Anti-apoptosis on Endothelial Cells
Yang Liu1, 2, 3, Qing Tang1, 2, 3, Siying Shao1, 2, 3, Yi Chen1, 2, 3, Weihai Chen4, Xiaoyu Xu1, 2, 3 ![]()
1. College of Pharmaceutical Sciences and Chinese Medicine, Southwest University, Chongqing 400715, China
2. Institute of Chinese Medicine, Southwest University, Chongqing 400715, China
3. Chongqing Engineering Research Centre for Pharmacological Evaluation, Chongqing 400715, China
4. Faculty of Psychology, Southwest University, Chongqing, 400715, China
Received 2016-9-28; Accepted 2016-12-12; Published 2017-2-25
Abstract

Catalpol and puerarin are two monomers of Rehmannia glutinosa and Lobed Kudzuvine Root, which are two herbs commonly used together in ancient prescriptions of traditional Chinese medicine for cerebral ischemia. Our previous study shows that the lyophilized powder of the two monomers improved the outcome of cerebral ischemia excellently in rodents. However, if it protects vessels from ischemia is unknown. The present research studied the protection of lyophilized powder of catalpol and puerarin (CP) on endothelial cells and the relative mechanism in vivo and in vitro. Middle cerebral artery occlusion (MCAO) rats were used to study the improvement of CP on neurological deficiency, regional cerebral blood flow (rCBF), and infarct volume. The morphology of vessels and the apoptosis of brain vascular endothelial cells (BVECs) were observed and detected by immunohistochemistry approaches. To study how CP protected primary BVECs (pBVECs) from ischemic penumbra, oxygen glucose deprivation (OGD)-damaged pBVECs were cultured in the condition of insufficient nutrition and low oxygen which recapitulate the low perfusion of ischemic penumbra. Using the cell model, the mechanism by which CP protected pBVECs was studied by shRNA and pathway inhibitors. CP at the dose of 65.4 mg/kg increased regional cerebral blood flow (rCBF), reduced infarct volume, protected vessel integrity and inhibited endothelial cell apoptosis in vivo. But it only improved rCBF, vessel integrity and BVECs apoptosis at the dose of 32.7 mg/kg. In vitro, the protection of CP on pBVECs was proved to be ERK/HIF-1a- and PI3K/AKT/mTOR/HIF-1a-dependent. This study indicates a possibility of CP being a new drug for cerebral ischemia. Besides, this research provides an alternative cell model for penumbra ECs study.
Keywords: Catalpol, Puerarin, vessel, apoptosis, cerebral ischemia, Endothelial cell
1. Introduction
Cerebral ischemia threatens human's health seriously because of poor efficient medicine and clinical management [1]. This clinic embarrassment is mainly due to the complex pathologies of cerebral ischemia. In recent years, combination use of drugs aiming to multiple targets is raised and highly suggested [2]. Notably, this multiple-target strategy has a long history in traditional Chinese medicine. Rehmannia glutinosa (Dihuang) and Lobed Kudzuvine Root (Gegen), for instance, are two Chinese herbs which have been applied in cerebral ischemia in China for hundreds of years. These two herbs were commonly used together in many ancient prescriptions of traditional Chinese medicine for stroke, such as Duzhong-Duhuo-Decoction and Qianghuo-Decoction which are respectively recorded in Wai Tai Mi Yao and Qian Jin Fang.
Concerning the poor quality control of Chinese herbs, the effective elements of herbs are used. Notably, combination use of monomers of traditional Chinese medicines has been successfully implicated into endometriosis [3] and promyelocytic leukemia [4]. Therefore, we combine catalpol and puerarin which are two bioactive monomers respectively isolated from Lobed Kudzuvine Root (Gegen) and Rehmannia glutinosa (Dihuang) to treat cerebral ischemia. We choose puerarin because puerarin treats acute stroke in clinical effectively [5]. The reason of choosing catalpol is that catalpol shows prospect in stroke [6] and depress [7] in pre-clinical research. Further, our previous studies show that the combination use of the two monomers well improved the outcome of middle cerebral artery occlusion (MCAO) mouse [8] and cerebral ischemia/reperfusion rats [9] in aspects of neurological deficiency, infarct volume, edema, inflammation, and oxidative stress. Besides, it increased cerebral CD31 positive cells in MCAO mouse [8] as well as protected human umbilical vascular endothelial cells (HUVECs) from oxygen glucose deprivation (OGD) [10], indicating that the protective effect of CP may be relative to vessel.
Vessel is physiologically and pathologically crucial for brain due to controlling blood perfusion. Structurally, vessel is the most important composition of blood-brain barrier (BBB) which regulates paracellular movement of solutes, ions, and water [11]. Around 2000, the concept of neurovascular unit (NVU) was raised. National Institute of Neurological Disorders and Stroke (NINDS) suggested that all components of NVU should be protected in cerebral ischemia. As controlling blood flow and secreting factors, vessel is regard as the one of the most important components of NVU [12]. Vessel is sensitive to ischemia and is typically damaged in the super acute stage of ischemia [13]. Once cerebral ischemia happens, regional cerebral blood flow (rCBF) in ischemic area is rapidly decreased, resulting in a series of pathological events including Ca2+ overload, oxidative stress, EAA toxicity, inflammation, and apoptosis [1, 12, 14]. Sequentially, BBB is damaged [1, 12]. Further, damaged vessels give rise to edema and hemorrhagic transformation [12]. As the most important role of BVECs in vessel and BBB, protecting them from ischemia is significant and promising [15, 16]. Moreover, maintaining more functional vessels would benefit post-ischemia blood perfusion and facilitate neovascularization and neurogenesis [17].
This study investigated the vascular protection of CP on ischemic brain in aspects of vascular morphology and rCBF. Mechanism was studied by shRNA and pathway inhibitors in primary brain vascular endothelial cells (pBVECs) in vitro in a penumbral culture condition.
2. Methods
2.1 The single component identification of CP by HPLC
CP consisted of catalpol (Liu bo bai niao Biological Technology Lot. NO. 08051009) and puerarin (Liu bo bai niao Biological Technology Lot. NO. 090602) with a ratio of 9:40 [22]. The catalpol and puerarin in CP were well characterized by HPLC (Agilent 1200, USA) according to previous report [8]. Chromatographic conditions were as following: a column Agilent Zorbax SB-C18 (4.6 mm × 250 mm, 5 μm) was used; the eluent for gradient elution was water and acetonitrile; sample size was 10 μl; flow rate was set at 1.0 ml/min and the column temperature was kept at 30°C. Catalpol and puerarin were detected at 210 nm. The HPLC analysis was validated and met the methodological requirements.
