Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(3):383-390. doi:10.7150/ijbs.17236 This issue Cite
Research Paper
Inhibition of Proteasome Activity Induces Aggregation of IFIT2 in the Centrosome and Enhances IFIT2-Induced Cell Apoptosis
Limin Chen1,2#, Shuyuan Liu1#, Fen Xu3, Yunyuan Kong1, Lagen Wan1, Yonglu zhang1, Zhanglin Zhang1 ![]()
1. Department of Clinical laboratory, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, China;
2. Center for Experimental Medicine, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, China;
3. Department of Clinical Laboratory Technology, Jiangxi Medical College, Shangrao, Jiangxi 334000, China.
#Contributed equally.
Received 2016-8-17; Accepted 2016-12-27; Published 2017-2-25
Abstract

IFN-induced protein with tetratricopeptide repeats 2 (IFIT2), one of the most highly responsive interferon-stimulated genes, inhibits the proliferation and migration of cancer cells and regulates viral replication. IFIT2 has been demonstrated to be a cytoskeleton-associated protein that becomes enriched in the mitotic spindle of cells. However, the molecular mechanisms by which IFIT2 executes biological functions are largely unclear. The findings of this study showed that inhibiting the activation of proteasome led to the enrichment of IFIT2 and induced the aggregation of IFIT2 protein in the centrosome. Microtubule inhibitor colchicine and dynein inhibitor ciliobrevin inhibited the proteasome inhibitor-induced aggregation of IFIT2 protein in the centrosome. Intriguingly, IFIT2 and proteasome inhibitor worked together to induce the apoptosis of cancer cells. The results of the present study revealed that the inhibition of proteasome activity blocked the degradation of IFIT2 and promoted the aggregation of IFIT2 in the centrosome, which in turn induced cell apoptosis. In short, IFIT2 may be a potential target for cancer therapeutics.
Keywords: Aggregation, apoptosis, centrosome, IFIT2, proteasome
Introduction
Interferons (IFNs), a group of cytokines, harbor multiple functions including inhibiting viral replication and regulating differentiation, proliferation, and survival of various cell types, which have been used clinically as antivirus, antitumor, and immunomodulatory agents [1, 2]. IFNs play biological roles through stimulating gene expression by activating the Janus kinase/signal transducers and activators of transcription pathway. Genes induced transcriptionally by IFNs were referred to as IFN-stimulated genes (ISGs). Notably, more than 300 ISGs have been identified in the IFN-treated cells. Proteins that code by ISGs mediate the activation of IFNs by regulating transcription, translation, and signal transduction [3, 4]. IFN-induced protein with tetratricopeptide repeats 2 (IFIT2), known as ISG54, is one of the ISGs most responsive to IFNs.
The IFIT2 gene codes for a protein with nine tetratricopeptide repeats (TPR) motifs, which has also been designated as IFIT2 [5, 6]. TPR, a 34-amino acid motif folding into a helix-turn-helix, is most often present in cassettes of multiple repeats and mediates protein-protein interactions [7, 8]. Many TPR-containing proteins have been shown to specifically recognize their partners and form complexes such as molecular chaperones, anaphase-promoting complexes, and protein transport complexes [9-11]. IFIT2 was reported to interact with different proteins and RNA. IFIT2 regulates the functions of cell cycle, apoptosis, tumor colonization, and viral replication, which confer cellular resistance to viral infections and regulates proliferation, apoptosis, and migration of cancer cells [6, 12-14]. Moreover, IFIT2 has been demonstrated to be a cytoskeleton-associated protein. Colocalization of IFIT2 and β-tubulin and enrichment of IFIT2 in the mitotic spindle were observed in cells undergoing mitosis, which regulate the growth and migration of cells [15]. However, how IFIT2 regulates cell proliferation and by which molecular mechanisms it executes biological functions remain to be examined.
