Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(5):574-587. doi:10.7150/ijbs.19150 This issue Cite
Research Paper
Aldehyde Dehydrogenases 1A2 Expression and Distribution are Potentially Associated with Neuron Death in Spinal Cord of Tg(SOD1*G93A)1Gur Mice
Huiting Liang1*, Chengsi Wu1*, Youqing Deng2*, Lei Zhu1, Jie Zhang1, Weiming Gan1, Chunyan Tang1, Renshi Xu1 ![]()
1. Department of Neurology, First Affiliated Hospital of Nanchang University, Nanchang 330006, Jiangxi, China;
2. Department of Neurology, Third Affiliated Hospital of Nanchang University, Nanchang 330008, Jiangxi, China.
* These authors contributed equally to this work.
Received 2017-1-12; Accepted 2017-2-20; Published 2017-4-10
Abstract

The pathogenesis of amyotrophic lateral sclerosis (ALS) has not been unclear yet, it might be associated with the abnormal expression and distribution of certain proteins. Aldehyde dehydrogenases 1A2 (ALDH1A2) was thought to be one of potential candidates. Therefore, in this study we observed and analyzed the alteration of the expression and distribution of ALDH1A2 in the spinal cord of wild-type (WT) and Tg(SOD1*G93A)1Gur mice. We compared the expression and distribution of ALDH1A2 in the different segments, anatomic regions and neural cells of spinal cord at the different stages of WT and Tg(SOD1*G93A)1Gur mice applied the methods of fluorescent immunohistochemistry and western blot. Results revealed that ALDH1A2 extensively expressed and distributed in the spinal cord of adult WT and Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 in the white matter including the anterior, posterior and lateral funiculus were more than that in the gray matter including the central canal, the anterior and dorsal horn. ALDH1A2 majorly expressed and distributed in the astrocyte, microglial, oligodendrocyte and neuron cells. The ALDH1A2 expression significantly decreased and redistributed in some anatomic regions of spinal cord at the onset and progression stages of Tg(SOD1*G93A)1Gur mice. The expression decrease of ALDH1A2 followed with the increase of neuron cells death. This study suggested that the alteration of expression and distribution of ALDH1A2 was potentially associated with the pathogenesis of ALS.
Keywords: Amyotrophic lateral sclerosis, aldehyde dehydrogenases 1A2, glial cell, neuron, pathogenesis.
Introduction
Amyotrophic lateral sclerosis (ALS) is a chronic neurodegenerative disease that selectively damages the motor neurons in cerebrum, brainstem and spinal cord [1]. The pathogenesis of ALS has not been known up to now [2-7], the recent studies suggested that the pathological variations of several proteins are closely associated with the pathogenesis of ALS, among them, the proteins commonly approved by investigators include superoxide dismutase 1 (SOD1) [8], Tar DNA binding protein-43 (TDP-43) [9, 10], fused in sarcoma/translocated in liposarcoma (FUS/TLS) [9], and the dipeptide repeat proteins produced by the repeat expansion of non-ATG translation in the chromosome 9 open reading frame 72 (C9ORF72) [11, 12]. The pathological variations of protein lead to the protein aggregation and the inclusion formation, generate the lesions of cellular components and the synaptic loss, ultimately result in the neuronal death in ALS [13].
In the conditions of physiology, the optimal prerequisite of cellular functions and survival is to maintain the stabilization of cellular proteome [14]. Furthermore, cells achieve the proteomic stabilization through keeping the normal state of molecular chaperones, the protein clearance pathways and the stress-associated signaling networks [14]. They exert the synergistic action to prevent proteins from misfolding, aggregating and accumulating in the subcellular compartments [13, 15, 16]. The above described processes are helpful to maintain the cellular proteostasis. The unbalance or damage of these processes directly lead to the ageing associated disorders, such as diabetes, cancer, stroke, the metabolic disorders, the pulmonary fibrosis, the inflammation and the neurodegenerative diseases [14].
The formation of protein inclusion containing the variant SOD1, TDP-43, FUS/TLS, and/or the dipeptide repeat proteins produced by the C9orf72 encoded non-ATG translation in the cytoplasm of degenerating motor neuron in ALS were due to the alterations of their structures or/and functions [17-20]. The recent studied evidences have demonstrated that the sporadic ALS (sALS) is closely linked to the abnormal alterations of structures and/or functions of some proteins, such as the abnormal expression, redistribution, misfold, unfold, the abnormal phosphorylation, the aberrant methylation and so on, which contribute to the abnormality of proteostasis in cell [14, 21-24]. The abnormal alterations of proteostasis result in their abnormal deposition in the neural cells including the neuron and glial cells, generate the toxicity to the neural cells, loss their normal functions, develop into the neuron death in sALS [14, 25]. However, the abnormality of currently discovered proteins associated with ALS only can explain a small part of the pathogenesis of ALS, much more of the possibly abnormal proteins have not been found yet. Therefore, in the future study, it needs screen and search the other potential proteins which participate in the pathogenesis of ALS.
Aldehyde dehydrogenases (ALDHs) are the members of NADP+-dependent enzymes family, they play some important roles in the NADP+-dependent oxidation of aldehydes into acids in a lot of biologic processes, such as the detoxification of alcohol-derived acetaldehyde, the metabolism of corticosteroids, the biogenic amines and neurotransmitters and the lipid peroxidation. In the physiologic conditions, ALDHs catalyze to oxide aldehydes into their corresponding carboxylic acids. In additional, they exert the cellular protective effects not only through preventing from the xenobiotic cytotoxic effects and the intracellular aldehydes like cyclophosphamide and ethanol, but also producing some important carboxylic acids like the retinoic acid (RA) and the γ-aminobutyric acid [26]. The high activity of ALDHs was reported to express and distribute in the hematopoietic and normal stem cells in a variety of tissues [27-29]. It implies that ALDHs might be a regulator of stem cells like the neural stem cells besides the above described important effects.
ALDH1A2 belongs to a member of ALDHs family, also has the similar effects and functions of ALDHs family. Although the study about the correlation between ALDH1A2 and the pathogenesis of ALS has not been reported at present, some studies suggested that ALDH1A2 participated in the damage of neurons in the lesions of spinal cord and brain [30, 31], and that it was an important regulator of proliferation and differentiation of endogenous neural stem cells in the adult spinal cord [31]. Furthermore, we found the down regulation of ALDH1A2 expression in the spinal cord of Tg(SOD1*G93A)1Gur mice during our preliminary experiment of proteomic analysis. Therefore, we hypothesized that ALDH1A2 might be associated with the neuron degenerative death of ALS, was a potential candidate disease-related proteins of ALS. In order to determine our hypothesis, identify whether or not ALDH1A2 is an ALS disease-related protein, and try to explore the potential mechanisms in the pathogenesis of ALS, we designed this study. In our study, we observed the expression and distribution features of ALDH1A2 in the different segments, the different anatomic regions and the different types of neural cells, and further analyzed the potential relationship between the expression and distribution of ALDH1A2 and the death of neurons at the different stages of Tg(SOD1*G93A)1Gur ALS-like transgenic mice. Our results found that the decrease of ALDH1A2 expression and distribution were potentially associated with the neuron death in the Tg(SOD1*G93A)1Gur mice, and suggested that ALDH1A2 was a candidate protein which participated in the pathogenesis of ALS.
Materials and Methods
Study approval
All experiments were approved by the Research Committee of First Affiliated Hospital of Nanchang University, and all experimental procedures involving animals were carried out in the strict accordance with the animal use guidelines of above committee (The approved project and protocol number: 2015-12-16). All experiments were performed under a full anesthesia, and made every effort in order to minimizing the animal suffering.