2.2 MCAO
All animal experiments obeyed the ARRIVE guidelines and were carried out in accordance with National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978). MCAO rats were prepared by electrocoagulation according to previous reports [18, 19]. Basically, male SD rats (200-250g) were anesthetized by 1% isoflurane. A 2cm incision was located above the right eyepit and temporalis was departed from harnpan to expose temporal fossa. A little hole was carefully drilled by a small drill on temporal fossa. The hole was broadened until the middle cerebral artery was exposed. The artery was blocked by a bipolar coagulator with power of 2 for 2-3 s per time for several times until the vessel got shrank and paled. Rats suffered the same surgery except occlusion were chosen as sham controls. Rectal temperatures and cerebral blood flow were monitored during the whole surgery. Successful MCAO rats were considered with scores of mNSS between 3 and 8 and more than 80% rCBF reduction in infarct area.
2.3 rCBF
rCBF in the core area (2 mm caudal to bregma and 6 mm lateral to midline) and peripheral area (2 mm caudal to bregma and 3 mm lateral to midline) of ischemic territory was detected by a laser-Doppler flowmetry (LDF) (ML191, Australia ADInstruments) before and after surgery [20]. During the whole testing, the LDF probe (DP3 optical, I -mm diameter) was fixed and kept from moving. 14 days after the surgery, rCBF in peripheral ischemic core was evaluated again.
2.4 Neurological deficiency
Neurological deficiency was evaluated by modified neurological severity score (mNSS). As previous reported, rats were trained with mNSS for 7 days before surgery [21]. 2-4 hours after the surgery, MCAO rats were tested for mNSS. 14 days after MCAO surgery, the neurological deficiency of each rat was retested.
2.5 Animal groups and CP administration
Qualified MCAO rats were randomly divided into a MCAO group and CP groups (65.4 mg/kg, 32.7 mg/kg and 16.4 mg/kg) by a random number table generated by SPSS software 13.0. Catalpol (Liu bo bai niao Biological Technology Lot. NO. 08051009) and puerarin (Liu bo bai niao Biological Technology Lot. NO. 090602) were mixed with a ratio of 9:40 to generate CP [22]. Rats were received CP infusion via tail vein daily at the doses of 65.4 mg/kg, 32.7 mg/kg and 16.4 mg/kg for 14 days. Meanwhile, MCAO rats received vehicle served as control.
2.6 Vascular morphology observation
Vascular morphology in ischemic brain was observed by the immunohistochemistry stain of CD31, and BVECs apoptosis was visualized by VIII/TUNEL/DAPI stain. Rats were sacrificed for brain sections on the 4th day. 20 μm frozen sections and 4 μm paraffin sections of brains were prepared for immunofluorescence and immunohistochemistry respectively. Specific protocols were conducted according to previous reports [23]. Primary antibodies were anti-VIII (1:300 Santa Cruz sc3657) and anti-CD31 (1:300 Sigma-Aldrich Inc.).
The vessel morphology in the right cerebral cortex of coronal sections (1 mm, 2 mm, and 3 mm from optical chiasm) was observed. Those areas were selected because they are damaged by MCAO. The number of degraded vessel and the number of apoptotic BVECs were counted in three regions at size of 500 × 500 μm2 in each section.
2.7 Infarct volume
14 days after MCAO surgery, brains were isolated for TTC stain. Each brain was cut in 2 mm thick for 7 pieces and then immerged in 2% TTC solution. The infarct volume was evaluated by Image Pro-Plus 5.1 analysis system (Media Cybernetics Inc., Rockville, MD, USA). Infarct volume was quantified by following equation: I = (CD + CT - IT). In the equation, the I means infarct area, CD means the area without cresyl violet, and CT and IT respectively mean the total area of the contralateral hemisphere and the ipsilateral hemisphere. The total infarct volume in mm3, therefore, were calculated by ΣI ∗ 0.4 in which the 0.4 means the distance between neighbor two sections in mm. This equation was used to correct for the increase in volume of the ipsilateral hemisphere due to edema-induced swelling [24].
2.8 Culture of pBVECs and CP administration in vitro
Primary brain vascular endothelial cells (pBVECs) were isolated, cultured, and identified according to our previous report [25]. For all the in vitro study, CP was added when cells were damaged.
2.9 Conditional cell culture
Oxygen and glucose deprivation (OGD): when grown to confluence, pBVECs were cultured in FBS-, Sodium Pyruvate- and Glucose-free F12/DMEM culture medium (Gibco) in a 0.2% O2 and 5% CO2 incubator (Binder) at 37˚C for 20 hours. Low oxygen and nutrition (LON): after damaged by OGD, ECs were cultured in hypoxia (2% O2) and inadequate nutrition culture medium for 24 hours (F12/DMEM medium containing 20% FBS was diluted to four times of volume with deionized water). For pathway study, cells were pre-treated with PD98059 (10 μmol/ml), Rapamycin (0.1 μmol/ml) and LY249002 (10 μmol/ml) for 1 hour to inhibit ERK, mTOR, and AKT respectively. And then, cell apoptosis and necrosis in the model of OGD+LON were detected.
2.10 Cell survival, apoptosis, and necrosis
Cellular survival was tested by MTT as previous reported [8] and calculated via the following equation: cell survival ratio = (OD (CP group) - OD (blank value)) / (OD (control group) - OD (blank value)) * 100%. Apoptosis and necrosis were analyzed by TUNEL and PI stain. TUNEL stain was conducted following the direction of the kit. 1 μl PI (1 mg/ml) was added into each well. Images were captured and analyzed by a fluorescent microscope (Carl Zeiss, Oberkochen, Germany).
2.11 shRNA
Control shRNA and 2 sequences of rat HIF-1α shRNA were purchased from GenePharm. Control shRNA consists of a scrambled sequence that will not lead to specific degradation of any known cellular mRNA. The shRNA1 sequence: 5'- GATCCGCAGTGACGAAGGACAATATATTCAAGAGATATATTGTCCTTCGTCACTGCTTTTTT-3'; the shRNA 2 sequence: 5'-GATCCGGAGGACGATGAACATCAAGTTTCAAGAGAACTTGATGTTCATCGTCCTCCTTTTTTG-3'. Sequences were cloned into the lentivirus-based RNAi vector pGPU6/GFP/Neo (GeneChem Co., Ltd.). A non-targeting stem-loop DNA was also inserted into pGPU6/GFP/Neo vector as a negative control. HIF-1α-shRNA-lentiviral plasmid and negative control (NC)-shRNA plasmid were respectively packaged in 293T cells using the ViraPowerTM Lentiviral Expression Systems (Invitrogen) according to the manufacturer's protocol. 24 h later, green fluorescent protein (GFP) was expressed in 293T cells. The culture medium was harvested at 48 hours and 72 hours after transfection. Primary ECs were infected with HIF-1α-shRNA-lentivirus or NC-shRNA-lentivirus. For each transfection, 1 ml of the lentiviral particles and 1 ml of the complete medium with 6 μg/μl of polybrene were added gently into the cells. After 24 hours of incubation, the culture medium was changed into fresh complete medium for another 36 hours. Fluorescent microscope was used to observe the GFP expression. Transfected cells were then cultured in hypoxia (3% O2) for 12 hours and then were harvested for evaluating the nuclear expression of HIF-1α via Western Blot.