This study found that IFIT2 degraded in a proteasome-dependent manner. It interacted with γ-tubulin and localized in the centrosome on inhibiting the activation of proteasome. IFIT2 aggregated in the centrosome required intact microtubules and dynein-mediated transport. Furthermore, IFIT2 and proteasome inhibitor synergistically induced the apoptosis of cancer cells. Taken together, proteasome was involved in the degradation of IFIT2, inhibition of proteasome activity resulted in the aggregation of IFIT2 in the centrosome in a microtubule- and dynein-dependent manner, and finally the centrosomal localization of IFIT2 induced the apoptosis of cancer cells. In short, inducing the centrosomal enrichment of IFIT2 would benefit the clinical treatment of cancer.
Materials and Methods
Cell culture and reagents
The 293T and HCT116 cells were cultured in the Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. All cells were incubated at 37ºC with 5% CO2. Ciliobrevin D (Merck, NJ, USA), MG132 (Sigma, MO, USA), and bortezomib (BSP Pharmaceutical SRL, Belgium) were dissolved in dimethyl sulfoxide as a stock solution at 10mM. Colchicine (Sigma) was dissolved in the phosphate-buffered saline (PBS) as a stock solution at 100 μg/mL. Protease inhibitor phenylmethanesulfonyl fluoride (PMSF, AMRESCO, OH, USA) and cocktail (Roche, Switzerland) were dissolved in isopropanol and PBS as a stock solution, respectively. All stock solutions were stored at -20°C. The antibodies against ubiquitin, γ-tubulin, β-actin, IFIT2, green fluorescent protein were purchased from Santa Cruz Biotechnology (CA, USA). The antibodies against caspase3 and poly (ADP-ribose) polymerase (PARP) were obtained from Cell Signaling Technology (Danvers, MA, USA). TRITC-conjugated and horseradish peroxidase-conjugated anti-mouse IgG (H+L) secondary antibodies were obtained from Beyotime Biotechnology (China). IFNγ was available from R&D Systems China Co. Ltd. Annexin V-PE and Annexin V-FITC/propidium iodide (PI) kits were purchased from Bestbio Biotechnology (China).
Plasmids and transfection
The IFIT2 cDNA was amplified from the leukemia cell line NB4 cells treated with all-trans retinoid acid for 72 h. The cDNA was subcloned into pEGFP-C1 and pIRES2-EGFP (Enhanced Green Fluorescent Protein) vectors to obtain pEGFP-IFIT2 and pIRES2-IFIT2-EGFP plasmids, respectively. The plasmids were then transfected into the 293T and HCT116 cells using the Lipofectamine 2000 system (Qiagen, Hilden, Germany), according to the manufacturer's instructions.
Immunofluorescence
The cells cultured on coverslips were washed three times with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. After permeabilizing with 0.5% Triton X-100 in PBS for 5 min at room temperature, the cells were incubated with primary antibody diluted in 1% bovine serum albumin for 2 h at 37°C. Coverslips were washed five times in PBS with 0.2% Tween 20 and incubated at 37°C for 1 h with secondary antibodies and 4',6-diamidino-2-phenylindole. Coverslips were washed as indicated earlier and mounted on slides in 9:1 glycerol/PBS with 0.1% p-phenylenediamine. Finally, fluorescence images were captured using an Olympus BX51 microscope (Olympus Optical Co., Ltd. Japan) with a cold charge-coupled device (CCD) camera at 1000 times magnification.
For live-cell imaging, pEGFP-IFIT2 plasmid was transfected into the 293T cells. At 48 h, the cells were treated with MG132. Images were taken at 37ºC using a High-Content Cell Imaging Analysis (ImageXpress Micro XL, Molecular Devices LLC, CA, USA) at 400 times magnification.
Coimmunoprecipitation
Cell lysates prepared in the buffer containing 150mM NaCl, 50mM Tris-HCl (pH 8.0), and 0.5% NP40 were mixed with protein A agarose (Santa Cruz) and the indicated antibodies at 4ºC overnight with rotation. The precipitated proteins were then eluted by boiling beads in sodium dodecyl sulfate (SDS)-loading buffer [4% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.2% bromophenol blue, 100mM Tris-HCl (pH 6.8)] and analyzed using Western blot.
