Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(4):458-470. doi:10.7150/ijbs.18644 This issue Cite
Research Paper
Characterization of Cytosine Methylation and the DNA Methyltransferases of Toxoplasma gondii
Haixia Wei, Shichen Jiang, Longfei Chen, Cheng He, Shuizhen Wu, Hongjuan Peng ![]()
Department of Pathogen Biology, Guangdong Provincial Key Laboratory of Tropical Disease Research, and Key Laboratory of Prevention and Control for Emerging Infectious Diseases of Guangdong Higher Institutes, School of Public Health, Southern Medical University, Guangzhou, Guangdong Province, 510515, P.R. China.
Received 2016-12-6; Accepted 2017-1-22; Published 2017-3-11
Abstract

DNA methylation is a key epigenetic modification which confers phenotypic plasticity and adaptation. Cyst-forming strains of Toxoplasma gondii undergo tachyzoite to bradyzoite conversion after initial acute infection of a host, and the reverse conversion may occur in immune-suppressed hosts. The formation of m5C is catalyzed by DNA methyltransferase (DNMT). We identified two functional DNA methyltransferases, TgDNMTa and TgDNMTb, in T. gondii that may mediate DNA methylation. The recombinant proteins showed intrinsic methyltransferase activity; both have higher transcription levels in bradyzoites than that in tachyzoites. We performed genome-wide analysis of DNA methylation in tachyzoites and bradyzoites. The results showed more methylation sites in bradyzoites than that in tachyzoites. The most significantly enriched GO-terms of genes with DNA methylation were associated with basal cellular processes such as energy metabolism and parasite resistance to host immunity. Tachyzoite proliferation in parasitophorous vacuoles (PV) can be inhibited by the DNA methyltransferase inhibitor 5-azacytidine, a chemical analogue of the nucleotide cytosine that can inactivate DNA methyltransferases. These findings provide the first confirmation of DNA methylation in T. gondii.
Keywords: Toxoplasma gondii, DNA methylation, tachyzoite, bradyzoite, 5-cytosine methyltransferase, 5-azacytidine.
Introduction
Toxoplasmosis is a major neglected parasitic infection [1]. Toxoplasma gondii is a type of intracellular protozoan with a taxonomic status of Eukaryota; Alveolata; Apicomplexa; Conoidasida; Coccidia; Eucoccidiorida; Eimeriorina; Sarcocystidae [2]. Toxoplasma is an important zoonotic pathogen that can infect all warm-blooded animals. It is estimated that 1/3 of the world population is infected with T. gondii [3]. After infecting intermediate hosts including humans, Toxoplasma gondii undergoes a two-stage conversion between tachyzoites, which can cause acute toxoplasmosis, and bradyzoites, which are surrounded by a carbohydrate-rich cyst wall and account for chronic infection [4, 5]. Skariah Sini et al reviewed important information about bradyzoite differentiation: the parasite's cell cycle is tightly correlated to differentiation, both host cell environmental stress independent of host immunity and host immunity can induce differentiation, and freshly isolated sporozoites and bradyzoites spontaneously trigger stage conversion [6]. T. gondii stage conversion has been associated with global mRNA modifications, which suggests that developmental switches are at least partially regulated at the transcriptional level [7-9]. The mechanism for the inter-conversion between tachyzoites and bradyzoites in T. gondii is still unclear.
Epigenetic modification of cytosine methylation works as a key process affecting phenotypic plasticity and adaptation and regulating gene transcription profiles [10]. In higher eukaryotes, cytosine methylation in the promoters can result in a wide range of processes, such as gene expression silencing, parental imprinting and chromosome X inactivation in females, DNA repair, and gene expression regulation [11-13]. Gene body methylation has also been reported to have effects on silencing repetitive DNA elements [14] or alternative splicing [15]. DNA methylation can occur as N6-methyladenine (m6A), N4-methylcytosine (m4C), and C5-methylcytosine (m5C), and the former two are mainly found in bacterial DNA [16, 17]. The percentage of m5C varies greatly among species, which can be as high as more than 30% in some plants, approximately 10% in fish and amphibians, 5% in mammals and birds, and as low as 0-1% in some insects [18]. The presence of m5C has been reported in several classes of unicellular eukaryotes such as Entamoeba histolytica [19, 20] and Plasmodium falciparum [21, 22]. In T. gondii, methodology involving the HpaII tiny fragment enrichment by ligation-mediated PCR (HELP) and mass spectrometry analysis suggests that T. gondii RH strain tachyzoites lack detectable DNA cytosine methylation [23]. However, it is important to evaluate the DNA methylation status of all life cycle stages before claiming that it is absent in an organism [24, 25]. Recently, a more sensitive method, MethylC-seq, for m5C methylation detection was developed and is regarded as a standard profiling method that could theoretically detect all cytosine methylation [26]; and this high-throughput sequencing method coupled with the bisulfite conversion of an un-methylated C to a T at the single-base resolution, makes it possible to accurately identify DNA cytosine methylation, even in non-CG contexts [27, 28].
The formation of m5C is catalyzed by DNA methyltransferase (DNMT) with the cofactor S-adenosylmethionine [19]. Mammalian DNMTs consist of DNMT1, DNMT3a and DNMT3b; DNMT1 prefers hemi-methylated DNA as substrates, while DNMT3a and DNMT3b are known as de novo DNA MTases that work on non-methylated DNA [25, 29]. It was reported that DNMT2 in humans is a tRNAAsp MTase rather than a DNA MTase [30]. By contrast, DNMT2 has been proposed to be a genuine DNMT in lower eukaryotes, as DNMT2 can catalyze DNA methylation in Entamoeba histolytica [19], Plasmodium falciparum [22], and Schistosoma mansoni [31]. DNMT2 also catalyzes tRNAAsp MTase in E. histolytica [32], which is consistent with the finding that both DNA and RNA can be used as substrates for DNMT2 methyltransferases [30, 33, 34]. Cytosine methylation catalyzed by DNMT2 is not limited to CpG, but can also occur in Cp(A/T/C) [19, 22, 31, 35].
In this study, we report the existence DNA methylation of T. gondii and characterize the methylomes of tachyzoites and bradyzoites, and we also identify two functional DNMTs that may mediate DNA cytosine methylation in T. gondii.
Materials and methods
Ethical statements
Specific pathogen free (SPF) Male Kunming mice (18-22 g, 7-9 weeks) were purchased from the Laboratory Animal Centre of Southern Medical University and raised in a SPF laboratory in the university. All animal experiments were performed following the guidelines for laboratory animal manipulation approved by the Guangdong Laboratory Animals Monitoring Institute and under the inspection of the Animal Welfare and Ethics Review Board of Southern Medical University.
T. gondii strain and culture conditions
The T. gondii ME49 strain was used in the study. Tachyzoites were cultured in human foreskin fibroblast (HFF) as described previously [36]. The ME49 strain bradyzoites were maintained in Kunming mice. Bradyzoites were isolated as follows: the brains of the infected mice were isolated, homogenized, and diluted in phosphate-buffered saline (PBS). Lymphocyte separation medium (Sigma-Aldrich, cat. #1077) was used to separate the cysts from the brain homogenate, and the cysts were collected from the bottom of the tubes. Trypsin (0.125% final) was then added to digest the cyst wall to release the bradyzoites.
