Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(4):471-479. doi:10.7150/ijbs.16290 This issue Cite
Research Paper
Dipalmitoylphosphatidic acid inhibits tumor growth in triple-negative breast cancer
Qian-Qian Zhang1 ![]() , Jian Chen1, Da-Lei Zhou1, You-Fa Duan1, Cui-Ling Qi1, Jiang-Chao Li1, Xiao-Dong He1, Min Zhang1, Yong-Xia Yang2, Lijing Wang1
, Jian Chen1, Da-Lei Zhou1, You-Fa Duan1, Cui-Ling Qi1, Jiang-Chao Li1, Xiao-Dong He1, Min Zhang1, Yong-Xia Yang2, Lijing Wang1 ![]()
1. Vascular Biology Research Institute, School of Basic Course, Guangdong Pharmaceutical University, Guangzhou 510006, China;
2. School of Basic Course, Guangdong Pharmaceutical University, Guangzhou 510006, China.
Received 2016-5-26; Accepted 2017-1-30; Published 2017-3-12
Abstract

Triple-negative breast cancer (TNBC) is a subtype of breast cancer with a poor prognosis, accounting for approximately 12-24% of breast cancer cases. Accumulating evidence has indicated that there is no effective targeted therapy available for TNBC. Dipalmitoylphosphatidic acid (DPPA) is a bioactive phospholipid. However, the function of DPPA in the growth of TNBC has not yet been studied. In this study, we employed TNBC cells and a subcutaneous tumor model to elucidate the possible effect of DPPA on tumor growth in TNBC. We showed that DPPA significantly inhibited tumor growth in the mouse subcutaneous tumor model and suppressed cell proliferation and angiogenesis in TNBC tumor tissues. This inhibition was mediated partly by suppressing the expression of cyclin B1 (CCNB1), which directly promoted the accumulation of cells in the G2 phase and arrested cell cycle progression in human TNBC. In addition, the inhibition of tumor growth by DPPA may also be mediated by the suppression of tumor angiogenesis in TNBC. This work provides initial evidence that DPPA might be vital as an anti-tumor drug to treat TNBC.
Keywords: Dipalmitoylphosphatidic acid, triple-negative breast cancer, growth, angiogenesis.
Introduction
Breast cancer is one of the most common malignant tumors and is the leading lethal type of malignant tumor in human females worldwide. Moreover, the incidence of breast cancer is increasing, and the number of young women diagnosed with breast cancer has continued to rise in recent years [1]. Research has indicated that breast cancers with similar histological features may have distinct clinical outcomes and disparate responses to therapy [2, 3]. To date, multiple studies have been conducted to subtype breast cancer at the molecular level and to further determine therapy modalities and the disease prognosis. Therefore, breast cancer is mainly classified according to the presence of molecular markers, including the estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2 or erbB2), for further treatment with the effective targeted therapies [2-7]. Triple-negative breast cancer (TNBC) is a type of breast cancer with a poor prognosis that is defined as the absence of the ER, PR and HER2 markers [2, 8, 9]; therefore, TNBC does not respond to hormonal therapy or targeted therapy. Due to the limited standard treatment options and targeted therapies, there is an urgent need to explore an efficient drug to treat TNBC.
Phosphatidic acid (PA) is generated from phosphatidylcholine (PC) via a hydrolysis reaction by phospholipase D, from lysophosphatidic acid (lyso-PA) through an acylation reaction by lyso-PA acyl-transferase or from diacylglycerol (DAG) through a phosphorylation reaction by DAG kinase [10]. Furthermore, PA can be converted to lyso-PA and arachidonic acid (AA) by phospholipase A2 or to DAG by lipid phosphatases [10-12]. PA, lyso-PA and DAG are well-characterized lipid second messengers that can interact with multiple cell signaling pathway targets that regulate cancer cell survival, proliferation and progression to metastasis [13-19]. Dipalmitoylphosphatidic acid (DPPA) is a minor phospholipid metabolite that is involved in lipid biosynthesis. DPPA has two palmitic acid moieties instead of AA and can convert to lyso-PA to increase Bcl-2 expression in Hela cells but not in B lymphoma cells [17, 20]. However, the biological role and the significance of DPPA in TNBC remain entirely unknown.
Uncontrolled cell proliferation in cancer is often due to a malfunction in the cell cycle regulatory process. The cell cycle consists of four main stages. In the G1 phase, cells enter the cell cycle and prepare to duplicate DNA, which occurs in the S phase; cells then enter the G2 phase and prepare for mitosis in the M phase, in which the cells divide into two daughter cells [21]. A G2/M cell cycle checkpoint arrest is critical for DNA integrity and cell proliferation of cancer cells [22, 23]. Moreover, evidence from studies has indicated that G2/M-transition-targeting agents may be used as novel anti-cancer therapeutics in TNBC [24, 25].