Materials
Animal
The Tg(SOD1*G93A)1Gur mice [32] (Jackson laboratory, Bar Harbour, Maine) were multiplied in the neurological lab of First Affiliated Hospital of Nanchang University. The transgenic mice were maintained by mating the male Tg(SOD1*G93A)1Gur mice with the female C57BL/6J mice. In order to detecting whether or not the positive Tg(SOD1*G93A)1Gur mice, the Tg(SOD1*G93A)1Gur mice were appraised by the PCR of genomic DNA isolated from the tail tissue. IL-2 was used as a negative control sample. The following primers were used, the human G93A SOD1 forward-primer was 5′-CAT CAG CCC TAA TCC ATC TGA-3′, the reverse-primer was 5′-CGC GAC TAA CAA TCA AAG TGA-3′, the IL-2 forward-primer was 5′-CTA GGC CAC AGA ATT GAA AGA TCT-3′, the reverse-primer was 5′-GTA GGT GGA AAT TCT AGC ATC ATCC-3′. The amplified conditions were 94 °C degeneration for 3 seconds, 60 °C annealing for 1 minute and 72 °C extension for 1 minute, a total of 35 cycles. The protocols were similar with our previous experimental description [33]. The non-transgenic mice also were used as the negative controls in concert with the WT mice in order to ensure that the overexpression of hSOD1 did not affect ALDH1A2 staining. The disease stages were divided into three stages based on the days of disease duration, the pre-onset (60-70 days), onset (90-100 days) and progressive or late (120-130 days) stages [32-34].
Methods
Fluorescent immunohistochemical staining of spinal cord
For the fluorescent immunohistochemical staining of spinal cord, slices were permeabilized with 0.2% Triton X-100 and blocked with 10% goat serum in 1×PBS after rehydrated in 1×PBS (pH 7.4), incubated with the following antibodies at 4 °C overnight: ALDH1A2 1:100 (Abcam, Hong Kong Ltd.), Glial fibrillary acidic protein (GFAP) (The biomarker of astrocyte) 1:500 (Abcam, Hong Kong Ltd.), RNA binding protein fox-1 homolog 3 (Fox3) (Neuronal nuclear antigen, NeuN, the biomarker of neuron, one of NeuN antibodies) 1:50 (Santa Cruz Biotechnology Inc.), claudin-11 (The biomarker of Oligodendrocyte) 1:50 (Santa Cruz Biotechnology Inc.) and Ionized calcium binding adapter molecule-1 (IBA-1) (The biomarker of microglia) 1:50 (Santa Cruz Biotechnology Inc.), then washed 6 times for each 5 minutes with 0.2% Triton X-100 in 1×PBS, incubated with the secondary antibody of polyclonal donkey anti goat (1:400) and polyclonal donkey anti rabbit (1:400) (Santa Cruz Biotechnology Inc.) conjugated to fluorescein isothiocyanate (Green) or/and rhodamine (Red) for 2 hours at room temperature, and stained by 4',6-diamidino-2- phenylindole (DAPI) (Blue). After being extensively washed 6 times for each 5 minutes, slices were mounted with the antifade medium, they were then examined and taken picture under a Nikon E800 fluorescent microscope with a spot digital camera (Diagnostic Instruments, Sterling Heights, Mich., USA) and a Photoshop software (Adobe Systems, San Jose, Calif., USA).
The multiple labeling histochemical method conjugated to ALDH1A2, Fox3, GFAP, claudin-11, IBA-1 antibodies and DAPI was used to doubly or trebly stain in one same sample using two or three different primary antibodies and secondary antibodies conjugated to the different color fluorescence, detected the overlapped staining of different color fluorescence, aimed to investigate the distribution of ALDH1A2 in the different neural cells. The negative controls of secondary antibody regularly were used for the ALDH1A2 antibody to assess its validity. The quenching agent of autofluorescence (AutoFluo Quencher C1212; Beijing APPLYGEN Technologies Inc., Beijing, China) was used in all samples.
Analysis of immunohistochemical positive cells
The analysis of immunohistochemical positive cells was performed by Image-Pro Plus 6.0 software (Media Cybernetics Co. Ltd., USA), counted the amount of positive cell and total cell amount in the different regions of spinal cord at 200 magnifications of slices, total cell amount each regions derived from the amount of DAPI positive cells. Each group consisted of 10 slices, and calculated the percentage (%) of positive cell of all slices. Three mice were used per group, the average percentages of positive cells were used for quantitatively analyzing.
Analysis of western blot
The WT and Tg(SOD1*G93A)1Gur mice were anesthetized and perfused with 20 ml of 0.9% saline. The spinal cord was excised and homogenized in the ice 1×RIPA lysis buffer (150 mM NaCl/1% Nonidet P-40/12 mM sodium deoxycolate/ 0.1% SDS/50 mM Tris-HCl, pH 7.2) supplemented with the protease inhibitor. The lysate was centrifuged at 12,000×g at 4°C for 10 minutes. The supernatant was used for Western blot analysis of ALDH1A2. The sample used for Western blot was measured the protein concentration using the BioRad protein assay kit (BioRad, Hercules, CA, USA). The solution of equal protein (20 μg protein/well) was loaded into each well of 10% SDS polyacrylamide gel and electrophoresed, then transferred onto the nitrocellulose membrane (Millipore, Bedford, MA, USA). The membrane was blocked with 5% albumin of bovine serum in TBST (10 mM Tris-HCl, pH 7.5/150 mM NaCl/0.1% Tween-20) for 1 hour at the room temperature on shaker, and then incubated with the specific antibodies of 1:2000 ALDH1A2 (Abcam, Hong Kong Ltd.) and 1:5000 beta-actin (Proteintech Group) overnight at 4 °C. Membrane was washed three times for 5 minutes each times with TBST at the room temperature, then incubated with the horseradish peroxidase-conjugated secondary antibody (KPL, Gaithersburg, MD) for 1 hour at the room temperature. The membrane was washed three times for each 5 minutes with TBST and visualized in the ECL reagents (Amersham Bioscience, Piscataway, NJ). The optic density (OD) of Western blot band was measured with Quantity One 1-D Analysis software (Bio-Rad Laboratories, Inc.), 10 samples (10 mice) per group were used, and repeated three times, the average OD of 30 bands each sample was compared and analyzed.
Data and statistical analysis
All experimental data were expressed using mean ± SEM. Statistical analysis was performed using the SPSS (Version 17.0) statistical software (SPSS, Chicago, IL, USA). Some specific comparisons between the control and experimental groups were analyzed by the analysis of variance (ANOVA) including a multivariate analysis of variance (MANOVA), two-ways ANOVAs as well as the eventual Post Hoc test used for paired comparisons, such as the group of the different anatomic regions and the group of different disease stages. Each data set of our groups passed a test for the Gaussian distribution. The major data were not Gaussian distribution. P<0.05 was considered as the statistically significant.