2.12 Western blot
Endothelial nuclei were extracted by kit and were used to determine the protein level of HIF-1α. Mitochondria were isolated by kit for detecting Cyt-c. Western blot was repeated in 3 independent experiments with 6 wells of each group. Specific steps were followed the previous report [9]. Respectively, primary antibodies were used in following concentrations: anti-HIF-1α :1500 Proteintech 12245), anti-cleaved caspase-3 (1:1000 CST 9662), anti-ERK (1:500 Proteintech 16443), anti-pErk (Thr202/Tyr204) (1:1000 CST 4370), anti-S6K (1:500 abcam ab36864), anti-pS6K (Thr389) (1:1000 CST9234), anti-P53 (1:1500 Proteintech 10442), anti-pP53 (Ser37) (Santa Cruz sc-9051), anti-pP53 (Ser15) (R&D AF1043), anti-AKT (1:1000, CST 9272), anti-pAKT (1:1000, CST 9271) p4EBP(1:1000, CST 2855), 4EBP(1:1000, CST 9955), VDAC (1:1000 CST 4866) and anti-β-actin (1:50000 Proteintech 60008).
2.13 Statistic analysis
Normal distribution data are present as mean ± standard deviation (S.D.), and were analyzed by an unpaired two-tail Student's t-test or a ANOVA test. Non-normal distribution data are present as medians ± range and analyzed by Kruskal-Wallis ANOVA on ranks followed by Bonferroni's test. Tukey's test was used as post-hoc test performed after the ANOVA. P < 0.05 was considered as statistical significance.
3. Results
3.1 CP ameliorated cerebral ischemia of rats
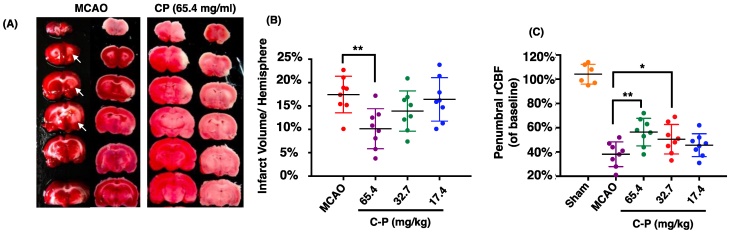
Catalpol and puerarin in CP compound were identified by HPLC (Figure 1). To test if CP protects brain form cerebral ischemia, neurological deficiency, rCBF and infarct volume in MCAO rats received CP (65.4 mg/kg, 32.7 mg/kg, and 16.4 mg/kg) or vehicle were evaluated. mNSS data show that most MCAO rats prepared by electrocoagulation were of mild neurological deficiency. To avoid a significant difference of neurological deficiency in MCAO rats, rats those with 3-8 scores of mNSS were used. At the same time, only MCAO rats those with rCBF reduction more than 80% in infarct area were considered to be occluded successfully. Before received CP infusion, MCAO rats were tested for mNSS and rCBF to check if there is a significant difference among groups at the initiation of experiment. Statistical analysis indicates that there was no significant different mNSS and rCBF among the groups (data are not shown). On the 14th day, infarct volume and rCBF in the peripheral region of MCA territory were well improved by CP infusion once per day at the dose of 65.4 mg/kg (Figure 2 A-C). But CP at the dose of 32.7 mg/kg only significantly increased rCBF (Figure 2 C). Unfortunately, no protective effect was observed in 16.4 mg/kg group in all tested indexes. Although no significant improvement of neurological deficiency was observed in CP treated MCAO rats (Figure S1), the infarct volume and rCBF data still indicate a dose-dependent protection of CP on ischemic rat brains.
Catalpol and puerarin were identified by HPLC. (A): the HPLC analysis of catalpol standard. (B): the HPLC analysis of puerarin standard. (C) and (D): the HPLC analysis of CP.

CP ameliorated the outcome of MCAO rats. CP infusion at the dose of 65.4 mg/kg for 14 days significantly decreased cerebral infarct volume (A and B). CP at doses of 65.4mg/kg and 32.7 mg/kg obviously increased the rCBF of MCAO rats (C). n = 8. mean ± SD. * p<0.05; ** p<0.01.
3.2 CP maintained vascular integrity and kept BVECs from apoptosis
To observe the morphology of vessel in ischemic brain, CD31 was used to label vessels. In figure 3 A and B, vessels in sham rats maintained integrity well. But in MCAO rats, they were present as semi-continuous BVECs or totally isolated BVECs instead of intact vessels (figure 3 A). Meanwhile, obvious basement membrane degradation in ischemic vessels was observed (figure 3 B). With CP infusion at doses of 65.4 mg/kg and 32.7 mg/kg for 4 days, vessel integrity was improved and the vessel degradation was decreased (figure 3 A and B). Simultaneously, statistical analysis indicates that the number of degraded vessels was decreased by the two doses of CP (figure 3 C).
CP protected vessel integrity via inhibiting the apoptosis of BVECs. 4 days after the surgery, vessels were present as semi-continuous or completely isolated BVECs, indicating the loss of vessel integrity in MCAO group. Meantime, CP at doses of 65.4 mg/kg and 32.7 mg/kg protected the integrity from lost (A). Vessel degradation in MCAO groups was obviously increased. With CP infusion (65.4 mg/kg and 32.7 mg/kg) for 4 days, the degradation was extenuated (as pointed out by black arrows in B). Statistical analysis indicates that the number of degraded vessel was significantly decreased by 65.4 mg/kg and 32.7 mg/kg of CP (C). Apoptotic BVECs were labeled by VIII/TUNEL and these cells were abundant in MCAO group on the 4th day. CP at doses of 65.4 mg/kg and 32.7 mg/kg significantly reduced the number of apoptotic BVECs (D and E). n = 7, mean ± SD。* p<0.05; ** p<0.01; *** p<0.001.