In this study, we explored the effect of DPPA on tumor growth in TNBC using human MDA-MB-231 cells and a subcutaneous tumor mouse model and found that DPPA significantly inhibited tumor growth, cell proliferation and tumor angiogenesis. Furthermore, DPPA strongly arrested the G2/M transition and further inhibited cell cycle progression in MDA-MB-231 cells. Then, we analyzed the key regulatory factors of the G2 phase and found that cyclin B1 (CCNB1) was the DPPA target that regulated cellular proliferation in human TNBC. These data demonstrated that DPPA, as an anti-cancer drug, inhibited the expression of CCNB1 to arrest the G2/M transition in human TNBC.
Materials and methods
Reagents and antibodies
DPPA (BML-LP103) was obtained from Enzo Life Sciences, Inc. (NY, USA) and was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10mM with 15 minutes of sonication at room temperature. Cell Counting Kit-8 (CCK8) was obtained from Beyotime (China). Rabbit anti-GAPDH (#2118, diluted at 1:1000 for western blotting) was obtained from Cell Signaling Technology, Inc. (USA), while rabbit anti-Ki67 (GT210102) was obtained from Gene Tech (Shanghai) Co., Ltd. (China), and rabbit anti-CCNB1 (BA0766) and rabbit anti-CD34 (BA0532) were obtained from Boster (Wuhan) Co., Ltd. (China). CCNB1 siRNA was synthesized by RiboBio Co., Ltd. (China) according a previous report [26].
Cell lines and transfection
The 4T1 mouse TNBC cell line and the MDA-MB-231 human TNBC cell line were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China). These cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100μg/mL streptomycin at 37ºC in a 5% CO2 humidified incubator. Cell transfection with siRNA (100 nM) was performed using Lipofectamine 3000 (Invitrogen).
Animal ethics
The BALB/c mice (8-week-old, female) and BALB/c athymic nude mice (5-week-old, male) were obtained from the Guangdong Medical Laboratory Animal Center. All the mice were maintained in a temperature-controlled environment with a 12-h light/dark cycle. The use and study of the animals in this project were approved by the Ethics Committee of the Laboratory Animal Center of Guangdong Pharmaceutical University. All the surgical procedures were performed according to the Guidelines for Animal Experiments that were drawn up by the Ethics Committee.
Subcutaneous tumor model and therapy
The BALB/c mice were inoculated subcutaneously with 4T1 cells (105 cells) at the second right mammary fat pad area, and the BALB/c athymic nude mice were inoculated subcutaneously with MDA-MB-231 cells (106 cells) at the same body site as the BALB/c mice. Each type of mouse subcutaneous tumor model was randomly divided into DMSO and DPPA groups, with eight mice in each group. At 9 days after the 4T1 cell injection or 7 days after the MDA-MB-231 cell injection, the mice received an intravenous tail injection of DPPA (3mg/kg body weight) or the same concentration of DMSO once every two days for two weeks. The length (L) and width (W) of tumors were measured with calipers, and the tumor volume (V) was calculated using the following formula: V = (L × W2) × 0.5.
Immunohistological analysis
Tumor tissues from the mice with subcutaneous tumors were fixed in 10% neutral formalin solution for 24 hours and then embedded in paraffin. The paraffin sections (4μm) were stained for Ki67 and CD34 expression according to a previously described method [27]. The proliferation index was calculated as the percentage of Ki-67-stained cells per field at 40× magnification. The microvessel density (MVD) was calculated as the number of CD34-positive vessels per field at 40×. Three fields were selected randomly in a section from a non-necrotic area of the tumor tissues.
Cell proliferation assay
MDA-MB-231 cells were plated at density of 1,000 cells/well in a 96-well plate. Then, the indicated concentration of DPPA was added 24 hours later, and each concentration was tested in quadruplicate. Cell viability and cell proliferation were evaluated using the Cell Counting Kit-8 (CCK8) assay. The CCK8 reagent was added at the indicated time, and the plate was incubated at 37ºC for 4 hours and then quantifiably measured at a wavelength of 570 nm. The data were obtained from three independent experiments.
A colony formation assay was also used to detect cell proliferation. Briefly, MDA-MB-231 cells (3,000 cells/well) and 4T1 cells (1,500 cells/well) were seeded into 6-well plates. Then, the indicated concentrations of DMSO and DPPA were added into the wells 24 hours later. The colonies were fixed and stained after 7 days of growth. The effect of each treatment was assessed from three replicates and three independent experiments.
Flow cytometric assays
MDA-MB-231 cells (with or without siRNA transfection) were plated at a density of 5 × 105 cells/dish in 60-mm dishes and incubated for 24 hours. Next, the cells were serum-deprived for 24 hours, followed by the re-addition of serum and DPPA or transfection with siRNA; the cells were then harvested 20 hours later. The cells were stained using BD PharmingenTM PI/RNase staining buffer (#550825), and the analysis was conducted using a flow cytometer (Becton Dickinson). Cell cycle modeling was performed using the Modfit LT software, version 3.2 (Verity Software House). The data were obtained from three independent experiments.