Enzymatic activity assay for endogenous and recombinant T. gondii DNMTa and DNMTb
Nuclear protein extraction for endogenous DNMT activity assay. When HFF cells ruptured, and the tachyzoites were released, the free parasites were harvested. The tachyzoite nuclear extract were prepared with the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, China, cat. #P0027) and were used immediately for DNMT activity assay following the manufacturer's instruction.
Expression and purification of the recombinant T. gondii DNMTs. Using ToxoDB, we searched for the coding sequences of TGME49_227660 for TgDNMTa and TGME49_243610 for TgDNMTb and the PCR primers were synthesized accordingly. As TgDNMTb expression in E. coli was undetectable, the DNMT conserved domain of TgDNMTb was expressed instead.
Total RNA isolation from tachyzoites was performed using the RNeasy Plus Mini Kit (Qiagen, cat. #74134), the cDNA library was immediately generated with the GoScript™ Reverse Transcription System (Promega, A5001). TgDNMTa was amplified with the primers: 5'-CCGGAATTCATGGAGATGATGGAGAAG-3' and 5'-AAATATGCGGCCGCTCAGTCTCGTCGGC-3'. The conserved DNMT domain of TgDNMTb was amplified with the primers: 5'-TGCAGGATATCCCTTGCTCGACATCTCCCAG-3' and 5'-CCGGAATTCAGTCACCGGCTGCTCAGTCT-3'. The PCR products were cloned into the pET32a (+) vector, which allowed the expression of the target protein fused to a 6×His tag. Two of the recombinant vectors with TgDNMTa and TgDNMTb inserts were sequenced to confirm that the cDNAs were in frame and no mutation had been introduced.
The recombinant plasmids were transformed into E. coli separately. Expression of the fusion proteins was initiated by isopropyl-beta-D-thiogalactopyranoside (IPTG). The fusion proteins were purified under native condition with the Ni-NTA Fast Start Kit (Qiagen, Cat. #30600). The purified proteins were used immediately for the DNMT activity assay.
DNMT activity assay. DNMT activity was measured using the EpiQuik DNA Methyltransferase Activity Kit (EpigenTek Cat. #P-3001, Colorimetric). Assays were conducted in triplicate on three independent preparations of detection samples (9 µg of purified recombinant protein of TgDNMTa, 5 µg of purified recombinant protein consisting of the TgDNMTb-converse domain, and 10 µg of nuclear protein), positive controls (0.5 μg of purified bacterial DNMT), and blanks (buffer alone).
RT-qPCR analysis for DNMT transcription level in T. gondii tachyzoites and bradyzoites
The total RNA and cDNA synthesis were performed as described above. The TgDNMTa and TgDNMTb gene transcription levels were analyzed by qPCR using the SYBR® Select Master Mix (Life Technologies, cat.4472908). Gene-specific primers were as follows: TgDNMTa-F: 5'-GTTGACGCCGTCCAGTC-3'; TgDNMTa-R: 5'-CTGCGTCTGGCTTGGTC-3'; TgDNMTb-F: 5'-GCACTTTCACCAAGGGATACA-3'; TgDNMTb-R: 5'-CGTCAGATGGTTCTGAAGAAGAG-3'. Reactions were carried out with a BioRad Applied Biosystems 7500 qPCR System. Relative transcription levels of the TgDNMTa and TgDNMTb genes were normalized to the transcription level of the housekeeping gene GAPDH (GPADH -F: 5'-ATTTTGCTTGGGATTCGAGGA-3'; GPADH -R: 5'-TGCAGGGTAACGATCAAAAAATG-3') and were calculated using the 2-ΔΔCt method [37]. Each qPCR was performed in triplicate, and the experiments were repeated for three times. The mean values were used as the relative transcription level of each DNMT.
Genome-wide cytosine methylation detection and methylation profile analysis
Preparation of T. gondii genomic DNA, MethylC-seq library construction and sequencing. The genomic DNA of T. gondii ME49 strain tachyzoites and bradyzoites was extracted using the DNeasy® Blood &Tissue Kit (Qiagen, cat. #69504). The library construction and sequencing were performed as previously described [38]. The reference DNA sequence was downloaded from ToxoDB (http://toxodb.org/common/downloads/release-11/TgondiiME49/fasta/). Three biological replicates were collected and sequenced following the processes mentioned above, and the mapped cytosine sites were used in further analysis.
Verification of m5C locus in the genomic DNA of Toxoplasma gondii. The tachyzoites and bradyzoites genomic DNA were digested using the EpiJETTM DNA Methylation Analysis Kit (MspI/HpaII) (Thermo Scientific Cat. #K1441), which can detect the methylation of the internal CpG in the CCGG sequences by digesting gDNA followed by PCR or qPCR. The primers for common PCR were as follows: forward: 5'-AGACTGCTGCTTGCGGCTT-3'; reverse: 5'- CGAACGAAGACGCTTTACAGG-3'. And the primers for qPCR are shown in Table S1. When △CT (CT Msp1-CTHpaII)≥1, the percent of methylation at the target site was evaluated according to the formula: 100/(1+E%)^(CT average of HpaII treated DNA minus CT average of undigested DNA), and E% is the qPCR efficiency value. All results were obtained from three repetitive experiments with three replicates. According to the manufacturer's instructions, when the △CT (CT Msp1-CTHpaII) is near 0.5, there is no methylation present in the examined site.
Gene ontology (GO) enrichment and KEGG pathway analysis. To further define the biological functions of the genes with different DNA methylation levels in tachyzoites and bradyzoites, GO term and KEGG pathway enrichments were performed using http://toxodb.org. The KEGG pathway was also determined with the DAVID bioinformatics database [39]. Benjamini multiple-testing correction of the EASE score was used for determining the statistical significances of GO term and KEGG pathway enrichments [40].
Transient inhibition of T. gondii endogenous DNMT activity with 5-AzaC and observation of T. gondii infection ability
Three groups of tachyzoites with equal cell numbers were treated with 0 μM (Control Group), 12.5 μM, and 100 μM 5-AzaC (Sigma-Aldrich, cat. #A2385) (Inhibitor Treatment Groups) in complete medium for 30 min and then washed with complete medium three times. The DNA was extracted to detect the methylation level after being treated with inhibitor. At the same time, the tachyzoites were used to infect HFF cells grown on coverslips to a MOI (multiplicity of infection) of 3 for 2 h and 48 h, respectively. The coverslips were washed with PBS three times to remove the uninvaded parasites and immobilized with 4% paraformaldehyde for 5 min followed by 100% methanol for 10 min. A Giemsa stock solution (DingGuo, cat. #AR-0751, China) was diluted 2:5 with Giemsa buffer (0.04 M KH2PO4 , 0.04 M Na2HPO4, pH 7.2) to create the working solution, which was added to each well for 1 h staining. The coverslips were washed three times with 1 ml of Giemsa buffer per well with gentle shaking for 5 min per wash. The coverslips were removed from the wells, air-dried and mounted with 100% glycerol for light microscopy. One hundred host cells from five visual fields (top, bottom, left, central, and right) of each coverslip were chosen to quantify the percentage of the infected host cells (invasion rate) under a 100x objective lens (1000x magnification). The proliferation efficiency was evaluated as the mean number of tachyzoites per parasitophorous vacuoles (PV) in the chosen 100 PVs for each coverslip.