Immunoblotting
Cell lysates were prepared with lysis buffer containing 1% Triton X-100, 50mM Tris-HCl (pH 8.0), 150mM NaCl, 1mM PMSF, 1mM Na3VO4, and protease inhibitor cocktail. Protein concentrations were determined using the Bio-Rad protein assay. Cell lysates with 20 µg total proteins were denatured in SDS and separated on 10% SDS-polyacrylamide gel electrophoresis gels. Proteins were transferred to nitrocellulose membranes, which were blocked for 1 h at room temperature with 5% nonfat dry milk. The membranes were incubated with primary antibodies overnight at 4ºC with rotation, and then incubated with peroxidase-conjugated secondary antibodies at room temperature for 1 h. Finally, protein bands were visualized using the enhanced chemiluminescence detection system (Amersham, NJ, USA).
Apoptosis analysis
The cell apoptosis was determined by dual staining with Annexin V-FITC and PI. The HCT116 cells treated or untreated with 5000 U/mL IFNγ for 12 h were incubated with 10μM MG132 for another 24 h, and then the cells were stained with Annexin V-FITC and PI, and analyzed by flow cytometry using an FC500 cytometer (Beckman, CA, USA). The apoptotic cells were identified as Annexin V+ and/or PI+ cells.
pEGFP-IFIT2 plasmid was transfected into the HCT116 cells for 48 h to observe apoptosis in the IFIT2-aggregated cells. The cells were digested with trypsin and stained with Annexin V-PE after treatment with bortezomib for 24 h. GFP-IFIT2 and Annexin V-PE were observed using an Olympus BX51 microscope with a cold CCD camera at 400 times magnification.
Statistical analysis
All data were presented as mean ± standard deviation (n ≥ 3). A statistical analysis was performed using the Student t test. A value of P <0.05 was considered to be statistically significant. The SPSS13.0 software (SPSS Inc., IL, USA) was used for all analyses.
Results
IFIT2 protein degradation in a proteasome-dependent manner
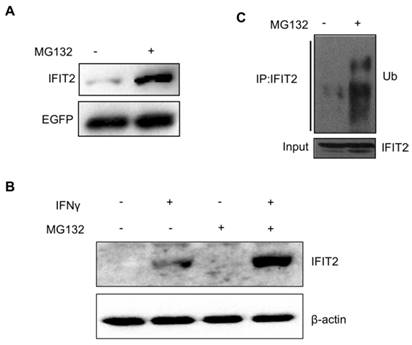
A plasmid pIRES2-IFIT2-EGFP containing the internal ribosome entry site (IRES) of the encephalomyocarditis virus between IFIT2 and EGFP coding region was constructed to investigate whether the degradation of IFIT2 is regulated in a proteasome-dependent mechanism. The IRES allows both IFIT2 and EGFP genes to be translated from a single bicistronic mRNA. The 293T cells transfected with pIRES2-IFIT2-EGFP were treated with MG132, a proteasome inhibitor, for 6 h to inhibit the activation of proteasome. The Western blot results showed that MG132 treatment induced the accumulation of IFIT2 protein significantly (Fig. 1A). IFNγ was used to induce the expression of endogenous IFIT2, and MG132 treatment increased the level of IFIT2 (Fig. 1B). Furthermore, a marked increase in the ubiquitinated IFIT2 was detected in the MG132-treated cells (Fig. 1C). These results suggested that proteasome regulates the degradation of IFIT2.
Degradation of IFIT2 was dependent on the proteasome. (A) MG132 treatment increased the level of IFIT2. (B) Endogenous IFIT2 increased in the MG132-treated cells. (C) MG132 treatment induced the accumulation of ubiquitinated IFIT2.