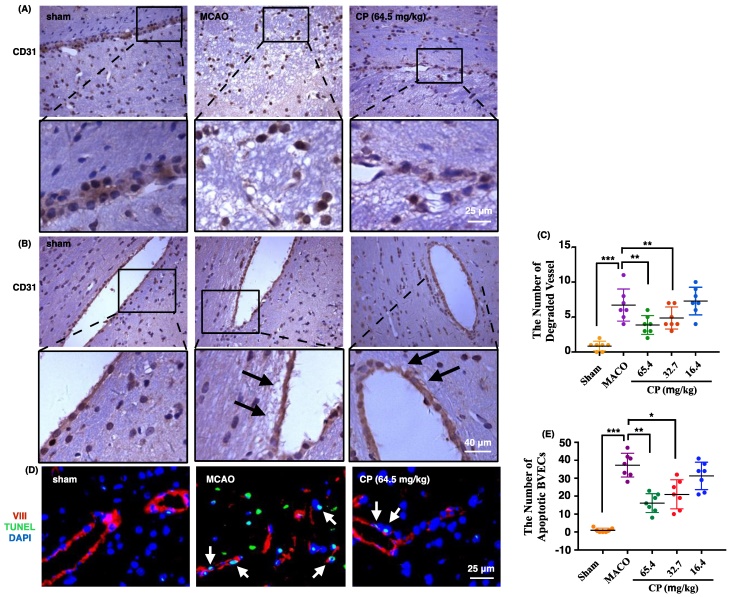
As apoptosis is the main manner of cell death occurring in penumbra, we deduced that CP protects BVEC from apoptosis. In sham group, very rare apoptotic BVECs which were identified by TUNEL stain were observed. But apoptotic BVECs were abundant in ischemic penumbra 4 days after the surgery (figure 3 D and E). As expected, apoptotic BVECs in penumbral area were significantly decreased by CP intervene (65.4 mg/kg and 32.7 mg/kg) (figure 3 D and E).
3.3 OGD+LON was established for simulating penumbra
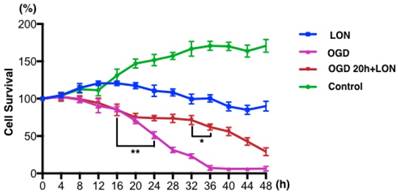
We tried to culture BMECs in low oxygen and nutrition (LON) to imitate the insufficient perfusion in penumbra. However, the survival ratio of pBVECs upon LON maintained at level of 90% ± 6.52% during the whole observed period (Figure 4), indicating that LON failed to duplicate the penumbra-induced injury. Since OGD is commonly used to imitate ischemic injury in vitro, we combined OGD with LON to model penumbra situation in vitro. In this model, cells were damaged by OGD and then cultured in LON. According to the curve (Figure 4), cell survival ratio decreased to 70% after damaged by OGD for 20 hours, and then it continuously decreased to below 10% at 36 hours. Hence, 20 hours was chosen for OGD damaging pBVECs. The OGD-damaged cells showed a stable survival curve in the prior 12 hours of LON culture (Figure 4), but it then decreased to 62.00% ± 4.00% at 16 hours and 29.00% ± 5.29% at 28 hours (Figure 4). This curve indicates that OGD-damaged pBVECs could survive in LON culture condition for a limited time, which resembles that cells in ischemic penumbra is curable in a limited time in vivo.
ODG+LON recapitulated the features of penumbra. In normal culture condition, pBVECs expanded and cell survival ratio was increased to 170.67% ± 8.62% at 48 hours. Under LON condition, cell survival ratio was maintained at a relative stable level (90% ± 6.52%), indicating that LON failed to damage pBVECs. With OGD, cell survival ration was significantly decreased after 16 hours and very few cells were kept alive at 36 hours. Considering that OGD differs from the insufficient perfusion in penumbra, OGD is unsuitable for penumbra study. Therefore, combination OGD with LON was used to imitate the cell damage and insufficient perfusion of penumbra. In such a model, cells were damaged by OGD for 20 hours and then cultured in LON (2% O2 and a low nutrition condition that 20%FBS F12/DMEM was diluted to one fourth with water). During the prior 12 hours of LON stage, cell survival ratio was not decreased obviously but then it reduced significantly. After cultured in LON for 28 hours, cells were damaged severely. This curve indicates that OGD-damaged pBVECs could live for a limited time upon LON. n = 3, mean ± SD. * p<0.05; ** p<0.01.
3.4 CP protected pBVECs from ischemic damage in vitro
Using OGD model which is typically used as an in vitro model for ischemia, we asked if CP protects pBVECs from ischemic injury. After damaged by OGD for 20 hours, cell viability was decreased significantly (Figure 5 B and C). With treatment of CP (49 μg/ml), the viability was significantly increased (0.74 ± 0.04 vs. 0.61 ± 0.04), indicating that CP protected pBVECs from ischemic damage.
CP protected pBVECs from OGD and OGD+LON. (A) is the scheme of the experiment. (B) and (C) show that CP protected pBVECs from OGD at dose of 49 μg/ml. (D) and (E) show that CP at doses of 24.5 μg/ml, 49 μg/ml and 98 μg/ml protected pBVECs from OGD+LON while it was added into cells at the beginning of OGD damage. n = 6, mean ± SD. * p<0.05; ** p<0.01; *** p<0.001.
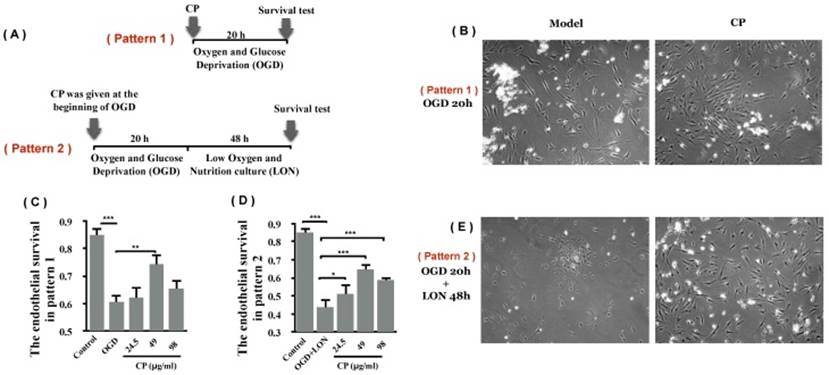
In addition to OGD, the protective effect of CP on pBVECs upon OGD+LON was observed. After damaged by OGD for 20 hours and then cultured in LON for 24 hours, the viability of pBVECs was reduced to 0.44 ± 0.03 (Figure 5 D and E). With CP infusion at doses of 24.5 μg/ml, 49 μg/ml and 98μg/ml, cell viability was obviously increased to 0.51 ± 0.04, 0.65 ± 0.04 and 0.59 ± 0.03 respectively (Figure 5 D and E).