Long-term treatment of intracellular tachyzoites with 5-AzaC and observation of the inhibition result
Four groups of tachyzoites were prepared. Two of them were treated with 0 μM 5-AzaC complete medium (No Inhibitor), and the other two were treated with 12.5 μM 5-AzaC (Inhibitor Treatment) complete medium for 30 min; then, 0 μM 5-AzaC culture medium or 12.5 μM 5-AzaC culture medium was added to the four groups of T25 flasks of 100% confluent HFF cells. After two hours post infection, the culture medium was changed with a pH 7.2 medium or a pH 8.2 medium, so the four groups were treated separately as follows: No Inhibitor (pH 7.2); No Inhibitor (pH 8.2); Inhibitor Treatment (pH 7.2); Inhibitor Treatment (pH 8.2). The medium for each group was refreshed daily. The cells and the parasites from the four groups were collected on days 0, 2, 4, 6, 8 and 10. The total RNA isolation and cDNA synthesis were performed as described above. Gene-specific primers for the BAG1 gene were as follows: BAG1-F: 5'-GGCCTCACTCACATTTCTCA-3'; BAG1-R: 5'-ACGGGCTTACTTCATCCAAATA-3'. The relative expression levels of the BAG1 gene were normalized with the same method as described above. Each real-time PCR reaction was performed in triplicate, and the experiments were repeated for three times.
Statistics
Statistical comparisons were conducted with independent t-test or one-way ANOVA with the SPSS13.0 software (Chicago, IL, USA).
Results
Identification of C5-DNA methyltransferase (DNMT) genes in the T. gondii genome
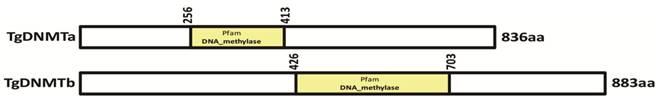
Two genes (TGME49_227660 and TGME49_243610) encoding the proteins with the conserved DNMT domain (GB accession number: PF00145) [41] were found in the T. gondii genome (www.toxodb.org). Both proteins encoded by these two genes lack the extended amino-terminal regulatory region present among DNMT1 and DNMT3 family members, and they are likely to belong to the DNMT2 family (Tables S2 and S3). These two genes were designated as Tgdnmta and Tgdnmtb, respectively. The Tgdnmta gene is located on chromosome X and is 2511 bp in length, contains no introns, and encodes a predicted protein of 836 amino acids (aa), with the DNMT2 conserved domain located in the center of the protein (256-413 aa) (Figure 1). Tgdnmtb is located on chromosome VI and is 2652 bp in length, contains one intron, and encodes a predicted protein of 883 aa in length, with the DNMT2 conserved domain located near the carboxyl terminus of the protein (426-703 aa) (Figure 1). These two putative proteins share limited homology (approximately 20% identity), and the homologous region is restricted to the DNMT2 domain.
Identification of the TgDNMT candidates. Two genes (TGME49_227660 and TGME49_243610) encoding the proteins with a DNMT domain (accession number PF00145) were found in the T. gondii genome and were designated as Tgdnmta and Tgdnmtb. The putative TgDNMTa and TgDNMTb contained DNMT domains at 256-413 aa and 426-703 aa, respectively.
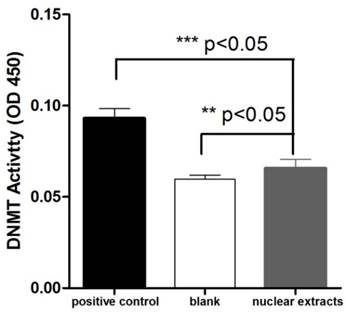
We investigated the presence of DNMT activity in T. gondii nuclear protein extract using an ELISA-like in vitro methylation assay. 10μg of nuclear tachyzoite extracts were used in the DNMT activity assay. The absorbance read at 450 nm on a microplate reader was used to evaluate the activity. The OD450 value of T. gondii nuclear protein extract was significantly higher than the blank (buffer), and it was also significantly different from the positive control (0.5 μg of purified bacterial DNMT) (Figure 2).
Detection of DNMT activity in T. gondii nuclear protein extracts. The OD450 values were measured for the reactions performed with 0.5 μg of purified bacterial DNMT (positive control), 10 μg of total T. gondii nuclear protein extract, and buffer only (blank). Three repetitive experiments with triplicate for each sample were conducted. The DNMT activity difference between the nuclear protein extract and negative control (or positive control) was analyzed with one-way ANOVA in SPSS13.0 software (Chicago, IL, USA), and a significant difference was found in both comparisons. T. gondii nuclear protein extract had apparent DNMT activity though it was lower than that of the bacterial DNMT provided by the manufacturer for the positive control. Error bars: standard deviation (SD).
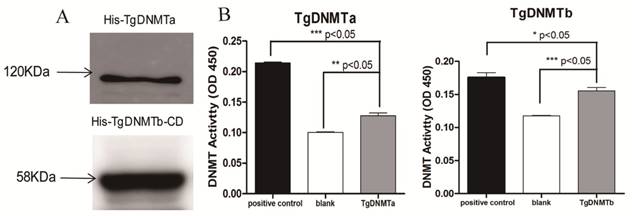
The purifications of the His-tagged TgDNMTa and the His-tagged TgDNMTb conserved domain were verified by Western blot (Figure 3A, Figure S1). The band sizes were consistent with the predicted ~120 kDa for TgDNMTa and ~58 KDa for TgDNMTb-conserved domain, respectively. A significant difference was found in the comparison of the recombinant TgDNMTa or TgDNMTb-conserved domain with the blank (negative control) (Figure 3B). The TgDNMTa and TgDNMTb-conserved domain had apparent DNMT activity, though this activity was lower than that of the positive control.
Validation of TgDNMTs as functional DNMTs by prokaryotic expression and activity assay. A: Western blot analysis of purified TgDNMTa and TgDNMTb-conserved domain expressed in E. coli with the His-tag antibody. B: The recombinant DNMT activity was assayed by the method illustrated in Figure 2. A significant difference was found between the recombinant TgDNMTa or TgDNMTb- conserved domain with the blank (negative control). TgDNMTa and TgDNMTb- conserved domain had apparent DNMT activity though it was lower than that of the bacterial DNMT provided by the manufacturer as a positive control. Error bars: Standard Error of Mean (SEM).
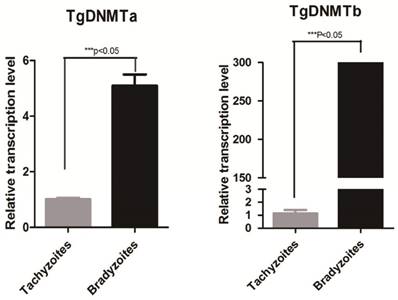
Using RT-qPCR, we found that the gene transcription levels of both Tgdnmta and Tgdnmtb in bradyzoites were significantly higher than the levels in tachyzoites; for Tgdnmtb, the relative transcription level in bradyzoites is approximately 300 times higher than that in tachyzoites (Figure 4). Both of TgDNMTa and TgDNMTb-conserved domain have the ability to methylate cytosine (Figure 3B). All of the findings suggest that the DNA methylation level of bradyzoites is higher than those of tachyzoites.