Inhibition of proteasome led to the aggregation of IFIT2
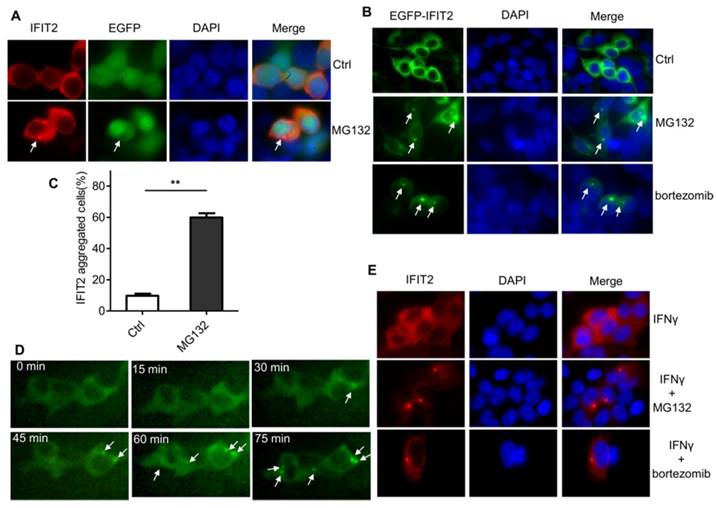
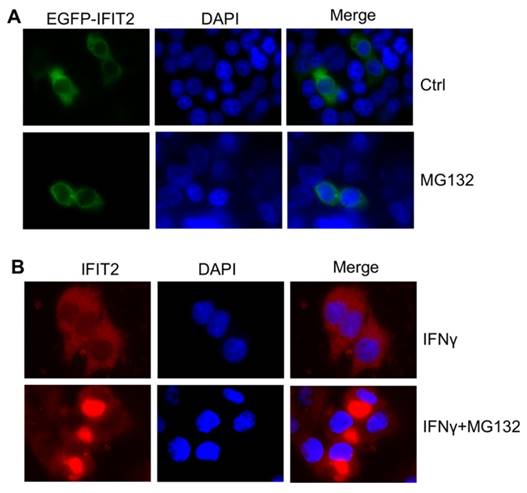
The IFIT2 localization was further examined in the cells. As shown in Figure 2A, IFIT2 was primarily dispersively distributed within the cytoplasm in the 293T cells when transfected with pIRES2-IFIT2-EGFP, which is consistent with previous studies [5]. However, MG132 treatment resulted in the aggregation of IFIT2 (Fig. 2A). To better detect the location of IFIT2 in the cells, a plasmid was generated that fused IFIT2 with EGFP and expressed an EGFP-IFIT2 fusion protein. The aggregation of EGFP-IFIT2 fusion protein was observed in 59.88% ± 2.73% cells treated with MG132, while in only 9.74% ± 1.36% cells without MG132 treatment (Fig. 2B and 2C). Moreover, another proteasome inhibitor bortezomib, an antitumor drug, also induced the aggregation of IFIT2 (Fig. 2B). The 293T cells transfected with EGFP-IFIT2 were treated with MG132 and images were continuously taken for 120 min at 15 min-intervals to dynamically observe the aggregation of IFIT2. The results showed that EGFP-IFIT2 distributed evenly in the cytoplasm at 0 min, and the fluorescent foci of EGFP-IFIT2 started forming when treated with MG132 for 30 min (Fig. 2D). At 75 min, almost all cells formed one or two foci (Fig. 2D). IFNγ was used to induce the expression of IFIT2 to determine whether inhibiting the activation of proteasome leads to the aggregation of endogenous IFIT2. MG132 and bortezomib also induced endogenous IFIT2 aggregation (Fig. 2E). Taken together, these results demonstrated that the inhibition of proteasome activity resulted in the aggregation of IFIT2.
Proteasome activity inhibition resulted in the aggregation of IFIT2. (A, B) IFIT2 protein aggregated in cells treated with MG132 or bortezomib. (C) Quantification of Figure 2B. **P < 0.01. (D) IFIT2 dynamically aggregated in cells treated with MG132. (E) MG132 or bortezomib treatment induced aggregation of endogenous IFIT2.
