Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(7):868-877. doi:10.7150/ijbs.19868 This issue Cite
Research Paper
Exposure to Concentrated Ambient Fine Particulate Matter Induces Vascular Endothelial Dysfunction via miR-21
Jianwei Dai1, 2* ![]() , Wensheng Chen1*, Yuyin Lin1, Shiwen Wang1, Xiaolan Guo1, Qian-Qian Zhang3
, Wensheng Chen1*, Yuyin Lin1, Shiwen Wang1, Xiaolan Guo1, Qian-Qian Zhang3 ![]()
1. GMU-GIBH Joint School of Life Sciences, Guangzhou Medical University, Guangzhou 510182, China;
2. The State Key Lab of Respiratory Disease, Guangzhou Institute of Respiratory Disease, The First Affiliated Hospital, Guangzhou Medical University, Guangzhou 510120, China;
3. Vascular Biology Research Institute, School of Basic Course, Guangdong Pharmaceutical University, Guangzhou 510006, China.
* These authors contribute equally to this work.
Received 2017-3-1; Accepted 2017-5-3; Published 2017-7-6
Abstract

Vascular endothelial permeability transition does not cause significant lesions, but enhanced permeability may contribute to the development of vascular and other diseases, including atherosclerosis, hypertension, heart failure and cancer. Therefore, elucidating the effect of Particulate Matter 2.5 (PM2.5) on vascular endothelial permeability could help prevent disease that might be caused by PM2.5. Our previous study and the present one revealed that PM2.5 significantly increased the permeability of vascular endothelial cells and disrupted the barrier function of the vascular endothelium in Sprague Dawley (SD) rats. We found that the effect occurred mainly through induction of signal transducer and activator of transcription 3 (STAT3) phosphorylation, further transcriptional regulation of microRNA21 (miR-21) and promotion of miR-21 expression. These changes post-transcriptionally repress tissue inhibitor of metalloproteinases 3 (TIMP3) and promote matrix metalloproteinases 9 (MMP9) expression. This work provides evidence that PM2.5 exerts direct inhibitory action on vascular endothelial barrier function and might give rise to a number of vascular diseases.
Keywords: Fine particulate matter, vascular endothelial cell, permeability, miR-21.
Introduction
Blood vessels are lined with a single layer of cells, called endothelial cells. This layer acts as a selective barrier, fulfilling a role in exchanging macromolecules between the blood and tissues. This property, called permeability, is essential for the ability of the body to respond to changes in blood pressure and inflammation. Any damage to the endothelial barrier results in pathological vascular hyperpermeability, and altered monolayer permeability occurs before every case of vascular disease or cancer [1-3]. Endothelial permeability can be enhanced via the transcellular and paracellular pathways. Blood components pass directly through the cell in the transcellular pathway, while in the paracellular pathway, components cross the endothelial barrier through intercellular cell-cell junctions [3]. Under physiological conditions, the endothelial barrier function is maintained by junctions among the endothelial cells, extracellular matrix and basal membrane. Vascular permeability, and thus the paracellular flow of fluids, electrolytes, proteins, and cells through the endothelial layer, is mainly controlled by molecules involved in cell adherence and tight junctions, which are highly relevant to the extracellular matrix. Degradation of the extracellular matrix could directly potentiate enhanced permeability of the endothelium [4].
The exchange of macromolecules between the blood and tissues is accomplished through the extracellular matrix and is regulated by endogenous and exogenous factors: gene expression and biomodulators. The stabilization of the dimensional structure of the extracellular matrix is due to small proteoglycans, which result in the formation of a three-dimensional lattice structure surrounding the cells. Matrix metalloproteinases (MMPs), a family of zinc-dependent endopeptidases, play vital roles in degrading basal membranes and the extracellular matrix [5]. MMP2 and MMP9, also known as collagenases, degrade type IV collagen (gelatin), degrade the extracellular matrix and junction proteins, and thereby increase endothelial permeability [6, 7]. The proteolytic activity of MMPs can be regulated at several levels, including gene expression, secretion, and conversion from the zymogen to the active enzyme [8, 9]. In addition, all activated MMPs (including MMP2 and MMP9) are specifically inhibited by endogenous tissue inhibitors of metalloproteinases (TIMPs) [5, 8].