Results
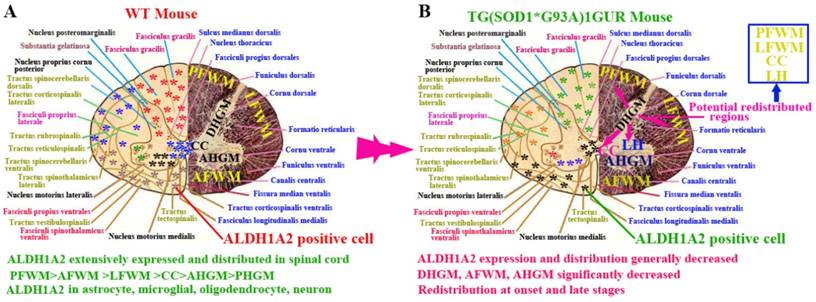
Distribution features of ALDH1A2 positive cells in different anatomic regions of different segments in spinal cord at the different stages of wild-type (WT) and Tg(SOD1*G93A)1Gur mice derived from single labeled analysis of fluorescent immunohistochemical experiments
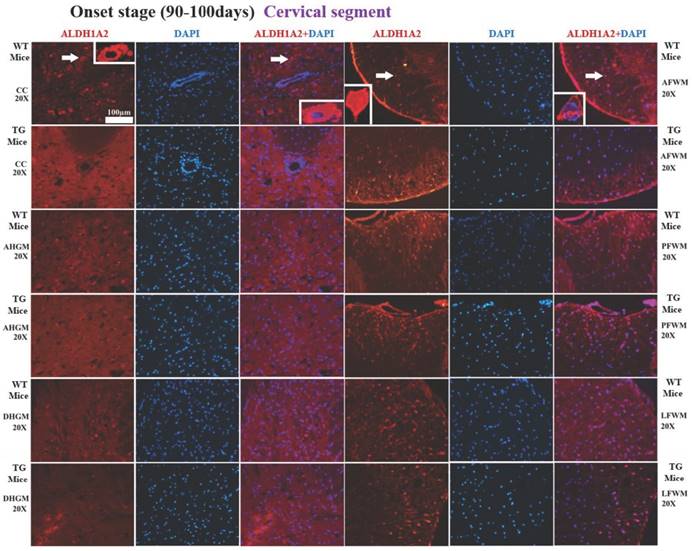
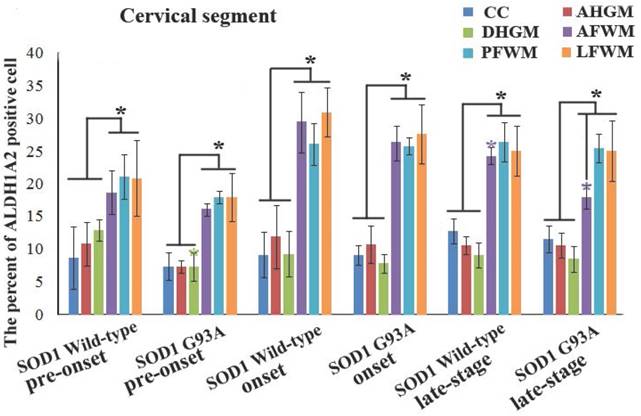
In the cervical segment of spinal cord, the ALDH1A2 positive cells in the anterior funiculus of white matter (AFWM), the posterior funiculus of white matter (PFWM) and the lateral funiculus of white matter (LFWM) were significantly more than that in the central canal (CC), the anterior horn of gray matter (AHGM) and the dorsal horn of gray matter (DHGM) at 60-70 days (Pre-onset stage), 90-100 days (Onset stage) and 120-130 days (Progression stage or late stage) of WT mice and Tg(SOD1*G93A)1Gur mice (Figure 1). There weren't significant difference between AFWM, PFWM and LFWM regions, and between CC, AHGM and DHGM regions at the pre-onset, onset and late stages of WT and Tg(SOD1*G93A)1Gur mice (Figure 2). Figure 1 only showed that the distribution features of ALDH1A2 positive cells in the different anatomic regions of cervical segments in spinal cord at the onset stage of WT and Tg(SOD1*G93A)1Gur mice, because the distribution features at the pre-onset and progression stages were the same with that at the onset stage. Figure 2 showed the compared details of ALDH1A2 positive cells in the different anatomic regions of cervical segments in spinal cord at the pre-onset, onset and late stages of WT and Tg(SOD1*G93A)1Gur mice. The results revealed the decrease of ALDH1A2 expression in the different anatomic regions of cervical spinal cord in the Tg(SOD1*G93A)1Gur mice, and that the significant decrease was detected in the DHGM (F=3.924, degrees of freedom (df)=10) at the pre-onset stage and in the AFWM (F=2.021, df=9) at the late stage (Figure 2). No redistribution was found in the cervical spinal cord.
The representative images of the expression and distribution of ALDH1A2 positive cells in the different anatomic regions of spinal cervical segment at the onset stage of the WT mice and the Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 generally exhibited a decreased tendency in the different anatomic regions of entire spinal cervical segment in the Tg(SOD1*G93A)1Gur mice. Among them, that in the DHGM at the pre-onset stage and the AFWM at the late stage significantly decreased. Abbreviation: WT, wild-type mice; TG, transgenic mice; PFWM, posterior funiculus of white matter; AFWM, anterior funiculus of white matter; LFWM, lateral funiculus of white matter; CC, central canal; AHGM, anterior horn of gray matter; DHGM, dorsal horn of gray matter.
The comparison of the percent of ALDH1A2 positive cells in the different anatomic regions of spinal cervical segment at the different stages between the WT mice and the Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 in the white matter including the anterior, posterior and lateral funiculus were significantly more than in the gray matter including the central canal, the anterior and dorsal horn at the pre-onset (60-70 days), onset (90-100 days) and progression stages (late-stage) (120-130 days) of the WT and Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 in DHGM and AFWM significantly decreased at the pre-onset and progression stages of the Tg(SOD1*G93A)1Gur mice respectively compared with the WT mice.
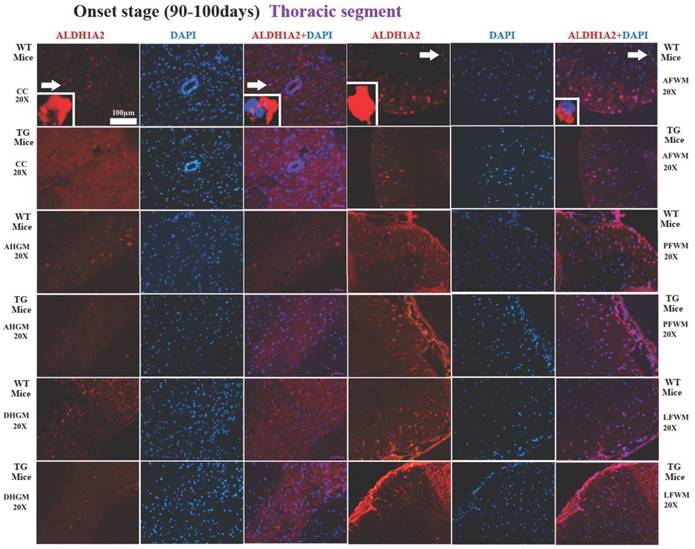
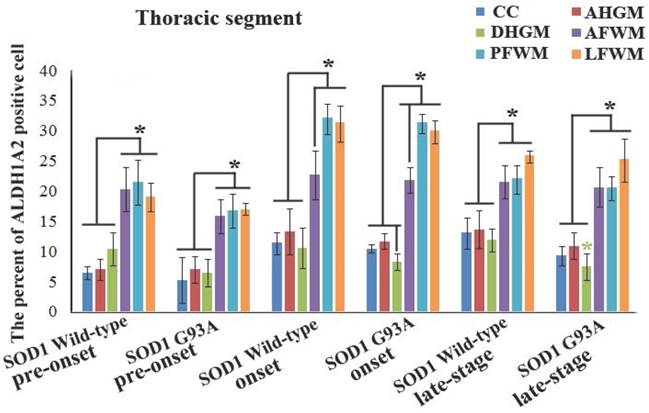
In the thoracic segment of spinal cord, the ALDH1A2 positive cells in AFWM, PFWM and LFWM were significantly more than that in CC, AHGM and DHGM at the pre-onset, onset and progression stages of WT mice and Tg(SOD1*G93A)1Gur mice (Figure 3). There weren't significant difference between AFWM, PFWM and LFWM regions, and between CC, AHGM and DHGM regions at the pre-onset, onset and late stages of WT and Tg(SOD1*G93A)1Gur mice. Figure 3 only exhibited that the distribution features of ALDH1A2 positive cells in the different anatomic regions of thoracic segments in spinal cord at the onset stage of WT and Tg(SOD1*G93A)1Gur mice, because the distribution features at the pre-onset and progression stages were consistent with that at the onset stage. Figure 4 showed the compared details of ALDH1A2 positive cells in the different anatomic regions of thoracic segments in spinal cord at the pre-onset, onset and late stages of WT and Tg(SOD1*G93A)1Gur mice. The results revealed the decrease of ALDH1A2 expression in the different anatomic regions of thoracic spinal cord in the Tg(SOD1*G93A)1Gur mice, and that the ALDH1A2 positive cells in the DHGM region at the progression stage of Tg(SOD1*G93A)1Gur mice were significantly less than that of WT mice (F=6.927, df=10) (Figure 4). No redistribution was detected in the cervical spinal cord (Figure 4).