Aggregated IFIT2 protein interacted with γ-tubulin and localized in the centrosome
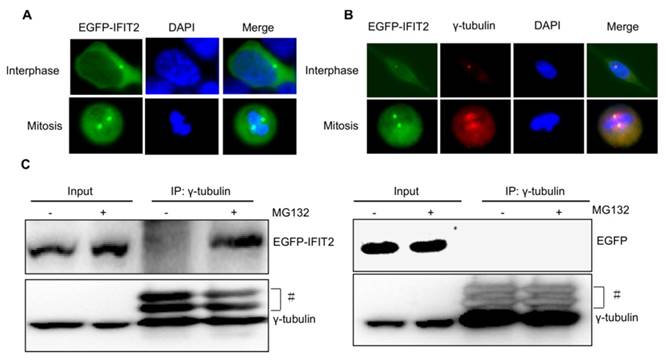
Considering that the IFIT2 aggresomes were localized close to the nucleus and one or two IFIT2 aggresomes were observed per cell (Fig. 2), this study hypothesized that the aggregation of IFIT2 might be associated with the centrosome. The fluorescent foci of EGFP-IFIT2 were detected in different phases of the cell cycle to validate this hypothesis. In the interphase, an IFIT2 aggresome was formed and localized close to the nucleus. However, two fluorescent foci of EGFP-IFIT2 on each side of the equatorial plate were detected in mitosis (Fig. 3A). These results suggested that IFIT2 might accumulate in the centrosome. γ-Tubulin is a marker of the centrosome; the colocalization of IFIT2 and γ-tubulin was further determined using the immunofluorescent assay. As shown in Figure 3B, IFIT2 and γ-tubulin were colocalized. This study found that the IFIT2 protein interacted with γ-tubulin in the MG132-treated cells using a coimmunoprecipitation assay, but the EGFP control could not interact with γ-tubulin (Fig. 3C). In short, proteasome inhibition leads to the interaction of IFIT2 and γ-tubulin, and the aggregated IFIT2 protein localizes in the centrosome.
Aggregated IFIT2 protein interacted with γ-tubulin and localized in the centrosome. (A) IFIT2 aggregation in cells at different cell-cycle phases. (B) Colocalization of IFIT2 and γ-tubulin. (C) IFIT2 interaction with γ-tubulin.
Proteasome inhibition mediated IFIT2 aggregation in the centrosome in a microtubule- and dynein-dependent manner
The accumulation of proteins in the centrosome is usually dependent on microtubule and dynactin [16]. The present study investigated whether proteasome inhibition-mediated IFIT2 enrichment in the centrosome was dependent on the microtubule. The 293T cells transfected with EGFP-IFIT2 were preincubated with different concentrations of colchicine for 4 h to depolymerize microtubules, which were then treated with MG132 for another 2 h. The data showed that the aggregation of IFIT2 in the centrosome reduced significantly with the increasing concentration of colchicine (Fig. 4A), indicating that the recruitment of IFIT2 to the centrosome was microtubule dependent.
IFIT2 aggregation in the centrosome was dependent on microtubule and dynein. (A) Colchicine inhibited the aggregation of IFIT2 in the centrosome in a dose-dependent manner. (B) Ciliobrevin D blocked the accumulation of IFIT2 in a concentration-dependent manner. *P < 0.05, **P < 0.01.