MicroRNAs (miRNAs) are a family of small non-coding RNA molecules that regulate the expression of protein-coding genes at the post-transcriptional and translational level. Each miRNA can repress multiple gene targets, and repression occurs by translational inhibition. Among the regulatory mechanisms that control gene expression patterns, microRNAs post-transcriptionally regulate gene expression by binding to sequences on the 3'untranslated region (3'UTR) with perfect or imperfect complementarity, resulting in cleavage or translational repression of the target mRNA. Recently, miRNAs have been proposed to play a role in regulating intestinal tight junction permeability [10]. Other reports demonstrated that microRNAs were up-regulated in patients with ulcerative colitis and that microRNA over-transcription led to intestinal epithelial barrier impairment [11, 12]. Furthermore, S-Z Chen reported that microRNA could lead to extracellular matrix remodeling through Adamts1 [13], resolving the question of whether microRNA can degrade the extracellular matrix by acting through a protein.
Recently, both epidemiological and clinical research has demonstrated that short- and long-term exposure to ambient particulate matter that is <2.5 μm in aerodynamic diameter (PM2.5, fine particles) is linked to an increase in cardiovascular disease. In the atmosphere, PM2.5 can reach the alveolar regions of the lung. Several mechanisms have been proposed to explain how inhalation of PM and diesel exhaust particles (DEPs) can cause cardiovascular health effects. However, there are few reports regarding the effects of PM on endothelial permeability. Previously, we collected PM2.5 from Beijing, consisting of heavy metals and polycyclic aromatic hydrocarbons (PAHs). Although we found that concentrated PM2.5 decreased membrane integrity and enhanced permeability in vascular endothelial cells (VECs) [14], the underlying mechanisms have yet to be fully determined. Herein, we focus on the role of miRNAs in increased vascular endothelial permeability resulting from PM2.5 exposure.
Materials and Methods
Chemicals and reagents
PM2.5 samples were collected from Beijing and analyzed as previously described [14]. Fluorescein isothiocyanate dextran (FITC-dextran) was obtained from Santa Cruz Biotechnology (CAS 60842-46-8 Dallas, Texas, USA). Evans Blue was obtained from Dingguo (AR-0762, Guangzhou, Guangdong, China). The following primary antibodies were used for the Western blotting (WB) assay and the immunohistochemistry (IHC) assay: rabbit anti-STAT3 (4904T, diluted to 1:2000, Cell Signaling Technology, Inc., MA, USA), rabbit anti-p-STAT3 (BA1709, diluted at 1:100 for IHC and 1:400 for WB, Boster, Wuhan, China), rabbit anti-TIMP3(ab39184, diluted at 1:100 for IHC and 1:1000 for WB, Abcam, Cambridge, USA), mouse anti-MMP9 (sc-21733, diluted at 1:100 for IHC and 1:500 for WB, Santa Cruz, Texas, USA), rabbit anti-VE-cadherin (BA3032, diluted at 1:100 for IHC and 1:400 for WB, Boster, Wuhan, China) and rabbit anti-β-actin(20536-1-AP, diluted at 1:2000 for WB, Chicago, USA). miR-21 mimics and inhibitor and negative control RNA (NC) were obtained from RiboBio Co., Ltd., China.
Animals and treatment
SD rats (6 weeks old, male) were purchased from the Guangzhou University of Chinese Medicine (Guangzhou, Guangdong, China). All the rats were housed on a 12:12-h day/night cycle at aroom temperature of 24 ± 2°Cin the Center of Laboratory Animals, Guangzhou Medical University [permitnumber: SYXK (Yue) 2015-0104]. All the experiments were carried out according to protocols approved by the Ethics Committee of the Center of Laboratory Animals at Guangzhou Medical University. All the surgeries were performed under 10% chloral hydrate, and all possible efforts were made to minimize suffering.
Cell lines and transfection
Human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical veins (Guangzhou Medical College Affiliated Hospital) as previous described [15] and were maintained in endothelial cell growth medium EBM (CC-3162, Lonza). HEK-293T cells were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China) and maintained in 1640 medium (12633012, GIBCO), which was supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 mg/mL). All the cells were incubated in a humidified chamber at 37°C supplemented with 5% CO2.
In vivo endothelial permeability assays
Twelve SD rats were randomly divided into two groups, namely, a normal saline (NS) group and a PM2.5 group. The rats were exposed to PM2.5 or normal saline via trachea drip at a dose of 4 mg/kg body weight every 3 days for 36 days. Then, 2% Evans Blue (solution in normal saline) at a dose of 50 mg/kg body weight was injected via the tail vein into the rats, which were sacrificed 90 min later. Normal saline was injected from the apex, after which the arcus aortae was removed. The dye was extracted from the arcus aortae into formamide at 60°C for 24 h, and the absorbance was measured at 620 nm.