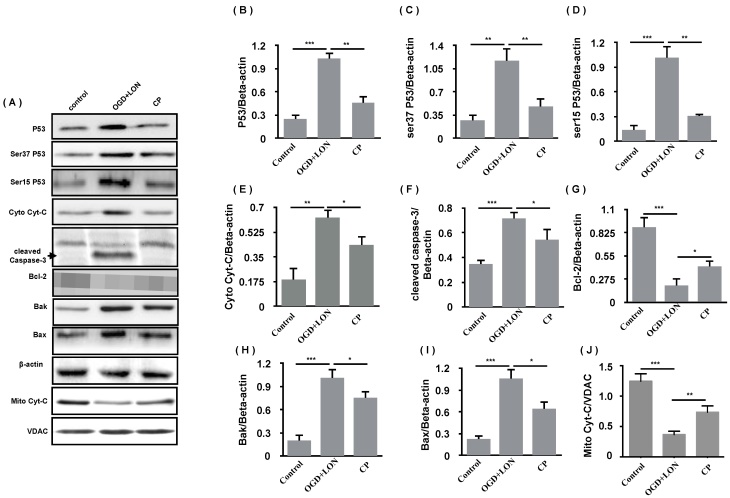
As 49 μg/ml was the most efficient doses of CP in the aspect of anti-apoptosis, we further asked if its mechanism is relative to p53/caspase-3 cascade. Using western blot, the anti-apoptosis of CP (49 μg/ml) was confirmed by the decreases in Bax and Bak and the increase in Bcl-2 in CP group. Meanwhile, P53, pP53(ser37, ser15) was decreased, indicating that P53 was less activated in CP group compared to vehicle group. As we know, damaged mitochondria which can be induced by P53/Bak-Bax result in the release of Cyt-C which activates caspase-3-dependent apoptosis. Giving this, we further tested if CP inhibits the release of Cyt-C and inhibited the activation of caspase-3. As shown in Figure 7, CP significantly decreased cytoplastic Cyt-C, increased mitochondrial Cyt-C, and reduced cleaved-caspase-3, indicating that CP significantly inhibited Cyt-C/caspase-3-dependent apoptosis.
CP inhibited the P53-dependent apoptosis of pBVECS cultured in OGD+LON. (A): bands show that CP at the dose of 49 μg/ml inhibited P53/caspase-3-dependent apoptosis in the models of OGD+LON. (B)-(I) are the statistical analysis of the OD values of the apoptotic proteins. n = 3, mean ± SD. * p<0.05; ** p<0.01; *** p<0.001.
HIF-1α was implicated in CP protecting pBVECs. CP (49 μg/ml) promoted the expression and nuclear relocation of HIF-1α in pBVECs in the model of OGD+LON (A). The loss of HIF-1α resulted in a decrease in cell viability (C) and increases in apoptotic and necrotic cells in CP-treated groups (B, D, E). Besides, the loss of HIF-1a impaired the inhibition of CP on Cyt-C release from mitochondria, showing increased cytoplastic Cyt-C and decreased mitochondrial Cyt-C (G-I). For apoptotic cell and necrotic cell, n = 6, mean ± SD; for western blot, n = 3, mean ± SD. * p<0.05; ** p<0.01; *** p<0.001.
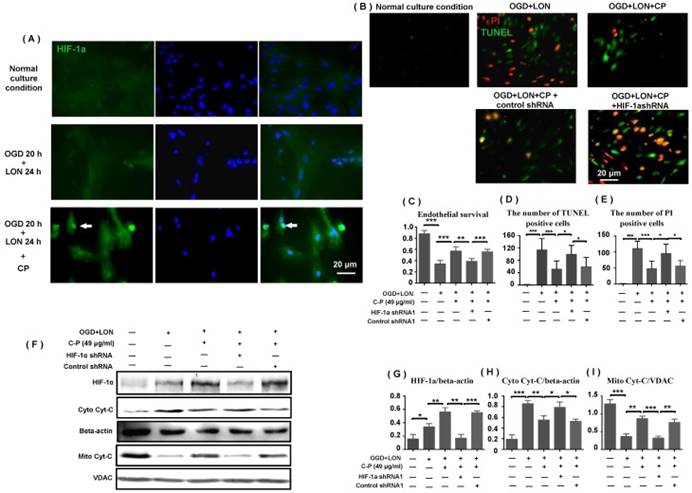
3.5 HIF-1α was implicated in CP preventing pBVECs from apoptosis and necrosis
As HIF-1α is essential for cell adapting to hypoxia, we further tested if CP impacts HIF-1α expression in pBVECs under OGD+LON. Western blot data show that CP at the dose of 49 μg/ml obviously increased HIF-1α protein level upon OGD+LON (Figure 7 F). At the same time, it promoted the nuclear recruitment of HIF-1α (Figure 7 A).
To confirm that HIF-1α is essential for CP protecting pBVECs from OGD+LON, shRNA was used to delete HIF-1α. Immunofluorescence shows that GFP was expressed in pBVECs (Figure S2 B), indicating that pBVECs were transfected by lentivirus particles. To confirm that HIF-1a is deleted by shRNA, pBVECs were cultured under 3% O2 for 12 hours to induce the expression of HIF-1α and then HIF-1a was checked by western blot. Western blot images show that HIF-1α was remarkably reduced by shRNA1 and shRNA2 at protein level (Figure S2 C and D). Then, we tested if control shRNA affects cell survival, apoptosis and necrosis under normal condition and OGD+LON. Cell survival was tested by MTT. Apoptotic cells were labeled by TUNEL and necrotic cells were labeled by PI. Under normal condition and OGD+LON, no significant difference was observed in cell survival, apoptosis, and necrosis between control group and control shRNA group (Figure S3).
Under OGD+LON, the loss of HIF-1α resulted in decreased survival, TUNEL positive cells and PI positive cells in CP-treated pBVECs (Figure 7 C-E). At the same time, the loss of HIF-1a led to increase in cytoplastic Cyt-C and decrease in mitochondrial Cyt-C in CP-treated pBVECs (Figure 7 F-I). Collectively, these experimental results indicate that CP protecting pBVECs from apoptosis was HIF-1α-dependent.