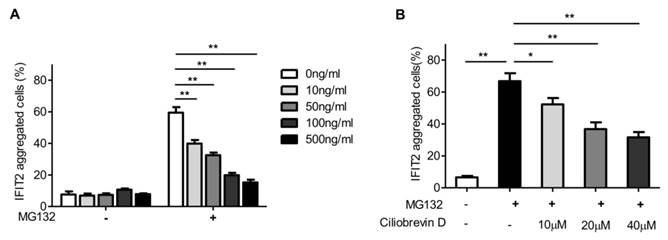
In the cells, the microtubule motor protein dynein carries out ATP-dependent movement along microtubules and transports proteins toward the minus end of the microtubules [17]. Ciliobrevin D is a dynein inhibitor, which competes with ATP at the binding site of dynein and inhibits dynein-mediated transport [18]. The 293T cells transfected with EGFP-IFIT2 were preincubated with different doses of ciliobrevin D, and then treated with MG132. The aggregation of IFIT2 was observed, and the results showed that ciliobrevin D inhibited proteasome inhibition-mediated IFIT2 accumulation in a dose-dependent manner (Fig. 4B).
Aggregation of IFIT2 induced the apoptosis of cancer cells
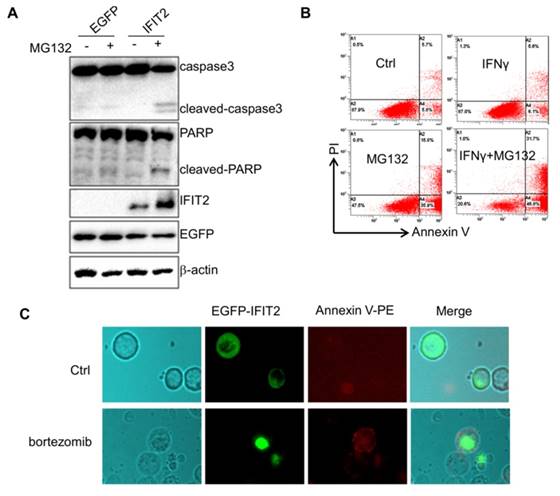
This study investigated whether inhibiting the activation of proteasome could result in the aggregation of IFIT2 in cancer cells. The HCT116 cells were transfected with EGFP-IFIT2 and treated with MG132, and fluorescent foci of EGFP-IFIT2 were formed in the cells (Fig. 5A). MG132 also induced significant aggregation of endogenous IFIT2 in the HCT116 cells (Fig. 5B). As IFIT2 accumulated in the centrosome has an essential role in cell proliferation, it was suggested that IFIT2 aggregation might be involved in cell growth. The level of IFIT2 in the HCT116 cells was upregulated, and cleaved caspase3 and PARP were detected upon treatment with MG132 (Fig. 6A). Furthermore, the HCT116 cells were preincubated with IFNγ to induce the expression of IFIT2 and treated with or without MG132 for 24 h. Massive apoptotic cells were detected in the MG132-treated cells (Fig. 6B). The results of the immunoflurescence assay showed that Annexin V was strongly stained in the cells with IFIT2 aggregation, suggesting that the aggregated IFIT2 led to the apoptosis of HCT116 cells (Fig. 6C). Taken together, proteasome inhibitors induce the aggregation of IFIT2, which results in the apoptosis of cancer cells.
Proteasome activity inhibition led to the aggregation of IFIT2 in cancer cells. (A) MG132 treatment induced the aggregation of IFIT2 in the HCT116 cells. (B) Endogenous IFIT2 significantly aggregated in the MG132-treated HCT116 cells.
Aggregation of IFIT2 induces the apoptosis of cancer cells. (A) Aggregation of IFIT2 induced cleavage of caspase3 and PARP in the HCT116 cells. (B, C) Aggregation of IFIT2 induced the apoptosis of HCT116 cells.
Discussion
This study found that the proteasome system participated in the degradation, transportation, and centrosomal localization of IFIT2 protein in cells, and facilitated the IFIT2-mediated cell apoptosis. Intriguingly, the findings of immunoblot and immunofluorescence assays revealed that the inhibition of the proteasome activity induced the accumulation of ubiquitinated IFIT2 protein and promoted an aggregate formation of IFIT2 beside the nucleus. The aggregated IFIT2 protein interacted with γ-tubulin and localized in the centrosome in different phases of the cell cycle, and the dynamic analysis of EGFP-IFIT2 in the live cells showed that proteasome inhibition promoted the movement and aggregation of IFIT2 in the centrosome. Microtubule inhibitor colchicine and dynein inhibitor ciliobrevin could inhibit the aggregation of proteasome inhibitor-induced IFIT2 proteins in the centrosome. Furthermore, IFIT2 worked together with the proteasome inhibitor to induce the apoptosis of cancer cells. These results suggested that proteasome regulated the degradation of IFIT2 protein and inhibition of proteasome activity leading to the aggregation of IFIT2 in the centrosome, which subsequently induced cell apoptosis.