The representative images of the expression and distribution of ALDH1A2 positive cells in the different anatomic regions of spinal thoracic segment at the onset stage of the WT mice and the Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 generally exhibited a decreased tendency in the different anatomic regions of entire spinal thoracic segment in the Tg(SOD1*G93A)1Gur mice. Among them, that in the DHGM at the late stage significantly decreased.
The comparison of the percent of ALDH1A2 positive cells in the different anatomic regions of spinal thoracic segment between the WT mice and the Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 in the white matter including the anterior, posterior and lateral funiculus were significantly more than in the gray matter including CC, the anterior and dorsal horn at the pre-onset, onset and progression stages of the WT and Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 in DHGM significantly decreased at the progression stages of the Tg(SOD1*G93A)1Gur mice compared with the WT mice.
In the lumbar segment of spinal cord, the ALDH1A2 positive cells in AFWM, PFWM and LFWM were significantly more than that in CC, AHGM and DHGM at the pre-onset, onset and progression stages of WT mice and Tg(SOD1*G93A)1Gur mice (Figure 5). There weren't significant difference between AFWM, PFWM and LFWM regions, and between CC, AHGM and DHGM regions at the pre-onset stage, onset stage and late stages of WT and Tg(SOD1*G93A)1Gur mice (Figure 5). Figure 5 only presented the distribution features that the ALDH1A2 positive cells in AHGM, DHGM and AFWM regions at the onset stage of Tg(SOD1*G93A)1Gur mice were significantly less than that of WT mice. Because the images that the ALDH1A2 positive cells in DHGM at the pre-onset stage of TG(SOD1*G93A)1Gur mice were significantly more than that of WT mice were not special, they were omitted. The results indicated that the ALDH1A2 positive cells in DHGM at the pre-onset stage of TG(SOD1*G93A)1Gur mice were significantly more than that of WT mice (F=0.145, df=10), that in AHGM (F=0.524, df=10), DHGM (F=1.894, df=10) and AFWM (F=0.494, df=10) regions at the onset stage of TG(SOD1*G93A)1Gur mice were significantly less than that of WT mice (Figure 6). The increased ALDH1A2 in DHGM during the pre-onset stage possibly redistributed to the other anatomic regions besides AHGM, DHGM and AFWM regions during the onset and late stage, based on that, we speculated that it occurred the redistribution among the different anatomic regions in the lumbar segment of TG(SOD1*G93A)1Gur mice (Figure 6).
The representative images of the expression and distribution of ALDH1A2 positive cells in the different anatomic regions of spinal lumbar segment at the onset stage of the WT mice and the Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 generally exhibited a decreased tendency in the different anatomic regions of entire spinal lumbar segment in the Tg(SOD1*G93A)1Gur mice, Among them, that in DHGM significantly increased at the pre-onset stage, that in AHGM, DHGM and AFWM significantly decreased at the onset stage.
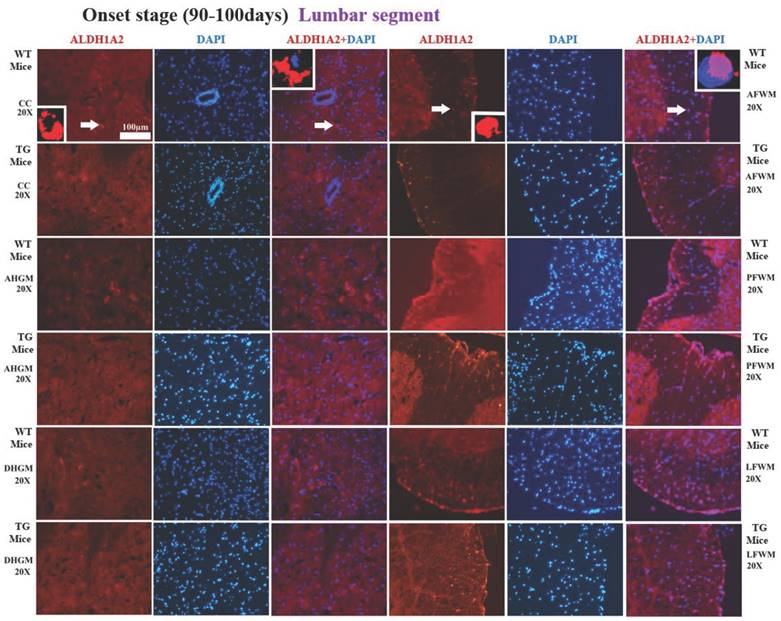
The comparison of the percent of ALDH1A2 positive cells in the different anatomic regions of spinal lumbar segment between the WT mice and the Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 in the white matter including the anterior, posterior and lateral funiculus were significantly more than in the gray matter including CC, the anterior and dorsal horn at the pre-onset, onset and progression stages of the WT and Tg(SOD1*G93A)1Gur mice. The expression and distribution of ALDH1A2 in DHGM significantly increased at the pre-onset stages, that in AHGM, DHGM and AFWM significantly decreased at the onset stage of the Tg(SOD1*G93A)1Gur mice compared with the WT mice.
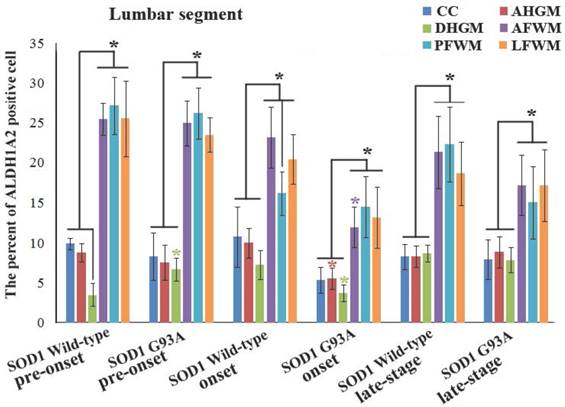
In the entire spinal cord, the distribution of ALDH1A2 positive cells in the different anatomic regions at the different stages of TG(SOD1*G93A)1Gur mice were significantly different. Among them, that in the major regions significantly decreased, only observed that that in the DHGM of lumbar spinal cord significantly increased at the pre-onset stage (Figures. 1-6), which implied that the ALDH1A2 redistributed in the some anatomic regions in the lumbar segment at the different stages of TG(SOD1*G93A)1Gur mice. The ALDH1A2 distribution exhibited a most significant decreased tendency in the multiple anatomic regions of lumbar spinal cord at the onset stages of TG(SOD1*G93A)1Gur mice. These results were consistent with the majorly damaged lumbar spinal cord in the ALS-like TG(SOD1*G93A)1Gur mice. The experiment aimed to observe and analyze the alteration features of ALDH1A2 expression and distribution in the different segments and the different anatomic regions of spinal cord at the different stages of WT and TG(SOD1*G93A)1Gur mice, to compare the difference between the WT and TG(SOD1*G93A)1Gur mice, to explore whether or not ALDH1A2 is a candidate ALS related protein and its potential mechanism in the pathogenesis of ALS.