Detection of Tgdnmta and Tgdnmtb transcription in tachyzoites and bradyzoites. Each qPCR reaction were performed in triplicate, and the detection were repeated for three times. Relative transcription levels of Tgdnmta and Tgdnmtb genes were normalized to the transcription level of housekeeping gene GAPDH and were calculated using the 2-ΔΔCt method. The differences of Tgdnmt transcriptional levels between tachyzoites and bradyzoites were analyzed with an independent t-test in SPSS13.0 software (Chicago, IL, USA). The transcription levels of both Tgdnmta and Tgdnmtb in bradyzoites were significantly higher than those in tachyzoites, and especially for Tgdnmtb, the relative transcriptional level in bradyzoites was approximately 300 fold higher than that of the transcription level found in tachyzoites. Error bars: SEM.
The presence of DNA methylation in the T. gondii genome
Genomic DNA was extracted from tachyzoites and bradyzoites, bisulfite treated, and sequenced using MethylC-Seq. In the sequencing of the three replicates, we generated approximately 3.54 Gb and 17.2 Gb of raw data for tachyzoite and bradyzoite genomic DNA, respectively. We aligned the sequences to the T. gondii ME49 strain reference sequences, which yielded 1.90 Gb data for tachyzoites and 0.5 Gb data for bradyzoites. The average read depth was 29.11 and 7.38 per strand for tachyzoites and bradyzoites, respectively. On average, approximately 96.80% of the total cytosines of the tachyzoites genome and 86.93% of the bradyzoites genome were covered by at least one sequence read. We used the Lambda genome as a non-methylated internal reference and applied the error rate of 0.0032 for tachyzoites and 0.0034 for bradyzoites to correct the mC identification. Corrected estimates resulted in a total of 2,660 high confidence methylated cytosines in tachyzoites and 4,402 high confidence methylated cytosines in bradyzoites (Tables S4 and S5), representing 0.27% and 0.41% of the total genomic cytosine loci, respectively. Digestion with the methylation-sensitive restriction enzyme and combination with common PCR or quantitative PCR (qPCR) were used to validate the results. For example, the cytosines in chromosome IV-1336110(+) were methylated in tachyzoites according to the sequencing data. As the methylation-sensitive enzyme HpaII could not completely digest the gDNA because of the methyl protection, the fragment containing this cytosine site could be partially amplified, and PCR products were present in the agarose gel. Whereas the methylation-insensitive enzyme (MspI) could digest the gDNA completely, no PCR products were present in the agarose gel (Figure 5A).
Verification of the cytosine methylation sites in the T. gondii genome using enzymes (HpaII/MspI) coupled with PCR. HpaII and MspI were used to digest gDNA followed by PCR or qPCR. A: At the T-IV-1336110+ site, where partial methylation was presented in tachyzoites, the methylation-sensitive enzyme HpaII did not fully cut the gDNA at this site, and PCR product appeared; however, methylation-insensitive enzyme MspI cut the gDNA at this site thoroughly, and no PCR products were obtained. B: qPCR validation was conducted after the gDNA was digested by HpaII and MspI, respectively. All results were obtained from three repetitive experiments with three replicates. T -tachyzoites, B -bradyzoites. Error bars: SEM.
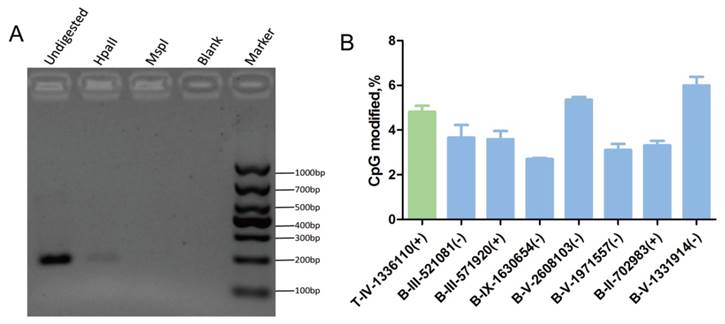
The qPCR showed that the percent of methylation level at the specific detecting sites from tachyzoites or bradyzoites DNA was approximately 5% (Figure 5B, Figure S2). For those select methylation sites in tachyzoites or bradyzoites DNA, the Ct value of the sample digested with HpaII was lower than that treated with MspI, which indicated that the sites were partially methylated. This result was consistent with the data of our BS-seq. These results confirmed the presence of methylcytosines in both the tachyzoite and bradyzoite genomes.
In the BS-seq data, no methylation of the cytosines of TGME49-chrIa 1295279 in both tachyzoites and bradyzoites DNAs was detected, that was consistent with the result of methylation-sensitive restriction enzyme digestion in combination with the qPCR experiment because the △CT between the CT average of MspI treated DNA and the CT average of HpaII treated DNA is near 0.5.
Characterization and analysis of the overall C-methylation in the genomes of tachyzoite and bradyzoite
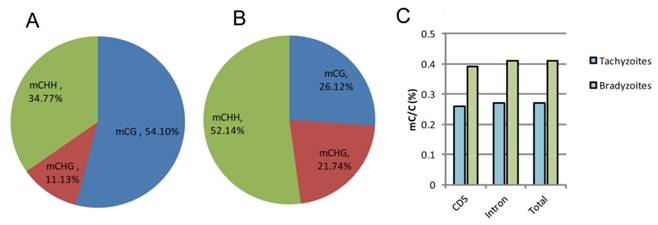
The highest number of mCs detected in tachyzoites were located in the CG regions, which occupied 54.10% of the total C-methylation, followed by the CHH regions (34.77%) and the CHG regions (11.13%); while in bradyzoites, the highest number of mCs detected were located in the CHH (H can be any nucleotide but G) regions, which occupied 52.14% of the total C-methylation, followed by the CG regions (26.12%) and the CHG regions (21.74%) (Figure 6A, B). For the non-CG methylated cytosine, the nucleotide H is always adenine, which is similar to Schistosoma mansoni [31]. Bradyzoites were shown to have greater proportion of methylated cytosines than tachyzoites in both the coding sequences (CDS) and the intron regions (Figure 6C), but the methylation levels varied little in the upstream regions, first exons, first introns, internal exons, internal introns, last exons, and downstream regions in both the tachyzoite and bradyzoite DNA.
Comparison of global DNA methylation in tachyzoite and bradyzoite genomes. A-B: Methylation context distribution of m5C in tachyzoites and bradyzoites; H represents any nucleotide A, T, and C. C: comparison of the proportion of methylated cytosines within the compartment of the genes.