Immunoblot analysis
Total proteins of the subcutaneous tumor tissues and the cells were extracted using RIPA lysis buffer, separated via 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto PVDF membranes and incubated with primary antibodies. The signals were detected using the ECL system (Cell Signaling Technologies, Danvers, MA).
Statistical analysis
The data are presented as the means ± standard deviation from at least 3 separate experiments. The differences between two groups were evaluated using a two-tailed Student's t test unless otherwise stated. Statistical significance was considered at P < 0.05. The corresponding relative integrated optical density (IOD) of protein expression levels in the IHC slices was analyzed using IPP software. The band intensities of the immunoblot were measured using Quantity One software.
Results
DPPA inhibits 4T1 subcutaneous tumor growth in vivo
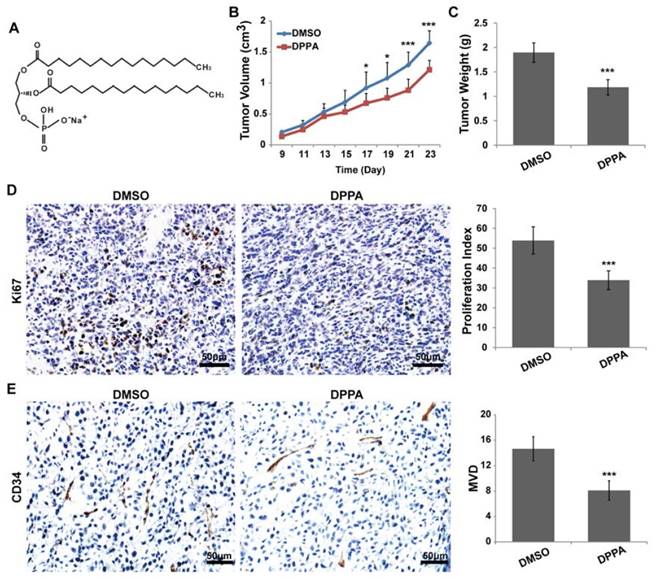
To evaluate the possible function of DPPA (Figure 1A) in TNBC, we employed a subcutaneous 4T1 TNBC tumor model and analyzed the effect of DPPA on tumor growth. DPPA (3 mg/kg) was injected every two days for two weeks beginning on the ninth day after the subcutaneous inoculation of 4T1 cells into the mouse mammary fat pad. The results revealed that DPPA significantly inhibited tumor growth because the tumor volume and tumor weight were markedly reduced compared with those of the control group (Figure 1B and 1C). In addition, the Ki67 staining results indicated that DPPA suppressed tumor cell proliferation (Figure 1D). Moreover, the results from the vascular endothelial marker CD34 staining showed that the tumor angiogenesis was inhibited by DPPA (Figure 1E). Therefore, these results indicated that DPPA acts as an anti-tumor drug by significantly inhibiting the tumor growth of mouse TNBC. However, whether DPPA can impede human TNBC tumor growth is yet to be confirmed.
DPPA inhibits 4T1 subcutaneous tumor growth in vivo. (A) The structure of DPPA. 4T1 cells were injected into the mammary fat pads of BALB/c mice, and 9 days later, DPPA (3 mg/kg body weight) or DMSO was injected once every two days for two weeks. DPPA significantly suppressed tumor volume (B) and tumor weight (C) in a mouse 4T1 subcutaneous tumor model. DPPA inhibited tumor cell proliferation and angiogenesis, which were measured by Ki67 (D) and CD34 (E) staining, respectively, using an IHC assay. N = 8, * P < 0.05, *** P < 0.001. Scale bars: 50 μm.
DPPA suppresses tumor growth in an MDA-MB-231 subcutaneous tumor model
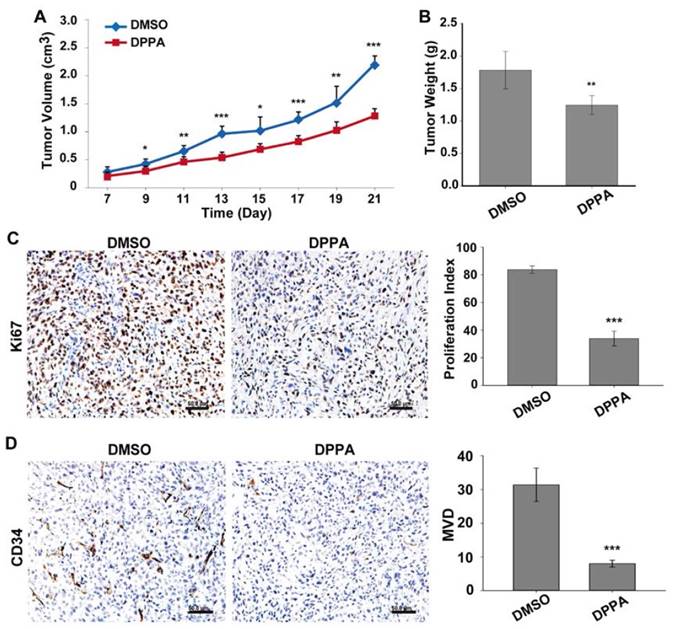
The inhibitory effect of DPPA on tumor growth was further studied in a subcutaneous tumor model of human TNBC cells. DPPA (3 mg/kg) was injected every two days for 14 days after viable MDA-MB-231 cells were subcutaneously inoculated into the mouse mammary fat pad. The tumor volume was measured and calculated, revealing that the tumor volume in the DPPA-treated group was significantly decreased compared with that in the control group (Figure 2A). After drug treatment, the mice were sacrificed, and the tumors were harvested and weighed. A comparison of the two treatment groups revealed that the tumor weight was also suppressed by DPPA treatment (Figure 2B). Moreover, DPPA remarkably inhibited the Ki67 staining for tumor cell proliferation and the CD34 staining for tumor angiogenesis in the tumor tissues (Figure 2C and 2D). All of these results suggested that DPPA inhibited the tumor growth of human TNBC.