Distribution features of ALDH1A2 positive cells in different neural cells of spinal cord in WT and TG(SOD1*G93A)1Gur mice derived from double or triple labeled analysis of fluorescent immunohistochemical experiments
The ALDH1A2 mainly distributed in the GFAP (Astrocyte cell) (Figure 7A), IBA-1 (Microglial cell) (Figure 7B), Fox3 (NeuN) positive cells (Neuron cell) (Figure 7C) and claudin-11positive cells (Oligodendrocyte cell) (Figure 7D) in the WT and TG(SOD1*G93A)1Gur mice. The distribution of ALDH1A2 positive cells in the different neural cells was not significantly different at the different stages of WT and TG(SOD1*G93A)1Gur mice, which indicated that the ALDH1A2 redistribution in the different neural cells didn't occur at the different stages of WT and TG(SOD1*G93A)1Gur mice. ALDH1A2 mainly expressed and distributed in the astrocyte in the dorsal horn, the microglia in the gray matter, the neuron in the anterior horn, the oligodendrocyte in the posterior horn.
The representative images of ALDH1A2 expression in the different neural cells at the progression stage of the WT and Tg(SOD1*G93A)1Gur mice. The ALDH1A2 positive cells partially overlapped with the GFAP, IBA-1, NeuN (Fox 3) and Claudin positive cells, which indicated that ALDH1A2 partially expressed in the astrocyte, microglia, neuron and oligodendrocyte cells. ALDH1A2 mainly expressed and distributed in the astrocyte of DH, the microglia of gray matter, the neuron of AH, the oligodendrocyte of PH. Abbreviations: DH, dorsal horn; AH, anterior horn; PF, posterior funiculus.
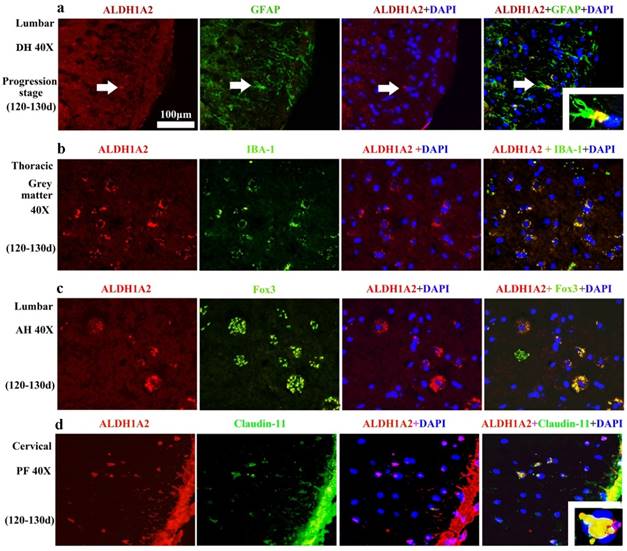
The control experiments to assess the potential autofluorescence and the specificity of primary and secondary antibody did not detected non-specific staining in the fluorescent immunohistochemical experiments (No data shown). The experiment aimed to observe and analyze the alteration features of ALDH1A2 expression and distribution in the different neural cells of spinal cord at the different stages of WT and TG(SOD1*G93A)1Gur mice, to compare the difference between the WT and TG(SOD1*G93A)1Gur mice.
Expression and distribution features of ALDH1A2 in entire spinal cord at different stages of WT and Tg(SOD1*G93A)1Gur mice derived from fluorescent immunohistochemical and west blot experiments
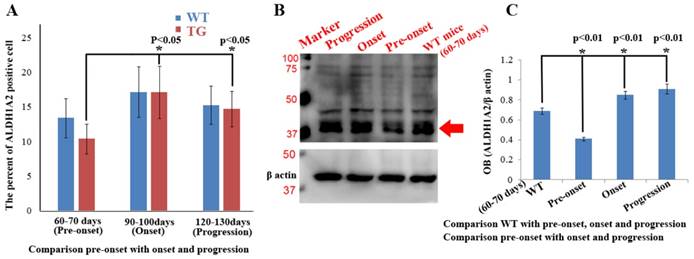
Total expression and distribution of ALDH1A2 in the entire spinal cord exhibited an increased tendency from 60 to 130 days in the WT and TG(SOD1*G93A)1Gur mice, especially in the onset stage. Total expression and distribution of ALDH1A2 at the different stages was not significantly different between the Tg(SOD1*G93A)1Gur mice and the WT mice, but that in the Tg(SOD1*G93A)1Gur mice were less than that in the WT mice, especially in the pre-onset stages (Figure 8A). Total expression of ALDH1A2 in the entire spinal cord at the pre-onset stage of Tg(SOD1*G93A)1Gur mice significantly decreased compared with the WT mice, that at the onset and progression stages of Tg(SOD1*G93A)1Gur mice significantly increased compared with that at the pre-onset stage of WT and TG(SOD1*G93A)1Gur mice (Figures. 8A, C). However, it was noteworthy that total ALDH1A2 positive cells at the pre-onset, onset and progression stages of Tg(SOD1*G93A)1Gur mice weren't significantly different, and exhibited a decreased tendency compared with that of WT mice, especially in the pre-onset stage (Figure 8A). The results suggest that the gradual decreased ALDH1A2 expression followed with the neuron damage at the different disease course in the entire spinal cord of ALS, which was a possible protective response. The ALDH1A2 protein was a possible protective factor in the neuron degeneration of ALS, in additional, the significant decrease of ALDH1A2 expression and distribution only occurred in the local damaged regions, total ALDH1A2 expression and distribution showed an increased tendency, and further confirmed that the local decrease of ALDH1A2 expression and distribution contributed to the damage of spinal cord. The experiment aimed to analyze the alteration features of ALDH1A2 expression and distribution in the entire spinal cord at the different stages of WT and Tg(SOD1*G93A)1Gur mice, to compare its difference in the entire spinal cord between the WT and Tg(SOD1*G93A)1Gur mice.
Total expression and distribution of ALDH1A2 in the entire spinal cord at the different stages of the WT and Tg(SOD1*G93A)1Gur mice. (A) Total ALDH1A2 positive cells significantly increased from the pre-onset, onset to progression stages of Tg(SOD1*G93A)1Gur mice, but there was not significantly difference between the WT and Tg(SOD1*G93A)1Gur mice, and followed with the increase of total ALDH1A2 positive cells of WT mice. (B, C) Total ALDH1A2 expression at the pre-onset stage of Tg(SOD1*G93A)1Gur mice significantly decreased compared with that of the WT mice, and significantly increased at the onset and progression stages of the Tg(SOD1*G93A)1Gur mice compared with the pre-onset of the WT and Tg(SOD1*G93A)1Gur mice.