The centrosome is the main microtubule-organizing center, comprising a pair of centrioles surrounded by amorphous pericentriolar material. The size of the centrosome is tightly regulated during the cell cycle progression [19]. Centrosomes contain an active 26S proteasome that degrades ubiquitinated proteins, which are transported to the centrosome [20-22]. It is reported that the inhibition of proteasome activity triggers the enrichment of ubiquitinated proteins in the centrosome and leads to aggresome formation [23, 24]. The present study revealed that the inhibition of the proteasome activity resulted in the accumulation of IFIT2 in cells. IFIT2 proteins are transported to the centrosome and aggregated in the centrosome on inhibiting proteasome activation.
Aggresomes are first found to transport misfolded proteins to the microtubule-organizing center facilitating their clearance [25]. Except for misfolded proteins, the impairment of proteasome also leads to the transport of the ubiquitinated proteins toward the microtubule-organizing center to form aggresome, and triggers autophagy [23, 26]. Previous studies suggested that targeting both the proteasome-dependent pathways and the aggresome pathway in tumor cells with bortezomib could induce marked accumulation of polyubiquitinated proteins and significant cell stress, promoting, in turn, the activation of autophagy and apoptosis [27, 28]. IFIT2, an IFN-induced protein containing nine TPR domains, forms complexes by interacting with virus RNAs, other ISG proteins, or other binding partners to execute various functions including antiviral, antitumor, and as a regulator of transcription and translation [6]. The formation of aggresome of IFIT2 resulted in cell apoptosis. IFIT2 and inhibitors of proteasome induce cell apoptosis synergistically. In conclusion, events that induce the expression or accumulation of IFIT2 may have antitumor effects.
Abbreviations
IFNs, Interferons; IFIT2, IFN-induced protein with tetratricopeptide repeats 2; IRES, internal ribosome entry site; ISGs, IFN-stimulated genes; TPR, tetratricopeptide repeats
Acknowledgements
The authors thank all members of the Department of Clinical Laboratory of the First Affiliated Hospital of Nanchang University for their support. This work was supported by the Natural Science Foundation of Jiangxi Province (20122BAB215012 and 20142BAB215068); the Science and Technology Support Program of Jiangxi Province (20121BBG70049); and the Educational Foundation of Jiangxi Province (GJJ14177).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Reich NC. A death-promoting role for ISG54/IFIT2. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2013;33:199-205
2. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405-14
3. Der SD, Zhou A, Williams BRG, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15623-8
4. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Current opinion in virology. 2011;1:519-25
5. Ulker N, Zhang X, Samuel CE. Mechanism of interferon action. I. Characterization of a 54-kDa protein induced by gamma interferon with properties similar to a cytoskeletal component. Journal of Biological Chemistry. 1987;262:16798-803
6. Sen GC, Fensterl V. Crystal structure of IFIT2 (ISG54) predicts functional properties of IFITs. Cell research. 2012;22:1407-9
7. Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends in biochemical sciences. 1995;20:257-9
8. Blatch GL, Lässle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. BioEssays. 1999;21:932-9
9. Cortajarena AL, Regan L. Ligand binding by TPR domains. Protein Science. 2006;15:1193-8