DPPA inhibits MDA-MB-231 subcutaneous tumor growth in vivo. MDA-MB-231 cells were injected into the mammary fat pads of athymic nude mice, and 7 days later, DPPA (3 mg/kg body weight) or DMSO was injected once every two days for two weeks. DPPA significantly suppressed tumor volume (A) and tumor weight (B). DPPA inhibited tumor cell proliferation and angiogenesis, which were measured using Ki67 (C) and CD34 (D) staining, respectively, in an IHC assay. N = 8, * P < 0.05, ** P < 0.01 and *** P < 0.001. Scale bars: 50 μm.
DPPA inhibits cell proliferation by inducing a G2 phase cell cycle arrest in human TNBC cells
Firstly, we assessed the toxicity of DMSO and found that a concentration of less than 15% in vitro did not produce a significant difference in the proliferation of the MDA-MB-231 cells compared with the untreated group (0%) (Supplementary Figure 1A), and the toxicity of DMSO was also found only at concentrations greater than 5% in the 4T1 cells (Supplementary Figure 1B). To explore the biological function of DPPA in TNBC cells, the MDA-MB-231 cells were treated with DPPA at the indicated concentration for 48 hours. Compared with the untreated cells (0μM), DPPA significantly inhibited the viability of the MDA-MB-231 cells at a concentration greater than 100μM (15% DMSO) (Figure 3A). Furthermore, the cells were treated with 100μM and 150μM DPPA, and DPPA markedly suppressed cell proliferation in a time-dependent manner (Figure 3B and 3C). However, DPPA did not inhibit cell proliferation in 4T1 cells as detected by the colony formation assay (Supplementary Figure 1B).
DPPA inhibits cell proliferation by inducing a G2 phase cell cycle arrest in MDA-MB-231 cells. MDA-MB-231 cells were seeded into 96-well plates, and the indicated dose of DPPA was added 48 hours later. The cell viability was measured using the CCK8 assay. (A) DPPA significantly inhibited cell viability in a dose-dependent manner according to the CCK8 assay. In addition, DPPA also inhibited cell proliferation in a time-dependent manner according to the CCK8 assay (B) and the colony formation assay (C). (D) A flow cytometric assay was used to detect the effect of DPPA on the regulation of cell cycle progression. DPPA arrested MDA-MB-231 cells at the G2/M transition. * P < 0.05, ** P < 0.01 and *** P < 0.001.
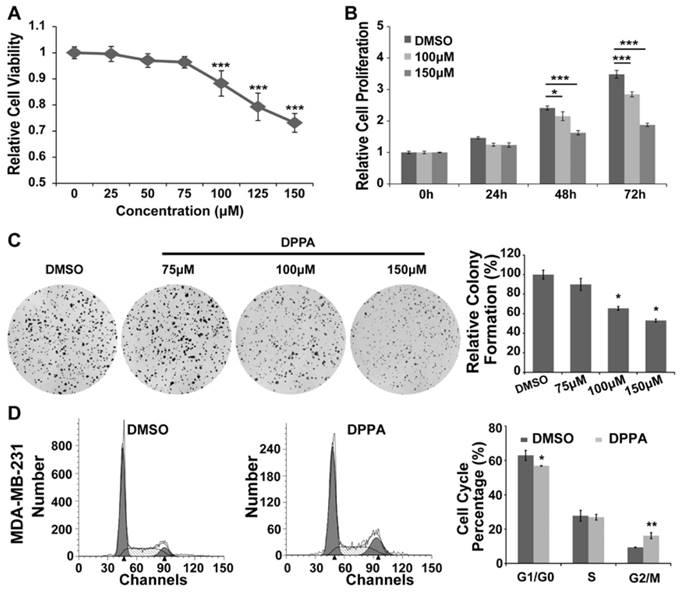
Because PA acts as a mediator of the ligand-induced inhibition of the G2/M transition in the human squamous cell carcinoma A431 cell line [13], we investigated whether DPPA suppresses cell cycle progression in human TNBC cells. The cells were synchronized by serum-deprivation for 24 hours, followed by the re-addition of serum; DPPA (100μM) was added, and the cells were harvested 20 hours later. The results indicated that DPPA significantly arrested the cell cycle at the G2/M transition, as indicated by a marked decrease in the number of cells in the G1 phase and the accumulation of cells in the G2 phase (Figure 3D). These results revealed that DPPA inhibited the proliferation of TNBC tumor cells, which may be achieved by the regulation of cell cycle progression.