Relationship between expression and distribution of ALDH1A2 and neuron cell death in spinal cord compared adult WT with Tg(SOD1*G93A)1Gur mice
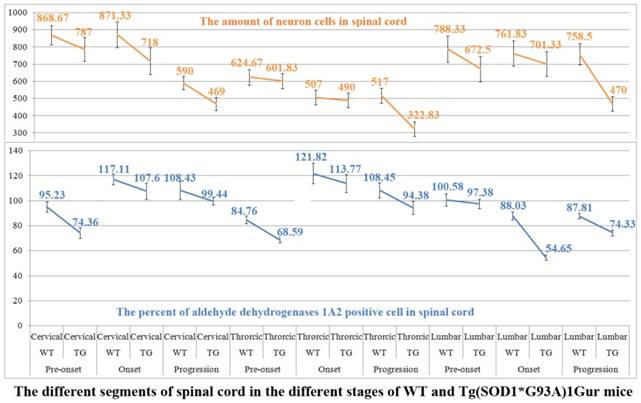
The systematic analysis about the relationship between the alteration of ALDH1A2 protein expression and distribution and the neuron cell death in the different segments of spinal cord during the different stages in the WT and Tg(SOD1*G93A)1Gur mice was conducted. The results demonstrated that the decrease of ALDH1A2 protein expression and distribution followed with the decrease of neuron cell amount in the cervical, thoracic and lumbar segments of adult spinal cord at the pre-onset, onset and progression stages. It suggested that the neuron cell death was potentially related to the decreased expression and distribution of ALDH1A2 protein, which implied that the more ALDH1A2 protein decrease followed with the more neuron cell death increase (Figure. 9). The results indicated that the decreased expression and distribution of ALDH1A2 protein possibly contributed to the death of neuron cell in the adult spinal cord of ALS-like mice. The experiment aimed to analyze the relationship between the expression and distribution of ALDH1A2 and the neuron death of both WT and Tg(SOD1*G93A)1Gur mice.
Discussion
In this study, we demonstrated the following four findings: 1). ALDH1A2 extensively expressed and distributed in the entire spinal cord of normal adult mice. Among them, the ALDH1A2 expression and distribution in the white matter was more than that in the gray matter, and were significantly different in the different anatomic regions of white and gray matter and the different segments. ALDH1A2 majorly expressed and distributed in the astrocyte, microglial, oligodendrocyte and neuron cells (Figure 10A). 2). The expression and distribution of ALDH1A2 generally decreased, the partial anatomic regions significantly decreased and occurred the redistribution at the onset and late stages of Tg(SOD1*G93A)1Gur mice. The redistribution in the different neural cells wasn't detected (Figure 10B). 3). The decrease of ALDH1A2 expression was potentially associated with the neuron death in ALS (Figure 9). 4). The ALDH1A2 might be a candidate disease-related protein, a protective factor of neurons, and modulate the neuron death in the pathogenesis of ALS.
The schematized representation of ALDH1A2 expression and distribution in spinal cord in the physiological and pathological condition. The diagram described that the alteration features of ALDH1A2 expression and distribution in the different anatomic regions including PFWM, AFWM, LFWM, CC, AHGM and DHGM, the different neural cells in the spinal cord of WT and Tg(SOD1*G93A)1Gur mice. Abbreviation: LC, lateral horn.
The relationship between the decrease of ALDH1A2 expression and distribution and the death of neuron cells in the cervical, thoracic and lumbar segments of spinal cord at the different stages of the WT and Tg(SOD1*G93A)1Gur mice. The decrease of ALDH1A2 expression and distribution followed with the decrease of neuron cell amount in the cervical, thoracic and lumbar segments of spinal cord at the pre-onset, onset and progression stages of the Tg(SOD1*G93A)1Gur mice compared with the WT mice, which implied the more decrease of ALDH1A2 protein followed with the more increase of neuron cell death.
ALDHs are a group of polymorphic enzymes that are responsible for the oxidation of aldehydes to carboxylic acids [30). There are three different types of ALDHs in mammals: type 1 (ALDH1) in cytosol, type 2 (ALDH1) in mitochondria and type 3 (ALDH1) in tumors, stomach and cornea. In all three types, ALDH1 and ALDH2 are two most important enzymes for the aldehyde oxidation, the two enzymes are the 54kDA tetrameric subunits. ALDH1 and ALDH2 exist in many tissues and organs, the most is in liver [35]. When ALDHs are transferred from liver to muscle and heart, they are dissolved. ALDH1 consists of ALDH1A1 and ALDH1A2. ALDH1A2 also is known as aldehyde dehydrogenase 1 family member A2, or retinaldehyde dehydrogenase 2 (RALDH2), is an enzyme encoded by the ALDH1A2 gene in the human beings [36]. ALDH1A2 is a cytosolic homotetramer expressed in a wide scale of embryonic and adult tissues like intestine, testis, lung, kidney, liver, brain, spinal cord and retina [37-39]. In our study, we found that ALDH1A2 extensively expressed and distributed in the entire spinal cord, and its expression and distribution showed the significant difference in the different anatomic regions and the different segments of adult normal spinal cord. It indicated that ALDH1A2 was an important protein (Enzyme) in the normal physiologic condition, and played some important physiologic functions in the different part of spinal cord. If the abnormality of ALDH1A2 expression and distribution occurs, would result in the spinal cord dysfunction related disease. In additional, we found that the decrease and redistribution of ALDH1A2 expression in the partial regions of spinal cord, which were potentially associated with the neuron death in the disease course of Tg(SOD1*G93A)1Gur mice. Our data suggested that the alteration of ALDH1A2 expression and distribution might be one of pathogenesis of ALS, the potential mechanisms might be related to the following literature reported information.
At present, ALDHs have been detected to involve in a lot of biological processes, including the detoxification of exogenously and endogenously produced aldehydes. The previous study suggested that ALDHs might modulate the proliferation and differentiation of stem cells [31]. ALDH2 might affect the regeneration of neural cells through modulating the proliferation and differentiation of endogenous neural stem cells, be an important regulator of neural repairing in the adult spinal cord [31]. The abnormal expression and distribution of ALDH1A2 in our study might be lead to the lesion and/or the recovering obstacle of neural cells in spinal cord, because of damaging the proliferation and differentiation of endogenous neural stem cells, subsequently develop ALS due to the excessive neuron loss of spinal cord.
ALDH1A2 are responsible for catalyzing the oxidation of both all-trans-retinal and 9-cis-retinal into RA [37]. It shows the highest specificity to all-trans-retinal compared with the other isozymes of ALDHs [40-42], participates in many developmental courses and is a crucial regulator of RA synthesis in the developing tissues [43]. The ALDH1A2-/- mouse reveals the defects of axial rotation, the incomplete neural tube closure and the reduction of trunk region in the development of spinal cord [37]. Several animal models have found that ALDH1A2 is a vital regulator during the development of many tissues like kidney, retina, lung, forebrain, pancreas and spinal cord [37, 44-48]. The recent studied data appeared that the spinal bifida in humans was associated with three distinct single nucleotide polymorphisms (SNPs) of ALDH1A2, one silent SNPs is A151A (c.453A>G), the SNPs of two introns are rs3784259 and rs3784260. ALDH1A2 might exert some important roles in the development of spinal cord [49]. It implies that ALDH1A2 might also participate in the development of neural cells in the adult spinal cord, its expression and distribution might be associated with the physiological development of adult spinal neural cells. The abnormal expression and distribution of ALDH1A2 in our study might interfere the development of normal renewed neural cells due to its function alteration catalyzing the oxidation of both all-trans-retinal and 9-cis-retinal into RA, lead to the imbalance of physiological shift of new and old neural cells, and the physiologic death of neural cells could not be replaced by the renewed neural cells in the adult spinal cord, produce the reduce of neural cells, lead to the loss of some spinal developmental functions, develop into the spinal cord diseases such as ALS.