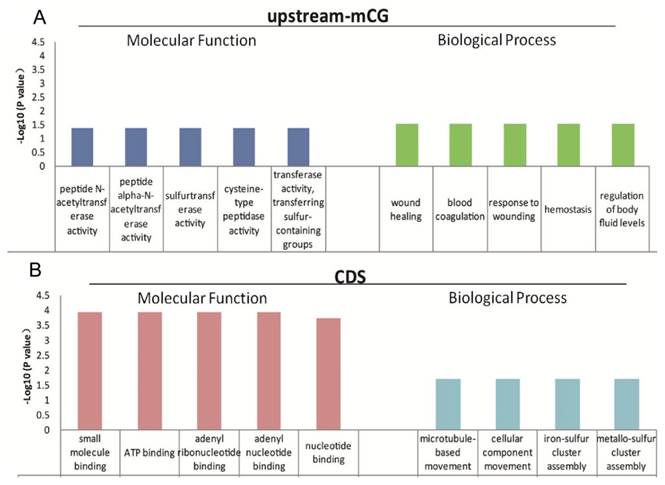
The function of mCG in the promoters is relatively clear [11-13], and gene body methylation, especially methylation in exons and near introns (the exon and intron boundaries), has also been reported to play important roles in gene expression regulation [14, 15]. The low genome cytosine methylation levels in both T. gondii stages enable us to analyze the function of the 624 genes with different CpG methylation levels in the upstream region and the 1550 genes with different cytosine methylation levels in the CDS between tachyzoites and bradyzoites (Table S6). GO and KEGG pathway enrichment were performed for these genes, and the data are shown in Tables S7 and S8. For those genes of tachyzoites and bradyzoites with different CpG methylation levels in the promoter region (the upstream region), the top five GO terms enriched in "molecular function" category were all related to "enzyme activity," "wound healing," "response to wounding," "blood coagulation," "hemostasis," and the "regulation of body fluid levels" were enriched in the "biological process" category (Figure 7A). Given this result, we proposed that the DNA methylation in the promoter region of these genes may be related to parasites' resistance to host immunity for wound healing. For the tachyzoite and bradyzoite genes with different cytosine methylation levels in the CDS region, the top five GO terms enriched in the “molecular function” category included "small molecule binding," “ATP binding,” and “nucleotide binding.” "Microtubule-based movement," "cellular component movement," "iron-sulfur cluster assembly" and "metallo-sulfur" were enriched in the “biological process” category (Figure 7B), the KEGG analysis in DAVID showed that these genes were enriched in the “DNA replication” and “Glycine, serine and threonine metabolism” pathways (Table S8). These genes associated with energy metabolism may participate in tachyzoite-bradyzoite interconversion.
Top five GO terms enriched for differentially abundant cytosine methylation regions (DMRs). A: the GO terms enriched in “molecular function” and “biological process” for the genes with different mCpG level in the upstream region; B: the GO terms enriched in “molecular function” and “biological process” for the genes with different mC levels in the CDS.
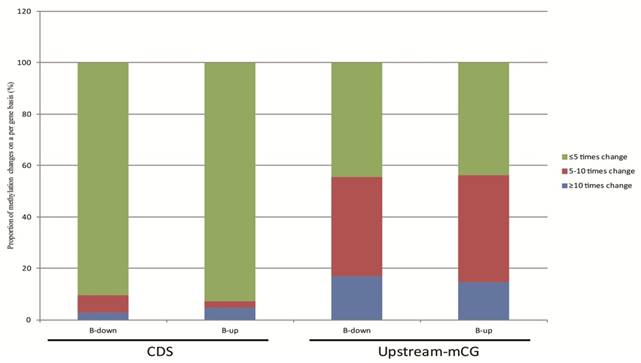
The relationship between DNA methylation and gene transcription were analyzed based on our RNA-seq data. The 624 genes of tachyzoites and bradyzoites with different CpG methylation levels in the upstream region and the 1550 genes with different cytosine methylation levels in the CDS between tachyzoites and bradyzoites were analyzed (Table S6). In these two groups of genes, we selected only the genes with a transcription level of "|log2 expression ratio |> 1 " for analysis. As a result, we obtained 190 genes with different CpG methylation levels in the upstream region and 576 genes with different cytosine methylation levels in the CDS region (Table S9). Among these 190 genes with different CpG methylation levels in the upstream region, the proportion of significantly higher methylation (bradyzoite/tachyzoites≥10) was slightly higher (approximately 1.15 times higher) in the down-regulated genes than in the up-regulated genes of bradyzoites; however, among the 576 genes with different cytosine methylation levels in the CDS region, the proportion of significantly higher methylation (bradyzoite/tachyzoites≥10) was apparently higher in the up-regulated genes than in the down-regulated genes of bradyzoites (Figure 8). The proportion of heavy methylation (bradyzoite/tachyzoites≥5) in the 190 genes group is more than that in the 576 genes group.
The relationship between DNA methylation and gene expression. 190 genes with different mCpG levels in the upstream region, and 576 genes with different mC levels in the CDS region, and all with a transcription level of "|log2 expression ration|>1" were included in the analysis. Among these 190 genes, the proportion of significantly higher methylation (bradyzoite/tachyzoites≥10) is slightly higher (approximately 1.15 times higher) in the down-regulated genes than in the up-regulated genes of bradyzoites, when compared to tachyzoite gene transcription; but among the 576 genes, the proportion of significantly higher methylation (bradyzoite/tachyzoites≥10) is apparently higher in the up-regulated genes than in the down-regulated genes of bradyzoites, when compared to tachyzoite gene transcription.
DNA methylation regulates T. gondii proliferation in the parasitophorous vacuole (PV)
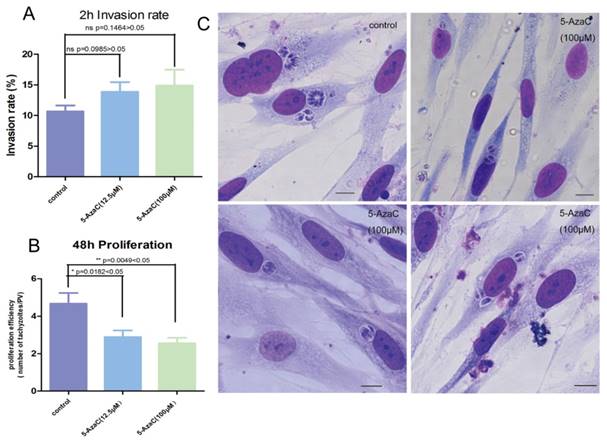
Whether the cytosine methylation of T. gondii DNA can function in the parasite's infection and proliferation was determined using an inhibitor of DNMT, 5-azacytidine (5-AzaC), which results in DNA demethylation or hemi-demethylation (Figure S2). Tachyzoites were treated with 5-AzaC for 30 min and washed with PBS to eliminate the inhibitor before HFF cells infection. At 2 h post infection, the infection rate was not significantly different between the host cells infected by 5-AzaC treated tachyzoites and normal control tachyzoites (p >0.05) (Figure 9A). The infection rate of the inhibitor-treated tachyzoites was even a little bit higher than the normal control tachyzoites, potentially a result of the stress reaction of the parasites to the drug. At 48 h post infection, the average number of tachyzoites in each PV was approximately five in the control group, while in the inhibitor-treated tachyzoites infection group, the average number of tachyzoites per PV was only three, which indicated that the proliferation of the tachyzoites in the PV was significantly inhibited (p<0.05) for the tachyzoites treated with 5-AzaC (Figure 9B, C).