CCNB1 was identified as a functional target of DPPA in the inhibition of human TNBC cell proliferation
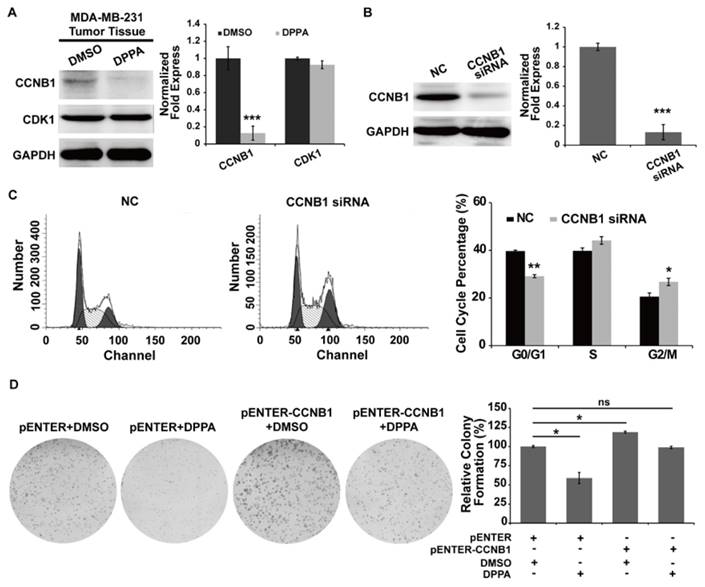
Because CCNB1 and CDK1 are essential for controlling the cell cycle at the G2/M transition, we detected the expression of these proteins in the tumor tissues of the DPPA-treated mice in the subcutaneous tumor model. As shown in Figure 4A, according to the immunoblot assay, DPPA dramatically inhibited the expression of CCNB1, but not CDK1, in the MDA-MB-231 subcutaneous tumor tissues compared with the control group. Furthermore, we inhibited CCNB1 expression using siRNA technology and found that suppressing the expression of CCNB1 also produced the accumulation of MDA-MB-231 cells in the G2 phase (Figure 4B and 4C). In addition, the inhibition of cell proliferation by DPPA was abolished by over-expressing CCNB1 (Figure 4D). All of these data revealed that DPPA suppressed cell cycle progression primarily through the inhibition of CCNB1 expression in human TNBC cells.
CCNB1 is involved in the G2 phase arrest by DPPA in MDA-MB-231 cells. Total proteins were extracted from the tumor tissues of mice in the MDA-MB-231 subcutaneous tumor model. (A) DPPA markedly inhibited the expression of CCNB1 in the tumor tissues of the MDA-MB-231 subcutaneous tumor models as assessed by an immunoblot analysis. (B) siRNA technology was used to knock down CCNB1 expression, and the inhibition efficiency in the MDA-MB-231 cells was determined using an immunoblot assay. The siRNA significantly inhibited the expression of endogenous CCNB1. (C) The inhibition of CCNB1 promoted the accumulation of cells in the G2 phase according to the flow cytometric assay. (D) The overexpression of CCNB1 promoted cell proliferation and abolished the inhibitory effect of DPPA on cell growth as assessed by a colony formation assay. ns: no significantly difference, * P < 0.05, ** P < 0.01 and *** P < 0.001.
Discussion
This study demonstrated the mechanism of the functional action of a bioactive lipid in TNBC. All the data support a model in which DPPA acts as an anti-tumor drug in TNBC by inhibiting tumor growth. The constitutive inhibition of CCNB1 leads to the accumulation of cells in the G2 phase, which results in the suppression of both cell cycle progression and abnormal cell proliferation in human TNBC (Figure 5). PA is the simplest membrane phospholipid and also functions as a lipid second messenger that regulates cell signal transduction, membrane trafficking, cytoskeletal rearrangement, and cell proliferation [20]. PA can be degraded into lyso-PA and AA by PLA2. Moreover, PA and lyso-PA are involved in the promotion of cell proliferation and cancer metastasis in multiple forms of human cancer [14, 15, 28-31]. DPPA (16:0 PA, without AA) can be converted to lyso-PA; however, the function of DPPA in tumor growth is not clear. Reports have indicated that PA induces the expression of Bcl-2 in both Hela cells and B lymphoma cells [17, 20]. Nevertheless, DPPA promotes Bcl-2 expression only in Hela cells [17, 20]. Based on this result, it can be postulated that DPPA might play an important role in solid tumorigenesis. In this work, we provide the first evidence that DPPA acted as an anti-tumor drug and significantly inhibited tumor growth in TNBC. Furthermore, other reports have indicated that DPPA can be employed to detect the effect of lyso-PA, an important metabolite of PA, on tumor growth in TNBC [17, 20]. Therefore, our results indicate that lyso-PA might be more important than AA regarding the inhibition of TNBC tumor growth as a metabolite of DPPA.