ALDH1A2 catalyzes the lipid peroxidation (LPO)-derived aldehydes and may eliminate the damage of oxidative stress. The ALDH1A2 of rat oxidizes the medium-chain saturated LPO-derived aliphatic aldehydes with the high affinity of hexanal, octanal and decanal [40]. The three isozymes of ALDHs that are responsible for synthesizing RA are ALDH1A1, ALDH1A2 and ALDH1A3 [50, 51]. These enzymes of ALDHs catalyze the final step of RA biosynthesis from retinaldehyde to RA. The RA synthesized by ALDH1A2 controls the differentiation, the growth arrest and the apoptosis of cell [30, 52, 53]. The ALDH1A2 protein is an enzyme that is responsible for synthesizing RA by the substrate of retinaldehyde. RA is the active derivative of vitamin A in retinol, is a paracrine hormone signaling molecule in the developing and adult tissues [54]. It suggests that RA might also be an important biological molecule in the development of adult spinal cord, the abnormal expression and distribution of its synthesized enzymes including ALDH1A1, ALDH1A2 and ALDH1A3 might affect the development of adult spinal cord, is the pathophysiological base of some spinal cord diseases like ALS.
In the spinal cord, the sonic hedgehog (SHH) and RA are necessary to form the ventral motor neurons [55-58]. The dorsally expressed Pax6, the ventrally expressed Nkx6.1 and the Olig2 expressed in the region of Pax6 and Nkx6.1 overlapped expression are required for the differentiation and development of motor neurons [59]. RA is synthesized by ALDH1A2 from the adjacent somitic mesoderm to the neuroectoderm of spinal cord. Mouse embryos lacking this source of RA can't express Pax6 and Olig2 in the spinal cord, whereas the expression of Nkx6.1 is unaffected [58]. In the defection of RA signaling, the undifferentiated neuroectoderm of spinal cord can't generate a ventral motor neuron cell [57, 58]. RA has been used as a differentiated agent along with SHH to generate motor neurons in the spinal cord of mice [60] and humans [61] embryonic stem cells, providing a potential source of replaced cells for the motor neuron diseases or the spinal cord injury [54]. According to the current studied information, we hypothesized that RA also might be a necessary biological molecule in the formation, differentiation and development of motor neurons in the adult spinal cord. The ALDH1A2 is the RA necessary synthesized enzyme, its expression and distribution might be potentially associated with the formation, differentiation and development of renewed motor neurons in adult spinal cord. The abnormal expression and distribution of ALDH1A2 in our study might interfere the potential source of replaced motor neurons, develop into ALS because of the RA synthesized deficiency. In those biological processes, might involve in the decrease of Pax6 and Olig2 expression due to the insufficiency of RA.
To the best of my knowledge, our study firstly explored the association between ALDH1A2 and the pathogenesis of ALS through observing and analyzing the alterations of ALDH1A2 expression and distribution in the different segments, the different anatomic regions and the different neural cells at the different stages of the WT mice and the Tg(SOD1*G93A)1Gur mice, and compared the alteration of ALDH1A2 expression and distribution between the WT and Tg(SOD1*G93A)1Gur mice. Our results revealed that ALDH1A2 was potentially associated with the neuron cell death in the Tg(SOD1*G93A)1Gur mice. The significant decrease and the redistribution of ALDH1A2 expression in some anatomic regions during the disease course of ALS-like Tg(SOD1*G93A)1Gur mice indicated that ALDH1A2 was a candidate ALS-related protein, participated in the pathogenesis of ALS and might be a protective factor of neurons, because the decrease of ALDH1A2 expression followed with the increase of neuron death in the spinal cord of ALS-like transgenic mice. The exact mechanisms in the pathogenesis of ALS are waiting for further studying. Up to now, we have not found the report about the relationship that ALDH1A2 is associated with ALS. According to the above cited previous reported literatures about the roles of ALDH1A2, we hypothesized that the ALDH1A2 might play some roles in the pathogenesis of ALS through the following biological processes. 1) Detoxify the exogenously and endogenously produced aldehydes through oxidizing aldehydes to carboxylic acids [30, 35], catalyze the oxidation of both all-trans-retinal and 9-cis-retinal into RA [37], eliminate the LPO-derived aldehydes damage of oxidative stress [40], prevent neuron cells from the aldehydes toxicity. 2) Modulate the formation, proliferation and differentiation of neural stem cells, promote the neuron regeneration from neural stem cells [31]. 3). Synthesize RA, modulate the level of RA, control the differentiation, the growth arrest and the apoptosis of neuron cells [30, 52, 53], develop the ventral motor neurons by the transcribed regulation of Pax6 and Olig2, modulate the regeneration of ventral motor neurons [58, 59].
In this study, we found that the significant expression decrease and redistribution of ALDH1A2 in some anatomic regions of adult spinal cord were potentially associated with the neuron death in the ALS-like disease (Figures 9, 10). The possible mechanisms that ALDH1A2 participated in the pathogenesis of ALS exerted through modulating and controlling the elimination of aldehydes toxicity, the formation, proliferation and differentiation of neural stem cells, the differentiation, the growth arrest and the apoptosis of neuron cells as well as the regeneration of ventral motor neurons of spinal cord.
Acknowledgements
This work was supported by the Committee of National Natural Science Foundation of China (30560042, 81160161 and 81360198), Education Department of Jiangxi Province (GJJ13198) and Jiangxi Provincial Department of Science and Technology ([2014]-47, 20142BBG70062).
Authors' contributions
H.L. R.X. and Y.D. designed the experiments and analyzed the data. C.W. isolated the spinal cord. C.W. L.Z. and W.G. bred the animal. H.L. C.W. J.Z. and C.T. performed all of the immunohistochemical experiments. H.L. and R.X. wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ludolph AC, Brettschneider J, Weishaupt JH. Amyotrophic lateral sclerosis. Current opinion in neurology. 2012;25:530-5
2. Carrì MT, D'Ambrosi N, Cozzolino M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochemical and biophysical research communications. 2016
3. Lee JM, Tan V, Lovejoy D. et al. Involvement of quinolinic acid in the neuropathogenesis of amyotrophic lateral sclerosis. Neuropharmacology. 2017;112(Pt B):346-64
4. Libro R, Bramanti P, Mazzon E. The role of the wnt canonical signaling in neurodegenerative diseases. Life sciences. 2016;158:78-88
5. Monahan Z, Shewmaker F, Pandey UB. Stress granules at the intersection of autophagy and ALS. Brain research. 2016;1649(Pt B):189-200
6. Rozas P, Bargsted L, Martínez F, Hetz C, Medinas DB. The ER proteostasis network in ALS: Determining the differential motoneuron vulnerability. Neuroscience letters. 2017;636:9-15
7. Bozzo F, Mirra A, Carrì MT. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neuroscience letters. 2017;636:3-8
8. Kaur SJ, McKeown SR, Rashid S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene. 2016;577:109-18
9. Ratti A, Buratti E. Physiological Functions and Pathobiology of TDP-43 and FUS/TLS proteins. Journal of neurochemistry. 2016;138:95-111
10. Sasaguri H, Chew J, Xu YF, Gendron TF, Garrett A, Lee CW. et al. The extreme N-terminus of TDP-43 mediates the cytoplasmic aggregation of TDP-43 and associated toxicity in vivo. Brain research. 2016;1647:57-64
11. Mis MS, Brajkovic S, Tafuri F, Bresolin N, Comi GP, Corti S. Development of Therapeutics for C9ORF72 ALS/FTD-Related Disorders. Molecular neurobiology. 2016
12. Todd TW, Petrucelli L. Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions. Journal of neurochemistry. 2016;138:145-62
13. Parakh S, Atkin JD. Protein Folding Alterations in Amyotrophic Lateral Sclerosis. Brain research. 2016;1648(Pt B):633-49