3.6 CP promoting HIF-1a was dependent on PI3K and ERK
As p4EBP and pS6K protect mRNAs including HIF-1a mRNA from degrading, we deduced that ERK and mTOR pathways which regulate the two proteins positively were relative to CP increasing HIF-1a and protecting pBVECs. To prove the hypothesis, we firstly tested if the two pathways are activated by CP. Western blot data show that pERK, pS6K, p4EBP were increased by CP, proving that CP activated ERK and mTOR (Figure 8 A, C-E). Then, we checked if blocking either ERK or mTOR impairs the increases of p4EBP and HIF-1a which are induced by CP. When either ERK or mTOR was blocked by PD98059 or rapamycin respectively, CP-increased p4EBP and HIF-1α were significantly reduced, indicating that CP up-regulated HIF-1a via ERK- and mTOR-regulated pS6K and p4EBP (Figure 8 A, E, and F).
ERK and PI3K/AKT/mTOR were involved in CP protecting pBVECs and up-regulating HIF-1a. CP (49 μg/ml) activated both ERK and mTOR to increase pS6K and p4EBP1. Inhibiting either ERK (by PD98059) or mTOR (by rapamycin) resulted in decreases in pS6K, p4EBP1, HIF-1a, mitochondrial Cyt-C and an increase in cytoplastic Cyt-C (A). (B) indicates that CP activating mTOR was dependent on PI3K/AKT but not ERK. (C)-(k) are statistical analysis of (A) and (B). (L) shows that inhibiting ERK or mTOR induced increases in apoptotic cells and necrotic cells as well as a decrease in cell viability. For apoptotic cell and necrotic cell, n = 6, mean ± SD; for western blot, n = 3, mean ± SD. * p<0.05; ** p<0.01; *** p<0.001.
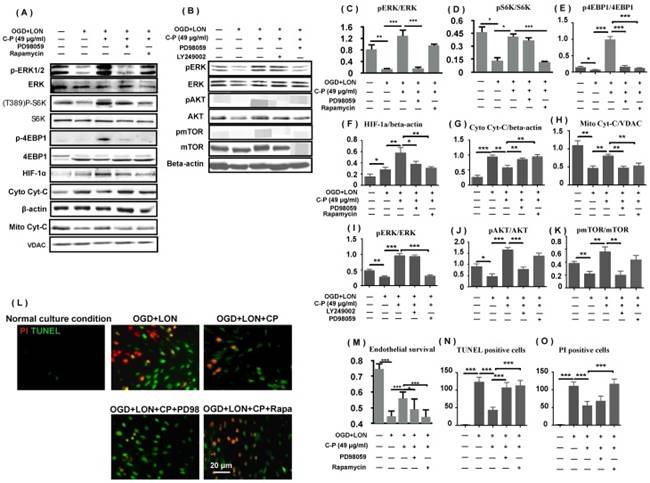
Then, we asked if blocking either ERK or mTOR attenuates the protection of CP on pBVECs. MTT data show that either PD98059 or rapamycin significantly reduced the cell viability and the mitochondrial Cyt-C and increased the cytoplastic Cyt-C in CP-treated pBVECs (Figure 8 M). Notably, inhibiting mTOR significantly increased TUNEL posive cells and PI positive cells in CP-treated cells, but inhibiting ERK only increased TUNEL positive cells (Figure 8 L, N, and O). These data indicate that ERK and mTOR were essential for CP protecting pBVECs, but they played different role in the antiapoptosis and necrosis of CP.
As mTOR was reported to be regulated by ERK and PI3K/AKT, we checked if CP activating mTOR is relative to ERK and PI3K/AKT. Western blot data show that blocking PI3K/AKT via LY249002 completely abolished CP-upregulated pmTOR in pBVECs, while inhibiting ERK via PD98059 led no significant change of pmTOR (Figure 8 B and I-K). These data indicate that CP activating mTOR via PI3K/AKT but not via ERK, which is consistent with that CP-increased pS6K was only reduced by rapamycin (mTOR inhibitor) but not by PD98059 (ERK inhibitor).
4. Discussion
4.1 CP protects brain cells from ischemic brain
Although we did not see significant improvement of neurological deficiency in CP-treated groups in the present study, we consider that CP was effective for neurological recovery as CP improved the neurological deficiency in our previous studies [8, 9]. The potential explanation for the difference is the different MCAO models, because neurological deficiency in MCAO rats generated by electrocoagulation was much weaker than it in MCAO rats prepared by nylon suture. Further, CP improved the outcome of cerebral ischemia including edema and infarct volume in MCAO mouse and ischemia/reperfusion rats [8, 9]. In the present study, we further proved that CP increased the rCBF in the ischemic penumbral area. In previous studies, CP protected several types of brain cells in vitro and in vitro, including PC12 cells [8], primary astrocytes [26], and HUVECs [10]. Here, we provide sound experimental evidence that CP protected BVECs from ischemia in vivo and in vitro. These findings highly suggest that CP protected NVU which mainly consists of neuron, astrocyte and endothelial cells.
4.2 BVECs are important target cells via which CP protects brain from ischemia
As vessel is one of the most important elements of neurovascular unit (NVU) [12], vascular protection is regarded as a promising therapy for cerebral ischemia [16]. Giving that catalpol and puerarin protect brain from ischemia via inhibiting oxidative stress and inflammation and promote neurogenesis, angiogenesis, and neural plasticity [27, 28], we wonder if combination use of the two monomers protects BVECs from ischemia-induced apoptosis.
In vitro study shows that CP protected HUVECs from OGD-induced apoptosis [10]. Further, report shows that CP protected NVU in which vessel is a core element from apoptosis in vitro and in vitro [9]. In addition to anti-apoptosis, CP improved cerebral circulation in the rat model of cerebral microcirculation disturbance induced by dextran infusion [29]. These data suggest that vessel is one of the important targets of CP. To confirm that if CP protects vessels from ishcemia, vascular morphology, rCBF, and the apoptosis of BVECs were carefully studied in the present study. Data show that CP improved all the indexes in a dose-dependent manner. As acknowledged widely neovascularization benefits the post-ischemic neural recovery [30]. There are studies showing that both catalpol and pueraine promoted neovascularization in vivo and in vitro [27, 28, 31]. In our previous report, we observed that CP significantly increased CD31 positive cells and VEGF positive cells in ischemic brain [8], suggesting that CP expanded angiogenesis in ischemic brains. Collectively, it is great potential that CP promoted the recovery of cerebral ischemia via inhibiting BVEC apoptosis and promoting neovascularization.