A schematic illustration of how DPPA might inhibit tumor growth in human TNBC. DPPA functions as an anti-tumor drug in TNBC by inhibiting tumor growth. The constitutive inhibition of CCNB1 by DPPA leads to the accumulation of cells in the G2 phase of the cell cycle, which results in the suppression of abnormal cell proliferation in human TNBC.
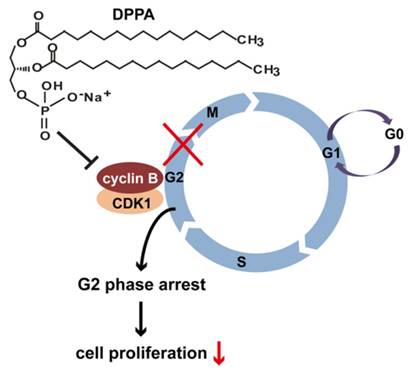
A previous report demonstrated that DPPA promotes the expression of anti-apoptosis signaling proteins in Hela cells [20]. The report suggested that DPPA may induce tumorigenesis in cervical cancer. However, we showed that DPPA markedly reduced tumor volume and weight in TNBC mouse models. This discrepancy implies that the action of DPPA is cell-type-dependent regarding tumorigenesis. Nevertheless, the functional mechanism of DPPA in the inhibition of tumor growth is currently unknown. A recent report showed that PA significantly induces cell cycle arrest in the G0/G1 phase in MDA-MB-231 cells [32]. However, whether DPPA is involved in cell cycle progression has not yet been explored. Therefore, we determined the cell cycle progression of MDA-MB-231 cells that were treated with DPPA and found that the cell cycle was significantly arrested in the G2 phase. These results suggest that DPPA inhibits tumor growth and cell proliferation by regulating cell cycle progression and suppressing the G2/M transition in human TNBC.
In all mammalian cells, cell proliferation is dependent on cell cycle progression. The cell cycle includes four continuous stages (G1, S, G2 and M) that are driven by the sequential activation of cyclin dependent kinases (CDKs) and cyclins [33]. The G2 phase is a key period of rapid cell growth and cell division. A previous study has shown that CCNB1, as a key regulatory protein, forms a complex with CDK1 that is essential for the regulation of mitosis and the transition from the G2 to the M phase [34]. CCNB1 and CDK1 are highly expressed in a variety of cancers [35-39]. Moreover, high levels of CCNB1/CDK1 are associated with the progression and survival of breast cancer cells [38, 40-42]. Our study revealed that DPPA arrested the MDA-MB-231 cell cycle in the G2 phase; however, the relationship between the DPPA-induced G2/M arrest and CCNB1 and CDK1 in MDA-MB-231 cells was still unclear. Therefore, we assessed the effect of DPPA on CCNB1 and CDK1 protein expression and showed that DPPA significantly inhibited the expression of CCNB1 but not CDK1 in tumor tissues. This result implied that CCNB1 is the main functional target of DPPA in the inhibition of cell cycle arrest in the G2 phase in human TNBC cells.
In this study, we found that DPPA inhibited the tumor growth of TNBC. Furthermore, DPPA suppressed cell proliferation and tumor angiogenesis in the tumor tissues. However, DPPA inhibited the proliferation of only the MDA-MB-231 cells but not the 4T1 cells in vitro. To date, we still do not understand the differences in the inhibitory effects between the in vivo and in vitro assays. Tumors grow in complex microenvironments in vivo, and multiple factors are involved in the progression of tumorigenesis, such as leukocytes and angiogenesis. Through an analysis of our data, we speculate that the inhibition of TNBC tumor growth by DPPA might occur through different mechanisms in humans and mice or not only by directly suppressing cell proliferation. Based on the inhibition of tumor angiogenesis by DPPA in the tumor tissues, these data suggest that DPPA may decrease tumor growth also via anti-angiogenesis. From another perspective, the metabolism of DPPA is a complex process and multiple factors are involved in the regulatory process. The in vitro condition might lack the necessary regulatory factors and may not completely simulate the in vivo environment. Therefore, we did not detect a direct inhibitory effect on 4T1 cell proliferation by DPPA in vitro. However, whether DPPA inhibits tumor angiogenesis must be further clarified in a future study.