14. Ruegsegger C, Saxena S. Proteostasis impairment in ALS. Brain research. 2016;1648(Pt B):571-9
15. Douglas PM, Cyr DM. Interplay between protein homeostasis networks in protein aggregation and proteotoxicity. Biopolymers. 2010;93:229-36
16. Scior A, Juenemann K, Kirstein J. Cellular strategies to cope with protein aggregation. Essays in biochemistry. 2016;60:153-61
17. Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nature Reviews Neurology. 2011;7:616-30
18. Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M. et al. Controversies and priorities in amyotrophic lateral sclerosis. lancet neurology. 2013;12:310-22
19. Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Experimental neurology. 2014;262(PtB):91-101
20. Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nature neuroscience. 2014;17:17-23
21. Ticozzi N, Ratti A, Silani V. Protein aggregation and defective RNA metabolism as mechanisms for motor neuron damage. CNS & neurological disorders-drug targets. 2010;9:285-96
22. Verma A, Tandan R. RNA quality control and protein aggregates in amyotrophic lateral sclerosis: a review. Muscle & nerve. 2013;47:330-8
23. Heutink P, Jansen IE, Lynes EM. C9orf72; abnormal RNA expression is the key. Experimental neurology. 2014;262(Pt B):102-10
24. Paez-Colasante X, Figueroa-Romero C, Sakowski SA, Goutman SA, Feldman EL. Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nature Reviews Neurology. 2015;11:266-79
25. Pasquali L, Lenzi P, Biagioni F, Siciliano G, Fornai F. Cell to cell spreading of misfolded proteins as a therapeutic target in motor neuron disease. Current medicinal chemistry. 2014;21:3508-34
26. Jackson B, Brocker C, Thompson DC, Black W, Vasiliou K, Nebert DW. et al. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Human Genomics. 2011;5:283-303
27. Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem cell reviews and reports. 2001;7:292-306
28. Marcato P, Dean CA, Giacomantonio CA, Lee PWK. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2001;10:1378-84
29. Hartomo TB, Van Huyen Pham T, Yamamoto N, Hirase S, Hasegawa D, Kosaka Y. et al. Involvement of aldehyde dehydrogenase 1A2 in the regulation of cancer stem cell properties in neuroblastoma. International journal of oncology. 2015;46:1089-98
30. Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert opinion on drug metabolism & toxicology. 2008;4:697-720
31. Moreb JS, Ucar D, Han S, Amory JK, Goldstein AS, Ostmark B. et al. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chemico-biological interactions. 2012;195:52-60
32. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD. et al. Motor neurondegeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772-5
33. Zhou Y, Lu Y, Fang X, Zhang J, Li J, Li S. et al. An astrocyte regenerative response from vimentin-containing cells in the spinal cord of amyotrophic lateral sclerosis's disease-like transgenic (G93A SOD1) mice. Neurodegenerative diseases. 2015;15:1-12
34. Henriques A, Pitzer C, Schneider A. Characterization of a novel SOD-1(G93A) transgenic mouse line with very decelerated disease development. PLoS One. 2010;5:e15445
35. Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proceedings of the nutrition society. 2004;63:49-63
36. Ono Y, Fukuhara N, Yoshie O. TAL1 and LIM-only proteins synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute lymphoblastic leukemia by acting as cofactors for GATA3. Molecular and cellular biology. 1998;18:6939-50
37. Niederreither K, Subbarayan V, Dolle P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nature genetics. 1992;21:444-8
38. Hsu LC, Chang WC, Yoshida A. Mouse type-2 retinaldehyde dehydrogenase (RALDH2): genomic organization, tissue-dependent expression, chromosome assignment and comparison to other types. Biochimica et biophysica acta. 2000;1492:289-93
39. Niederreither K, Fraulob V, Garnier JM, Chambon P, Dollé P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mechanisms of development. 2002;110:165-71
40. Wang X, Penzes P, Napoli JL. Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. Journal of biological chemistry. 1996;271:16288-93
41. Zhao D, McCaffery P, Ivins KJ, Neve RL, Hogan P, Chin WW. et al. Molecular identification of a major retinoic-acid-synthesizing enzyme, a retinaldehyde-specific dehydrogenase. European journal of biochemistry. 1996;240:15-22
42. Gagnon I, Duester G, Bhat PV. Kinetic analysis of mouse retinal dehydrogenase type-2 (RALDH2) for retinal substrates. Biochimica et biophysica acta. 2002;1596:156-62
43. Maden M. Heads or tails? Retinoic acid will decide. Bioessays. 1999;21:809-12
44. Mic FA, Molotkov A, Molotkova N, Duester G. Raldh2 expression in optic vesicle generates a retinoic acid signal needed for invagination of retina during optic cup formation. Developmental dynamics. 2004;231:270-7
45. Martín M, Gallego-Llamas J, Ribes V, Kedinger M, Niederreither K, Chambon P. et al. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Developmental biology. 2005;284:399-411
46. Cartry J, Nichane M, Ribes V, Colas A, Riou JF, Pieler T. et al. Retinoic acid signalling is required for specification of pronephric cell fate. Developmental biology. 2006;299:35-51
47. Desai TJ, Chen F, Lü J, Qian J, Niederreither K, Dollé P. et al. Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Developmental biology. 2006;291:12-24
48. Ribes V, Wang Z, Dolle P, Niederreither K. Retinaldehyde dehydrogenase 2 (RALDH2)-mediated retinoic acid synthesis regulates early mouse embryonic forebrain development by controlling FGF and sonic hedgehog signaling. Development. 2006;133:351-61
49. Deak KL, Dickerson ME, Linney E, Enterline DS, George TM, Melvin EC. et al. Analysis of ALDH1A2, CYP26A1, CYP26B1, CRABP1, and CRABP2 in human neural tube defects suggests a possible association with alleles in ALDH1A2. Birth defects research part a-clinical and molecular teratology. 2005;73:868-75
50. Nya-Ngatchou JJ, Arnold SL, Walsh TJ, Muller CH, Page ST, Isoherranen N. et al. Intratesticular 13-cis retinoic acid is lower in men with abnormal semen analyses: a pilot study. Andrology. 2013;1:325-31
51. Amory JK, Arnold S, Lardone MC, Piottante A, Ebensperger M, Isoherranen N. et al. Levels of the retinoic acid synthesizing enzyme aldehyde dehydrogenase-1A2 are lower in testicular tissue from men with infertility. Fertil Steril. 2014;101:960-6
52. De Luca LM. Retinoids and their receptors in differentiation, embryogenesis, and neoplasia. FASEB J. 1991;5:2924-33
53. Kim H, Lapointe J, Kaygusuz G, Ong DE, Li C, van de Rijn M. et al. The retinoic acid synthesis gene ALDH1a2 is a candidate tumor suppressor in prostate cancer. Cancer research. 2005;65:8118-24
54. Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921-31
55. Sockanathan S, Jessell TM. Motor neuron-derived retinoid signaling specifies the subtype identity of spinal motor neurons. Cell. 1998;94:503-14
56. Diez del Corral R, Olivera-Martinez I, Goriely A, Gale E, Maden M, Storey K. Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron. 2003;40:65-79
57. Novitch BG, Wichterle H, Jessell TM, Sockanathan S. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron. 2003;40:81-95
58. Molotkova N, Molotkov A, Sirbu IO, Duester G. Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation. Mechanisms of development. 2005;122:145-55
59. Marquardt T, Pfaff SL. Cracking the transcriptional code for cell specification in the neural tube. Cell. 2001;106:651-4
60. Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385-97
61. Li XJ, Du ZW, Zarnowska ED, Pankratz M, Hansen LO, Pearce RA. et al. Specification of motoneurons from human embryonic stem cells. Nature biotechnology. 2005;23:215-21
Author contact
![]() Corresponding author: Renshi Xu, Department of Neurology, the First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, China. E-mail: xurenshiedu.cn or 13767015770com.
Corresponding author: Renshi Xu, Department of Neurology, the First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, China. E-mail: xurenshiedu.cn or 13767015770com.