Effect of the transient inhibition of T. gondii endogenous DNMT activity with 5-AzaC A: 5-AzaC treatment of T. gondii had no apparent influence on infection rate at 2 h post infection, and the infection rates in the inhibitor treatment groups were a little bit higher than that in the control group, but no significant difference was shown (p>0.05). Error bars: SEM. B: at 48 h post infection, the average number of parasites in each PV was approximately 5 in host cells infected with normal tachyzoites; while, in the cells infected with 5-AzaC treated tachyzoites, the number of tachyzoites per PV was approximately 3; a significant difference was found between the control group and the 5-AzaC treatment group (p<0.05), but no significant difference was observed between the groups infected with the tachyzoites treated with different concentrations (12.5 µM, 100 µM) of 5-AzaC. Error bars: SEM. C: the light microscopy of the HFF cells infected by tachyzoites treated or untreated with 5-AzaC, stained with Giemsa, and observed under a 100x objective lens (1000x magnification) at 48 h post infection. In the control group, the mean number of tachyzoites per PV was significantly more than that in the 5-AzaC treatment group. Bar: 10µm
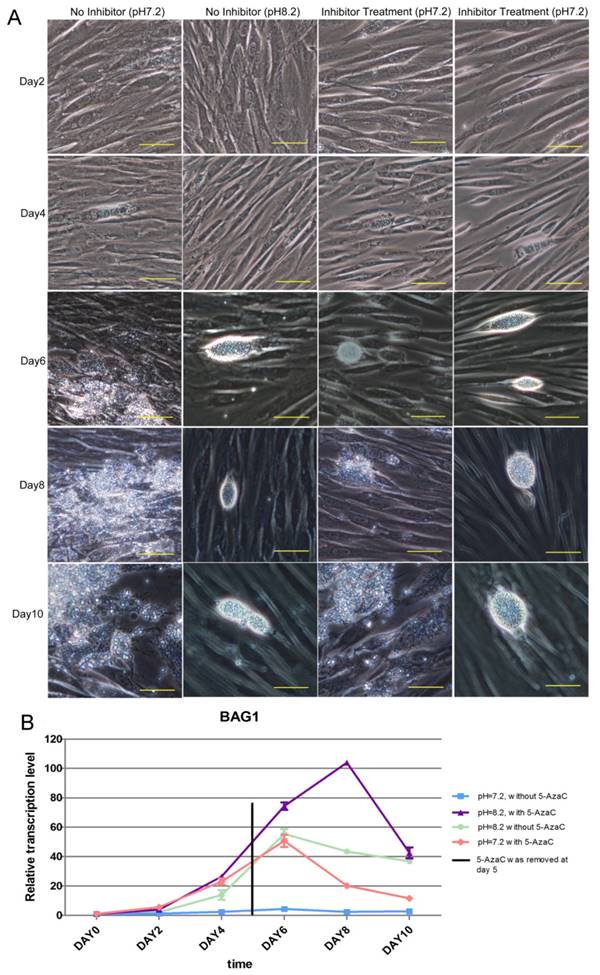
The result of the long-term inhibition of intracellular tachyzoites' DNMT activity was also detected. However, 5-AzaC was found to be toxic to HFF cells, and the cells began to shrink on day 2 in the Inhibitor Treatment (pH 7.2) and Inhibitor Treatment (pH 8.2) groups. Because the cells were almost detached in the Inhibited Treatment groups, the inhibitor 5-AzaC was removed on day 5. On day 6, the cells in the inhibitor-treated group were still shrunken, even though the inhibitor had been removed, and cyst-like structures could be found in the No Inhibitor (pH 8.2), Inhibitor Treatment (pH 7.2), and Inhibitor Treatment (pH 8.2) groups; however, in the No Inhibitor (pH 7.2) group, cells were ruptured and tachyzoites were released. On days 8 and 10, because the inhibitor was removed and the cells recovered, cyst-like structures were almost absent in the Inhibitor Treatment (pH 7.2) group, and the tachyzoites were released from PV. In the other two groups, No Inhibitor (pH 8.2) and Inhibitor Treatment (pH 8.2), cyst-like structures still were present (Figure 10A). The RT-qPCR detection of the BAG 1 transcription level to evaluate the extent of conversion to bradyzoites showed a similar result as the microscopic observation described above (Figure 10B).
Result of the long-term treatment of intracellular tachyzoites with 5-AzaC A: 5-AzaC is toxic to HFF cells, and the cells began to shrink on day 2 in the Inhibitor Treatment (pH 7.2), Inhibitor Treatment (pH 8.2) groups. On day 4, clear PVs were found in the four groups. Because the cells began to detach in the 5-AzaC treatment groups, the inhibitor was removed on day 5. On day 6, cyst-like structures were observed in three groups: No Inhibitor (pH 8.2); Inhibitor Treatment (pH 7.2); and Inhibitor Treatment (pH 8.2). In the No Inhibitor (pH 7.2) group, the cells were ruptured, and the tachyzoites were released. On days 8 and 10, because the inhibitor had been removed and the cells recovered, cyst-like structures were almost absent in Inhibitor Treatment (pH 7.2) group, and tachyzoites were released, which is similar to the No Inhibitor (pH 7.2) group. In the other two groups, Inhibitor Treatment (pH 8.2) and No Inhibitor (pH 8.2), the cyst-like structures could still be observed. All of the images were taken with a 20x objective lens (200 x magnification). Bar: 50µm. B: BAG 1 transcription levels were used to evaluate the conversion process, and on day 0 and day 2, the transcription level of BAG1 was low in the four groups; from day 4 to day 6, the transcription levels of BAG1 in the other three groups were significantly higher than those in the No Inhibitor (pH 7.2) group, and the Inhibitor Treatment (pH 8.2) group showed the highest level. On day 8, because the inhibitor had been removed and the cells recovered, the transcription level of BAG1 decreased greatly in the Inhibitor Treatment (pH 7.2) group; but although the inhibitor had been removed, the transcription level of BAG1 in the Inhibitor Treatment (pH 8.2) group remained at a high level. On day 10, the transcriptional level of BAG1 in the Inhibitor Treatment (pH 8.2) group greatly decreased, but remained at a high level, followed by the No Inhibitor (pH 8.2) group and the Inhibitor Treatment (pH 7.2) group. Error bars: SD.
Discussion
As an important epigenetic modification DNA methylation is widely distributed in prokaryotes and eukaryotes. The status of DNA methylation in protozoan genomes remains unclear. The detection of methylated cytosine (m5C) is highly dependent on the sensitivity of the applied methodology. Plasmodium falciparum, Toxoplasma gondii and Cryptosporidium parvum were reported to lack m5C in the genome by using the LC/ESI-MS method [23, 42], and the HELP method followed by bisulfite sequencing for two bradyzoites gene loci also detected no cytosine methylation in both RH and PLK strains [23]. However, by using an Agilent 1200 capillary HPLC pump interfaced with an LTQ linear ion trap mass spectrometer, analysis of m5C in the DNA hydrolysates of Plasmodium falciparum genome performed by online capillary HPLC-ESI-MS/MS revealed that this parasite genome was methylated asymmetrically (only one DNA strand was methylated) [22].
Our study confirmed the existence of DNA methylation in the two T. gondii life stages by using DNA bisulfite conversion combined with genome-wide sequencing, and this finding was also verified by digesting gDNA with MspI/HpaII followed by PCR or qPCR. It was found that the positions of methylated cytosines were different in bradyzoites and tachyzoites, and the global methylation level was higher in bradyzoites than that in tachyzoites, which may suggest a potential role of DNA methylation in the inter-conversion between tachyzoites and bradyzoites. In addition to the methylation in CG context, DNA methylation also occurred at CHG and CHH sites in T. gondii tachyzoite and bradyzoite genomic DNA.