The above analysis demonstrated that DPPA inhibited tumor growth partly by suppressing cell proliferation, which primarily occurs by blocking the cell cycle in the G2 phase by down-regulating the expression of CCNB1, which might further lead to a reduced formation of the CCNB1/CDK1 complex in human TNBC. In addition, DPPA might decrease tumor angiogenesis to further inhibit tumor growth in TNBC.
Abbreviations
DPPA: Dipalmitoylphosphatidic Acid; TNBC: Triple-negative breast cancer; CCNB1: cyclin B1; H&E: hematoxylin & eosin.
Supplementary Material
Supplementary figure 1.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 31200896 and 31471290), the Natural Science Foundation of Guangdong Province, China (2014A030313582), Science and Technology Planning Project of Guangdong Province, China (2013B021800271 and 2015A030302083), Pearl River S&T Nova Program of Guangzhou, China (201610010045) and Training Program for Excellent Young Teachers in Guangdong Province, China (YQ2015100).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bouchardy C, Fioretta G, Verkooijen HM. et al. Recent increase of breast cancer incidence among women under the age of forty. Br J Cancer. 2007;96:1743-1746
2. Kumar P, Aggarwal R. An overview of triple-negative breast cancer. Arch Gynecol Obstet. 2015;293:247-269
3. Colombo PE, Milanezi F, Weigelt B, Reis-Filho JS. Microarrays in the 2010s: the contribution of microarray-based gene expression profiling to breast cancer classification, prognostication and prediction. Breast Cancer Res. 2011;13:212
4. Sorlie T, Perou CM, Tibshirani R. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869-10874
5. Subramaniam S, Bhoo-Pathy N, Taib NA. et al. Breast Cancer Outcomes as Defined by the Estrogen Receptor, Progesterone Receptor, and Human Growth Factor Receptor-2 in a Multi-ethnic Asian Country. World J Surg. 2015;39:2450-2458
6. Weigelt B, Baehner FL, Reis-Filho JS. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: a retrospective of the last decade. J Pathol. 2010;220:263-280
7. Parise CA, Bauer KR, Brown MM, Caggiano V. Breast cancer subtypes as defined by the estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2) among women with invasive breast cancer in California, 1999-2004. Breast J. 2009;15:593-602
8. Engebraaten O, Vollan HK, Borresen-Dale AL. Triple-negative breast cancer and the need for new therapeutic targets. Am J Pathol. 2013;183:1064-1074
9. Peddi PF, Ellis MJ, Ma C. Molecular basis of triple negative breast cancer and implications for therapy. Int J Breast Cancer. 2012;2012:217185
10. Foster DA. Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim Biophys Acta. 2009;1791:949-955
11. Erickson RW, Langel-Peveri P, Traynor-Kaplan AE, Heyworth PG, Curnutte JT. Activation of human neutrophil NADPH oxidase by phosphatidic acid or diacylglycerol in a cell-free system. Activity of diacylglycerol is dependent on its conversion to phosphatidic acid. J Biol Chem. 1999;274:22243-22250
12. Billon-Denis E, Tanfin Z, Robin P. Role of lysophosphatidic acid in the regulation of uterine leiomyoma cell proliferation by phospholipase D and autotaxin. J Lipid Res. 2008;49:295-307
13. Kaszkin M, Richards J, Kinzel V. Phosphatidic acid mobilized by phospholipase D is involved in the phorbol 12-myristate 13-acetate-induced G2 delay of A431 cells. Biochem J. 1996;314( Pt 1):129-138
14. Xu X, Prestwich GD. Inhibition of tumor growth and angiogenesis by a lysophosphatidic acid antagonist in an engineered three-dimensional lung cancer xenograft model. Cancer. 2010;116:1739-1750
15. Shiozaki K, Takahashi K, Hosono M. et al. Phosphatidic acid-mediated activation and translocation to the cell surface of sialidase NEU3, promoting signaling for cell migration. FASEB J. 2015;29:2099-2111
16. Mazie AR, Spix JK, Block ER, Achebe HB, Klarlund JK. Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J Cell Sci. 2006;119:1645-1654
17. Oh KJ, Lee SC, Choi HJ. et al. Role of phospholipase D2 in anti-apoptotic signaling through increased expressions of Bcl-2 and Bcl-xL. J Cell Biochem. 2007;101:1409-1422
18. Han S, Huh J, Kim W, Jeong S, Min do S, Jung Y. Phospholipase D activates HIF-1-VEGF pathway via phosphatidic acid. Exp Mol Med. 2014;46:e126
19. Mahankali M, Farkaly T, Bedi S, Hostetler HA, Gomez-Cambronero J. Phosphatidic Acid (PA) can Displace PPARalpha/LXRalpha Binding to The EGFR Promoter Causing its Transrepression in Luminal Cancer Cells. Sci Rep. 2015;5:15379