10. Wang J, Dye BT, Rajashankar KR, Kurinov I, Schulman BA. Insights into anaphase promoting complex TPR subdomain assembly from a CDC26-APC6 structure. Nat Struct Mol Biol. 2009;16:987-9
11. Keiski C-L, Harwich M, Jain S, Neculai AM, Yip P, Robinson H. et al. AlgK Is a TPR-Containing Protein and the Periplasmic Component of a Novel Exopolysaccharide Secretin. Structure. 2010;18:265-73
12. Stawowczyk M, Van Scoy S, Kumar KP, Reich NC. The Interferon Stimulated Gene 54 Promotes Apoptosis. Journal of Biological Chemistry. 2011;286:7257-66
13. Lai KC, Liu CJ, Chang KW, Lee TC. Depleting IFIT2 mediates atypical PKC signaling to enhance the migration and metastatic activity of oral squamous cell carcinoma cells. Oncogene. 2013;32:3686-97
14. Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC. et al. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS pathogens. 2012;8:e1002712
15. Lai KC, Chang KW, Liu CJ, Kao SY, Lee TC. IFN-induced protein with tetratricopeptide repeats 2 inhibits migration activity and increases survival of oral squamous cell carcinoma. Molecular cancer research: MCR. 2008;6:1431-9
16. Dammermann A, Merdes A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. The Journal of Cell Biology. 2002;159:255-66
17. Wu J, Misra G, Russell RJ, Ladd AJC, Lele TP, Dickinson RB. Effects of dynein on microtubule mechanics and centrosome positioning. Molecular biology of the cell. 2011;22:4834-41
18. Roossien D, Miller K, Gallo G. Ciliobrevins as Tools for Studying Dynein Motor Function. Frontiers in Cellular Neuroscience. 2015:9
19. Blagden SP, Glover DM. Polar expeditions-provisioning the centrosome for mitosis. Nat Cell Biol. 2003;5:505-11
20. Wan Y, Yang Z, Guo J, Zhang Q, Zeng L, Song W. et al. Misfolded G[beta] is recruited to cytoplasmic dynein by Nudel for efficient clearance. Cell research. 2012;22:1140-54
21. Ouyang H, Ali YO, Ravichandran M, Dong A, Qiu W, MacKenzie F. et al. Protein Aggregates Are Recruited to Aggresome by Histone Deacetylase 6 via Unanchored Ubiquitin C Termini. Journal of Biological Chemistry. 2012;287:2317-27
22. Didier C, Merdes A, Gairin JE, Jabrane-Ferrat N. Inhibition of proteasome activity impairs centrosome-dependent microtubule nucleation and organization. Molecular biology of the cell. 2008;19:1220-9
23. Bang Y, Kang BY, Choi HJ. Preconditioning stimulus of proteasome inhibitor enhances aggresome formation and autophagy in differentiated SH-SY5Y cells. Neuroscience Letters. 2014;566:263-8
24. Ardley HC, Scott GB, Rose SA, Tan NGS, Markham AF, Robinson PA. Inhibition of Proteasomal Activity Causes Inclusion Formation in Neuronal and Non-Neuronal Cells Overexpressing Parkin. Molecular biology of the cell. 2003;14:4541-56
25. Chin LS, Olzmann JA, Li L. Aggresome Formation and Neurodegenerative Diseases: Therapeutic Implications. Current Medicinal Chemistry. 2008;15:47-60
26. Jänen SB, Chaachouay H, Richter-Landsberg C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia. 2010;58:1766-74
27. Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RHI, Dunner K. et al. Aggresome Disruption: A Novel Strategy to Enhance Bortezomib-Induced Apoptosis in Pancreatic Cancer Cells. Cancer Research. 2006;66:3773-81
28. Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL. et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8567-72
Author contact
![]() Corresponding author: Zhanglin Zhang, Email: zhzl1984com, Tel: +86-791-88697032, Mail Address: No. 17 Yongwai Street, Donghu District, Nanchang, Jiangxi, 330006, China.
Corresponding author: Zhanglin Zhang, Email: zhzl1984com, Tel: +86-791-88697032, Mail Address: No. 17 Yongwai Street, Donghu District, Nanchang, Jiangxi, 330006, China.