The DNA methylation varies among different organisms. For example, in Arabidopsis, the methylation level is approximately 24% at CpG, 7% at CHG and 2% at CHH [43] and 0.67% at CpG, 0.21% at CHG and 0.24% at CHH in the silkworm [44]. Non-CG methylation plays important roles in plants, for example this type of modification could silence exogenous DNA and imprint gene expression [45, 46]. Remarkably, non-CG methylations are also found in our study, and the functions of the genes found non-CG methylation and CG methylation in CDS were associated with energy metabolism (Figure 7B). This methylation profile was also found in Aspergillus flavus [47], Drosophila [35], Plasmodium [22], Schistosoma mansoni [31], and Trichinella spiralis [38].
Though we intended to describe the genome-wide mapping of DNA methylation in T. gondii as we wanted to reveal the real nature of the bradyzoite DNA methylation, the bradyzoites used in our study were purified from the brain homogenate of the infected mice, but not induced in vitro. It was very difficult to get rid of the mouse tissue contamination. The sequencing depth for the T. gondii gene was low because of the mouse DNA contamination, so only those fragments with at least four reads were selected for further analysis. The sequence information of the T. gondii ME49 strain from ToxoDB was used as a reference, and only the mapped sequences were used for methylation analysis. It was inevitable that some methylation information of bradyzoites may be lost due to this technical restriction of sample purification.
The DNMTs presented in T. gondii belong to the DNMT2 family. The role of DNMT2 is controversially discussed in the past few years on the catalytic substrate and about its function [19, 22, 24, 30, 31, 33, 34]. For example, DNMT2 confers resistance to nitrosative stress in Entamoeba histolytica through the regulation of tRNA methylation [48]. In our study, we found that both TgDNMTa and TgDNMTb had DNA methyltransferase activity. DNA methylation has a relationship with histone modification. It had been reported that the histone remodels the de novo DNA methylation in the mammalian oocyte [49]. Usually, DNA methylation exerts its influence via proteins that could bind to methylated DNA and then recruit histone-modifying enzymes [35, 50, 51]. We searched the ToxoDB and found a gene (TGME49_210778) that encodes a protein with a hemimethylated DNA binding domain. Taken together, these data indicate that T. gondii has all the machinery necessary for DNA methylation to happen and function.
The T. gondii bradyzoite in the cyst is in a quiescent stage with low metabolism, and as found in our study, the genes with different methylation levels in the CDS region of bradyzoites were primarily involved in metabolic processes. These findings may suggest that the DNA methylation found in the CDS region was related to the regulation of the low metabolism to keep the parasite at the bradyzoite stage. DNA methylation has also been reported to be related to metabolic processes in silkworm [44], Trichinella spiralis [38], Nasonia vitripennis [52], Pea aphid and honeybee [53], and even in mammals such as pigs [54] and preterm neonates [54]. All of the results suggest that the role of DNA methylation is relatively conserved in different organisms. Also, this study found that the genes with different CpG methylation levels in promoters between tachyzoites and bradyzoites were related to the parasites' resistance to host immunity (Figure 7A), which suggested that DNA promoter methylation may regulate gene transcription related to host immune evasion.
In this study, some correlation between DNA methylation and gene transcription was shown. Globally, when we compared the bradyzoites with the tachyzoites, the CpG methylation in the promoter region showed only slight evidence in the down-regulation of gene transcription; conversely, the cytosine methylation in the CDS region showed more apparent up-regulation of gene transcription (Figure 8). Complicated mechanisms exist for regulating gene transcription in organisms including microRNA, lncRNA, and histone modification. DNA methylation may work together with these other mechanisms to regulate gene transcription.
5-AzaC treatment resulted in a shift from a virulent to a non-virulent phenotype in Entamoeba histolytica trophozoites [19], inhibited egg production and maturation in Schistosoma mansoni [31], and prohibits in vitro development in Plasmodium falciparum [22]. In our study, 5-AzaC treatment of T. gondii tachyzoites inhibited their proliferation in PVs (Figure 9B, C). Together, these results suggested that 5-AzaC can disrupt parasite differentiation and development through the inhibition of DNMT activity. In our study, more methylated cytosine sites were detected in bradyzoite DNA, and higher DNMT transcription levels were found in bradyzoites, so we hypothesized that DNMTs might function in the regulation of the inter-conversion between tachyzoites and bradyzoites. However, as a potent DNMT inhibitor that can disrupt many organisms' growth and development, 5-AzaC can affect not only T. gondii but also host cells. When we tried a long-term of treatment of 5-AzaC for intracellular T. gondii tachyzoites, apparent cytotoxicity was observed in the host cells, and the intracellular tachyzoites transformed to bradyzoites; when the inhibitor was removed, host cells were refreshed, and the intracellular tachyzoites recovered. The inhibition of DNMT activity in HFF cells may deprive the nutrients for the intracellular tachyzoites, and as a result, the tachyzoites transformed to bradyzoites because of denutrition. Interestingly, we found a new drug that can function on the intracellular T. gondii tachyzoites and can be used as an inducer for tachyzoite transformation to bradyzoites in vitro.
Abbreviations
m6A: N6-methyladenine; m4C: N4-methylcytosine; m5C: C5-methylcytosine; DNMT: DNA methyltransferase; HELP: HpaII tiny fragment en-richment by ligation-mediated PCR; SPF: specific pathogen-free; HFF: human foreskin fibroblasts; PBS: phosphate-buffered saline; CDS: coding sequence; GO: Gene ontology; PV: parasitophorous vacuole; IPTG: isopropyl-beta-D-thiogalactopyranoside; 5-AzaC: 5-Azacytidine.
Supplementary Material
Additional File 1Figures S1-S2, Tables S1-S3.
Table S4.
Table S5.
Table S6.
Table S7.
Table S8.
Table S9.
Acknowledgements
This work was supported by the funding from the National Natural Science Foundation of China (No. 81271866, 81572012), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2014), the Guangdong Provincial Natural Science Foundation Key Project (2016A030311025), and Guangzhou health and medical collaborative innovation major special project (201604020011) to HJP.