20. Choi HJ, Lee JH, Park SY, Cho JH, Han JS. STAT3 is involved in phosphatidic acid-induced Bcl-2 expression in HeLa cells. Exp Mol Med. 2009;41:94-101
21. DiPaola RS. To arrest or not to G(2)-M Cell-cycle arrest: commentary re: A. K. Tyagi et al, Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G(2)-M arrest, and apoptosis. Clin. cancer res, 8: 3512-3519, 2002. Clin Cancer Res. 2002;8:3311-3314
22. Zhang J, Zhu X, Li H. et al. Piperine inhibits proliferation of human osteosarcoma cells via G2/M phase arrest and metastasis by suppressing MMP-2/-9 expression. Int Immunopharmacol. 2015;24:50-58
23. Ujiki MB, Ding XZ, Salabat MR. et al. Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol Cancer. 2006;5:76
24. Tate CR, Rhodes LV, Segar HC. et al. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012;14:R79
25. Kong Y, Chen J, Zhou Z, Xia H, Qiu MH, Chen C. Cucurbitacin E induces cell cycle G2/M phase arrest and apoptosis in triple negative breast cancer. PLoS One. 2014;9:e103760
26. Fang Y, Yu H, Liang X, Xu J, Cai X. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol Ther. 2014;15:1268-1279
27. Wang LJ, Zhao Y, Han B. et al. Targeting Slit-Roundabout signaling inhibits tumor angiogenesis in chemical-induced squamous cell carcinogenesis. Cancer Sci. 2008;99:510-517
28. Hurst JH, Hooks SB. Lysophosphatidic acid stimulates cell growth by different mechanisms in SKOV-3 and Caov-3 ovarian cancer cells: distinct roles for Gi- and Rho-dependent pathways. Pharmacology. 2009;83:333-347
29. Hu YL, Tee MK, Goetzl EJ. et al. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J Natl Cancer Inst. 2001;93:762-768
30. Park MH, Ahn BH, Hong YK, Min do S. Overexpression of phospholipase D enhances matrix metalloproteinase-2 expression and glioma cell invasion via protein kinase C and protein kinase A/NF-kappaB/Sp1-mediated signaling pathways. Carcinogenesis. 2009;30:356-365
31. Sliva D, Mason R, Xiao H, English D. Enhancement of the migration of metastatic human breast cancer cells by phosphatidic acid. Biochem Biophys Res Commun. 2000;268:471-479
32. Sliva D, Harvey K, Mason R, Lloyd F Jr, English D. Effect of phosphatidic acid on human breast cancer cells exposed to doxorubicin. Cancer Invest. 2001;19:783-790
33. Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138:255-271
34. Gao SY, Li J, Qu XY, Zhu N, Ji YB. Downregulation of Cdk1 and cyclinB1 expression contributes to oridonin-induced cell cycle arrest at G2/M phase and growth inhibition in SGC-7901 gastric cancer cells. Asian Pac J Cancer Prev. 2014;15:6437-6441
35. Zhou L, Li J, Zhao YP. et al. The prognostic value of Cyclin B1 in pancreatic cancer. Med Oncol. 2014;31:107
36. Kushner J, Bradley G, Young B, Jordan RC. Aberrant expression of cyclin A and cyclin B1 proteins in oral carcinoma. J Oral Pathol Med. 1999;28:77-81
37. Wang A, Yoshimi N, Ino N, Tanaka T, Mori H. Overexpression of cyclin B1 in human colorectal cancers. J Cancer Res Clin Oncol. 1997;123:124-127
38. Kawamoto H, Koizumi H, Uchikoshi T. Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in nonmalignant and malignant human breast lesions: immunocytochemical and quantitative image analyses. Am J Pathol. 1997;150:15-23
39. Yuan J, Kramer A, Matthess Y. et al. Stable gene silencing of cyclin B1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo. Oncogene. 2006;25:1753-1762
40. Dutta A, Chandra R, Leiter LM, Lester S. Cyclins as markers of tumor proliferation: immunocytochemical studies in breast cancer. Proc Natl Acad Sci U S A. 1995;92:5386-5390
41. Li Y, Chen YL, Xie YT. et al. Association study of germline variants in CCNB1 and CDK1 with breast cancer susceptibility, progression, and survival among Chinese Han women. PLoS One. 2013;8:e84489
42. Ding K, Li W, Zou Z, Zou X, Wang C. CCNB1 is a prognostic biomarker for ER+ breast cancer. Med Hypotheses. 2014;83:359-364
Author contact
![]() Corresponding authors: Lijing Wang, Phone: 86-20-39352231, Fax: 86-20-39352397, Email: wanglijing62com. Qian-Qian Zhang, Phone: 86-20-39352126, Fax: 86-20-39352397, Email: vinny223com.
Corresponding authors: Lijing Wang, Phone: 86-20-39352231, Fax: 86-20-39352397, Email: wanglijing62com. Qian-Qian Zhang, Phone: 86-20-39352126, Fax: 86-20-39352397, Email: vinny223com.