4.3 CP protects vessels via multiple pathologies and pathways
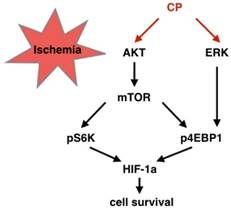
As acknowledged widely, edema, oxidative stress, or inflammation results in vessel damage. Apoptosis and necrosis are two ways by which these pathologies lead cells to death. According to our studies, CP ameliorated edema, oxidative stress, and inflammation very well. In vivo, water content was significantly reduced in CP-treated MCAO mice, indicating that CP reduced ischemia-induced edema [8]. Both in MCAO mouse and cerebral ischemia/reperfusion rats, CP decreased the levels of MDA, NO, and LDH, and enhanced the activity of SOD [8, 9], suggesting that it weakened oxidative stress. Furthermore, it declined IL-1β, IL-6, and TNF-α as well as down-regulating NF-KB pathway, implicating that inflammation was inhibited by CP. In this research, anti-apoptosis of CP on BVECs was evidenced in vivo and in vitro. By knock down HIF-1a via shRNA, this anti-apoptosis was further proved to be relative to HIF-1a which adjust cells to hypoxia. ERK and mTOR are two pathways protecting HIF-1α mRNA from degradation. This protection is dependent on phosphorylation of S6K and 4EBP1which protects HIF-1a mRNA from degradation [32]. In the present study, we proved that CP up-regulating HIF-1a was dependent on this manner (Figure 8 and 9). Notably, although CP activated ERK and PI3K/AKT both of which locate in up-stream of mTOR, we proved that CP activated mTOR via PI3K but not ERK since blocking PI3K/AKT but not ERK reduced the levels of pS6K and pmTOR in CP-treated pBVECs (Figure 8 A-D and I-K).
Graphical summary of the CP-activated signaling cascade. Under ischemic situation, CP activated PI3K/AKT/mTOR and ERK pathways, resulting in phosphorylation of S6K and 4EBP1. Then, the increased pS6K and p4EBP1 enhanced HIF-1a protein level which protected cells from ischemia.
4.4 OGD+LON can be regarded as an alternative penumbral model in vitro
Penumbra is a hot topic in research of stroke diseases. Apoptotic cells are one of the most cell death type in penumbra which is induced by insufficient blood perfusion [33]. How to rescue curable apoptotic cells in penumbra is a long term question. OGD, starvation, and OGD/reperfusion are popular cell modes for ischemic study in vitro. However, none of them meets the features of penumbra very well. Cell models specially designed for ischemic penumbra are rarely reported. There is a reported one which was designed for studying the neurotoxicity of microglia in penumbra. The model consists of three stages: at the first stage, the co-culture of neurons and astrocytes is damaged by OGD, which simulates the infarct core; at the following stage, the OGD-damaged co-culture was co-incubated with microglia to stimulate the microglia; and at the final stage, microglia were washed and incubated with naive neuron/astrocyte co-culture. In such a system, the neurotoxicity of microglia in penumbra was duplicated [34]. Another penumbral cell model was applied in B104 rat neuroblastoma cell line. In this model, cell culture condition was set as 1% FBS (decreased from 10%), 1% O2, and normal high glucose medium. In the cell model, the researchers observed a significant reduction in cell viability after the cells were cultured for 4-6 hours. Meanwhile, increased apoptotic and necrotic cells were observed in the model [35].
According to the second cell model stated above [34], we cultured pBVECs under low oxygen and low nutrition condition. Unfortunately, we failed to observe a cell damage in pBVECs. Potential explanation is the greatly different ischemic tolerance between cell types. Then, we combined OGD and LON to imitate penumbra. In the model, pBVECs were damaged by OGD for 20 hours and then cultured in LON (2% O2 and diluted 20% FBS culture medium) which recapitulates the insufficient perfusion of penumbra. In the model, OGD-damaged cells maintained a relative stable survival ratio in the first 12 hours of LON culture. But the ratio was decreased remarkably from 16 hours. The change of the cell survival in the model is similar to that cells in penumbra could only live for a very limited time [33]. Besides, we observed abundant apoptotic in the model, which resembles to the phenotyping of penumbra [33]. However, there is still a concern if this model can be used for other types of brain cells.
5. Conclusions
This research proved that CP protected brain from ischemia via protecting endothelial cells from apoptosis. This protection was relative to HIF-1a which was dependent on ERK, PI3K/AKT/mTOR pathways.
Supplementary Material
Supplementary figures.
Acknowledgements
This research was funded by the Fundamental Research Funds for the Central Universities (No. XDJK2015D016, No. XDJK2016C070), the National Natural Science Foundation of China (No. 81473549, No. 81402441, NO. 81302757) and the China Postdoctoral Science Foundation funded project (No.2014M562272).
Author's contribution
Yang L. performed the experiments, analyzed the data, and wrote the article. Qing T. performed the experiments and analyzed the data. Siying S. analyzed the data and wrote the article. Yi C. and Weihai C. funded the study and reviewed the article. Xiaoyu X designed the study, funded the study, and reviewed the article.
Conflict of interest
All authors declare that there are no conflicts of interest.