Authors' contributions
H.W. performed the experiments, analyzed the data and wrote the manuscript. S.J. performed animal experiments. L.C. performed RNA preparing. C.H., cell culture. S.W., cell culture. H.P. designed the study, funding support, and wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hotez PJ. Neglected parasitic infections and poverty in the United States. PLoS Negl Trop Dis. 2014;8(9):e3012
2. Adl SM, Simpson AG, Lane CE. et al. The revised classification of eukaryotes. J Eukaryot Microbiol. 2012;59(5):429-93
3. Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363(9425):1965-76
4. Lyons RE, McLeod R, Roberts CW. Toxoplasma gondii tachyzoite-bradyzoite interconversion. Trends Parasitol. 2002;18(5):198-201
5. Kim K, Weiss LM. Toxoplasma: the next 100 years. Microbes Infect. 2008;10(9):978-84
6. Skariah S, McIntyre MK, Mordue DG. Toxoplasma gondii: determinants of tachyzoite to bradyzoite conversion. Parasitol Res. 2010;107(2):253-60
7. Singh U, Brewer JL, Boothroyd JC. Genetic analysis of tachyzoite to bradyzoite differentiation mutants in Toxoplasma gondii reveals a hierarchy of gene induction. Mol Microbiol. 2002;44(3):721-33
8. Radke JR, Behnke MS, Mackey AJ. et al. The transcriptome of Toxoplasma gondii. BMC Biol. 2005;3:26
9. Cleary MD, Singh U, Blader IJ. et al. Toxoplasma gondii asexual development: identification of developmentally regulated genes and distinct patterns of gene expression. Eukaryot Cell. 2002;1(3):329-40
10. Wenzel MA, Piertney SB. Fine-scale population epigenetic structure in relation to gastrointestinal parasite load in red grouse (Lagopus lagopus scotica). Mol Ecol. 2014;23(17):4256-73
11. Attwood JT, Yung RL, Richardson BC. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci. 2002;59(2):241-57
12. Boyko A, Kovalchuk I. Epigenetic control of plant stress response. Environ Mol Mutagen. 2008;49(1):61-72
13. Goto T, Monk M. Regulation of X-chromosome inactivation in development in mice and humans. Microbiol Mol Biol Rev. 1998;62(2):362-78
14. Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13(8):335-40
15. Maunakea AK, Chepelev I, Cui K. et al. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23(11):1256-69
16. Ehrlich M, Gama-Sosa MA, Carreira LH. et al. DNA methylation in thermophilic bacteria: N4-methylcytosine, 5-methylcytosine, and N6-methyladenine. Nucleic Acids Res. 1985;13(4):1399-412
17. Ratel D, Ravanat JL, Berger F. et al. N6-methyladenine: the other methylated base of DNA. Bioessays. 2006;28(3):309-15
18. Field LM. Methylation and expression of amplified esterase genes in the aphid Myzus persicae (Sulzer). Biochem J. 2000;349(Pt 3):863-8
19. Fisher O, Siman-Tov R, Ankri S. Characterization of cytosine methylated regions and 5-cytosine DNA methyltransferase (Ehmeth) in the protozoan parasite Entamoeba histolytica. Nucleic Acids Res. 2004;32(1):287-97
20. Ali IK, Ehrenkaufer GM, Hackney JA. et al. Growth of the protozoan parasite Entamoeba histolytica in 5-azacytidine has limited effects on parasite gene expression. BMC Genomics. 2007;8:7
21. Pollack Y, Kogan N, Golenser J. Plasmodium falciparum: evidence for a DNA methylation pattern. Exp Parasitol. 1991;72(4):339-44
22. Ponts N, Fu L, Harris EY. et al. Genome-wide mapping of DNA methylation in the human malaria parasite Plasmodium falciparum. Cell Host Microbe. 2013;14(6):696-706
23. Gissot M, Choi SW, Thompson RF. et al. Toxoplasma gondii and Cryptosporidium parvum lack detectable DNA cytosine methylation. Eukaryot Cell. 2008;7(3):537-40
24. Raddatz G, Guzzardo PM, Olova N. et al. Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc Natl Acad Sci U S A. 2013;110(21):8627-31
25. Harony H, Ankri S. What do unicellular organisms teach us about DNA methylation? Trends Parasitol. 2008;24(5):205-9
26. Yong WS, Hsu FM, Chen PY. Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016;9:26
27. Lister R, Pelizzola M, Dowen RH. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315-22
28. Lister R, O'Malley RC, Tonti-Filippini J. et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133(3):523-36
29. Jeltsch A. Molecular enzymology of mammalian DNA methyltransferases. Curr Top Microbiol Immunol. 2006;301:203-25
30. Goll MG, Kirpekar F, Maggert KA. et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311(5759):395-8
31. Geyer KK, Rodríguez López CM, Chalmers IW. et al. Cytosine methylation regulates oviposition in the pathogenic blood fluke Schistosoma mansoni. Nat Commun. 2011;2:424
32. Tovy A, Hofmann B, Helm M. et al. In vitro tRNA methylation assay with the Entamoeba histolytica DNA and tRNA methyltransferase Dnmt2 (Ehmeth) enzyme. J Vis Exp. 2010 (44)
33. Schaefer M, Lyko F. Solving the Dnmt2 enigma. Chromosoma. 2010;119(1):35-40
34. Jeltsch A, Nellen W, Lyko F. Two substrates are better than one: dual specificities for Dnmt2 methyltransferases. Trends Biochem Sci. 2006;31(6):306-8
35. Marhold J, Kramer K, Kremmer E. et al. The Drosophila MBD2/3 protein mediates interactions between the MI-2 chromatin complex and CpT/A-methylated DNA. Development. 2004;131(24):6033-9
36. Liu M, Miao J, Liu T. et al. Characterization of TgPuf1, a member of the Puf family RNA-binding proteins from Toxoplasma gondii. Parasit Vectors. 2014;7:141
37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402-8
38. Gao F, Liu X, Wu XP. et al. Differential DNA methylation in discrete developmental stages of the parasitic nematode Trichinella spiralis. Genome Biol. 2012;13(10):R100
39. Dennis G Jr, Sherman BT, Hosack DA. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3
40. Hosack DA, Dennis G Jr, Sherman BT. et al. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4(10):R70
41. Finn RD, Mistry J, Tate J. et al. The Pfam protein families database. Nucleic Acids Res. 2010;38(Database issue):D211-22
42. Choi SW, Keyes MK, Horrocks P. LC/ESI-MS demonstrates the absence of 5-methyl-2'-deoxycytosine in Plasmodium falciparum genomic DNA. Mol Biochem Parasitol. 2006;150(2):350-2
43. Cokus SJ, Feng S, Zhang X. et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452(7184):215-9
44. Xiang H, Zhu J, Chen Q. et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol. 2010;28(5):516-20
45. Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204-20
46. Hsieh TF, Ibarra CA, Silva P. et al. Genome-wide demethylation of Arabidopsis endosperm. Science. 2009;324(5933):1451-4
47. Liu SY, Lin JQ, Wu HL. et al. Bisulfite sequencing reveals that Aspergillus flavus holds a hollow in DNA methylation. PLoS One. 2012;7(1):e30349
48. Hertz R, Tovy A, Kirschenbaum M, Geffen M. et al. The Entamoeba histolytica Dnmt2 homolog (Ehmeth) confers resistance to nitrosative stress. Eukaryot Cell. 2014;13(4):494-503
49. Stewart KR, Veselovska L, Kim J. et al. Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev. 2015;29(23):2449-62
50. Wang Y, Jorda M, Jones PL. et al. Functional CpG methylation system in a social insect. Science. 2006;314(5799):645-7
51. Bird AP, Wolffe AP. Methylation-induced repression-belts, braces, and chromatin. Cell. 1999;99(5):451-4
52. Wang X, Wheeler D, Avery A. et al. Function and evolution of DNA methylation in Nasonia vitripennis. PLoS Genet. 2013;9(10):e1003872
53. Hunt BG, Brisson JA, Yi SV. et al. Functional conservation of DNA methylation in the pea aphid and the honeybee. Genome Biol Evol. 2010;2:719-28
54. Li M, Wu H, Wang T. et al. Co-methylated genes in different adipose depots of pig are associated with metabolic, inflammatory and immune processes. Int J Biol Sci. 2012;8(6):831-7
Author contact
![]() Corresponding author: E-mail: floriapengcom
Corresponding author: E-mail: floriapengcom