References
1. Doyle K. P, Simon, R. P, Stenzel-Poore, M. P. Neuropharmacology - special issue on cerebral ischemia mechanisms of ischemic brain damage - review article. Neuropharmacology. 2008;55(3):310-318 doi:10.1016/j. neuropharm.2008.01.005
2. Marc F. New approaches to neuroprotective drug development. Stroke. 2011;42:S24-27 doi: 10.1161/STROKEAHA.110.592394
3. Tang Q, Shang F, Wang X. et al. Combination use of ferulic acid, ligustrazine and tetrahydropalmatine inhibits the growth of ectopic endometrial tissue: a multi-target therapy for endometriosis rats. Journal of Ethnopharmacology. 2014;151(3):1218-25
4. Wang L, Zhou G, Liu P et al. Dissection of mechanisms of chinese medicinal formula realgar-indigo naturalis as an effective treatment for promyelocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(12):4826-31
5. Tan Y, Liu M, Wu B. Puerarin for Acute Ischemic Stroke. Stroke. 2008;39:2188. doi: 10.1161/STROKEAHA.107.512228
6. Yan-Ru L, Peng-Wei L, Jian-Jun S. et al. Catalpol provides protective effects against cerebral ischaemia/reperfusion injury in gerbils. Journal of Pharmacy & Pharmacology. 2014;66:1265-1270 doi: 10.1111/jphp.12261
7. Wang J.M, Yang L.H, Zhang Y.Y. et al. BDNF and COX-2 participate in anti-depressive mechanisms of catalpol in rats undergoing chronic unpredictable mild stress. Physiology & Behavior. 2015;151:360-368 doi:10.1016/j. physbeh.2015.08.008
8. Yang L, Qiang X, Xu L. et al. Amelioration of stroke-induced neurological deficiency by lyophilized powder of catapol and puerarin. International Journal of Biological Sciences. 2014;10:448-456 doi:10.7150/ijbs.8571
9. Xue Q, Liu Y, He R. et al. Lyophilized Powder of Catalpol and Puerarin Protects Neurovascular Unit from Stroke. Int J Biol Sci. 2016;12(4):367-380 doi:10.7150/ijbs.14059
10. Zhang C, Qiang M.A, Xue Q. et al. Protective effect of Zige lyophilized powder on hypoxia/reoxygenation injury of human umbilical vein endothelial cells. China journal of Chinese materia medica. 2012;37:997-1001
11. Daneman R. The blood-brain barrier in health and disease. Annals of Neurology. 2012;72(5):648-672
12. Zoppo G. J. D. The neurovascular unit in the setting of stroke. Journal of Internal Medicine. 2010;267(2):156-71
13. Hsu Y. C, Chang, Y. C, Lin, Y. C, et al. Cerebral microvascular damage occurs early after hypoxia-ischemia via nnos activation in the neonatal brain. Journal of Cerebral Blood Flow & Metabolism Official Journal of the International Society of Cerebral Blood Flow & Metabolism. 2014;34(4):668-76
14. Woodruff T. M, Thundyil, J, Tang, S. C, et al. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Molecular Neurodegeneration. 2011;6(1):1-19
15. Zachary I, Mathur A, Yla-Herttuala S. et al. Vascular protection: a novel nonangiogenic cardiovascular role for vascular endothelial growth factor. Arteriosclerosis Thrombosis & Vascular Biology. 2000;20(6):1512-20
16. Rodríguez-Yáñez M, Castellanos M, Blanco M. et al. Vascular protection in brain ischemia. Cerebrovascular Diseases. 2006;21(Suppl 2):21-9
17. Liu J, Wang Y, Akamatsu Y. et al. Vascular remodeling after ischemic stroke: mechanisms and therapeutic potentials. Progress in Neurobiology. 2014;115(2):138-156
18. Liu H, Hu Y, Zhang X. et al. Improved electrocoagulation method for establishing rat cerebral apoplexy model. Third Mil Med Univ. 2011;33(17):1798-2011
19. Llovera G, Roth S Plesnila N. et al. permanent coagulation of the distal middle cerebral artery. J. Vis. Exp. 2014:e51729
20. Jiang T, Gao L, Guo J. et al. Suppressing inflammation by inhibiting the NF-kappaB pathway contributes to the neuroprotective effect of angiotensin-(1-7) in rats with permanent cerebral ischaemia. British Journal of Pharmacology. 2012;167:1520-1532
21. Chen J, Sanberg P. Y, Wang, L, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32(11):2682-8
22. Fen Z. The Treatment of ZG Compatibility to Cerebral Edema. [D]. Chongqing: Southwest University. 2011:11-16
23. Shi H, Gao Y, Liang W. Assessment of Angiogenesis in Models of Focal Cerebral Ischemia. Springer Protocol Handbooks. In: Jun C. Xiao-Ming Xu. Zao C. X. John H.Z. (Eds) Animal models of acute neurological injuries II. Humana Press, New York, pp. 2012:181-185 doi: 10.1007/978-1-61779-782-8
24. Li L, Saliba P, Reischl S. et al. Neuronal deficiency of HIF prolyl 4-hydroxylase 2 in mice improves ischemic stroke recovery in an HIF dependent manner. Neurobiology of disease. 2016;91:221-235
25. Yang L, Qiang X, Tang Q. et al. A simple method for isolating and culturing the rat brain microvascular endothelial cells. Microvascular Research. 2013;90:199-205 doi:10.1016/j. mvr.2013.08.004
26. Tong J, Zeng J, Wei S. et al. Neuroprotective Effect of Lyophilized Powder of Catalpol and Puerarin on Oxygen-glucose Deprivation/ reperfusion Injury in Astrocytes of Rat Cerebral Cortex in Vitro. Chin Pharm J. 2016:51 (13)
27. Zhu H, Wan D, Zhang F. Progress in studies of pharmacological action and mechanisms of catalpol on brain diease. China journal of Chinese materia medica. 2009;34(34):2983-6
28. Wei S. Y, Chen, Y, and Xiao-Yu, X. U. Progress on the pharmacological research of puerarin: a review. Chinese Journal of Natural Medicines. 2014;12(6):407-14
29. Shang Y.H, Wang H.J, Tian J.F. et al. Effect of Zige lyophilized powder for injection in improving acute cerebral microcirculation disturbance in rats. China Journal of Chinese Materia Medica. 2014;39:733-737
30. Jiang T, Yu J, Zhu X. et al. Angiotensin-(1-7) induces cerebral ischemic tolerance by promoting brain angiogenesis in a mas/enos-dependent pathway. British Journal of Pharmacology. 2014;171(18):4222-4232
31. Wan D, Yang X, Wang Y et al. Catalpol stimulates VEGF production via the JAK2/STAT3 pathway to improve angiogenesis in rats' stroke model. Journal of Ethnopharmacology. 2016;191:169-179 doi.org/10.1016/j.jep.2016.06.030
32. Yee K. M, Spivakkroizman, T. R, and Powis, G. Hif-1 regulation: not so easy come, easy go. Trends in Biochemical Sciences. 2008;33(11):526-534
33. Ferrer I, Friguls B, Dalfó E. et al. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathology & Applied Neurobiology. 2003;29(5):472-481
34. Kaushal V, Schlichter L.C. Mechanismsof microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221-2230
35. Datta A, Park J. E, Li, X, et al. Phenotyping of an in vitro model of ischemic penumbra by itraq-based shotgun quantitative proteomics. Journal of Proteome Research. 2010;9(9):472-84
Author contact
![]() Corresponding author: Professor Xiaoyu Xu, Laboratory of Molecular Pharmacology, College of Pharmaceutical Sciences and Chinese Medicine, Southwest University, Chongqing 400715, China. Tel.: +0086-23-6825-0761; Fax: +0086-23-6825-1225; E-mail: xuxiaoyuedu.cn
Corresponding author: Professor Xiaoyu Xu, Laboratory of Molecular Pharmacology, College of Pharmaceutical Sciences and Chinese Medicine, Southwest University, Chongqing 400715, China. Tel.: +0086-23-6825-0761; Fax: +0086-23-6825-1225; E-mail: xuxiaoyuedu.cn