In vitro FITC-dextran transwell assay
HUVEC monolayers were planted on transwell inserts, which were washed with EBM medium, then cultured until confluent. The cells were exposed to PM2.5 or vehicle for 24 h, and FITC-dextran was added to the top chamber. Samples were removed from the bottom chamber 24 h later and analyzed with a fluorometer at an excitation of 485 nm and an emission of 620 nm. Data represent the mean of three independent experiments.
miRNA expression analysis
Total RNAs were extracted using TRIzol reagent (15596-018, Invitrogen) from HUVECs or aortic arches of SD rats treated with normal saline, PM2.5(100 μg/mL), NC or miR-21 inhibitor. The miRNA expression was analyzed using the Affymetrix GeneChip miRNA 1.0 Array (RiboBio Co., Ltd., China).The data analysis was carried out according to previously reported procedures[16]. The expression of miR-21 was analyzed using a real-time quantitative PCR (qRT-PCR) assay. The total RNA (500 ng) was reverse transcribed using a PrimeScriptTM RT Reagent Kit (RR014A, TaKaRa, Japan), and the real-time PCR was performed on a 7500 Real-Time PCR System (ABI) using the SYBR® Premix ExTaq™ II (TliRNaseH Plus) PCR Kit (RR820A, TaKaRa, Japan). All the primers were obtained from RiboBio Co., Ltd., China. The data were analyzed using the 2-ΔΔCT methodology as previously described [17].
Luciferase assay
The TIMP3 3'UTR was predicted using RNA22, while the human miR-21 promoter was predicted using the Promoter 2.0 prediction server, and both were then amplified by PCR from the cDNA or genomic DNA of HUVECs. The PCR products were digested and inserted into psiCHECK-2 or pGL3-Basic vectors. Mutations were introduced within the TIMP3 3'UTR or STAT3 and miR-21 binding sites via PCR with mutation site primers. The pRL-TK vector was co-transfected with the pGL3-Basic constructs and used as a spike-in control to normalize transfection efficiency. After transfection for 24 h, the activity was measured using a Dual-Glo™ Luciferase Assay System (E2920, Promega, USA) according to the manufacturer's protocol. All primer sequences are provided in the supplementary material (Table S1).
Immunoblot analysis
Proteins were collected using lysis buffer (50 mM Tris-HCl, pH 8.8, 150 mM NaCl, 0.1% SDS, 1% NP-40, 1% sodium deoxycholate, 1% PMSF, 1% phosphotransferase inhibitor), and the concentration was estimated by BCA quantification. Total protein (30 μg) was separated on an SDS-PAGE gel to analyze the expression levels of TIMP3, MMP9, STAT3, and p-STAT3 according to previously described procedures [18]. β-Actin was used as a loading control to normalize protein levels.
Histological and immunohistological staining
The arcus aortaesamples from NS- and PM2.5-treated rats were fixed in 4% paraformaldehyde overnight and then embedded in optimal cutting temperature (OCT) compound and sectioned. The sections were stained with hematoxylin & eosin (H&E) and IHC. For IHC staining, the sections were incubated with primary antibodies against TIMP3, MMP9 or VE-cadherin at 4°C overnight, washed with PBS and incubated with HRP-conjugated second antibody, then stained with DAB and counterstained with hematoxylin.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using the ChIP-IT Express Enzymatic Kit (53035, Active Motif, China) according to the manufacturer's instructions. In brief, chromatin from cells was cross-linked with 1% formaldehyde (10 min at 22°C), sheared to an average size of ~500 bp, and then immunoprecipitated with anti-STAT3 antibody. The ChIP-PCR primers were designed using Primer Premier 5.
Statistical analysis
Statistical significance was assessed using SPSS 16.0. All data are presented as the mean ± standard deviation (S.D.) from 3 independent experiments. Pairwise differences between groups were analyzed using a two-sided Student's t-test. Statistical significance was defined as P<0.05. The intensities of immunoblotting bands were quantitated and normalized to β-Actin using Quantity One software (Bio-Rad, USA). IPP software (Media Cybernetics, Inc., USA) was used to quantify the protein expression levels in the IHC staining slices by measuring the cumulative integrated optical density (IOD).
Results
Exposure to PM2.5 alters the permeability of the aortic arch in SD rats
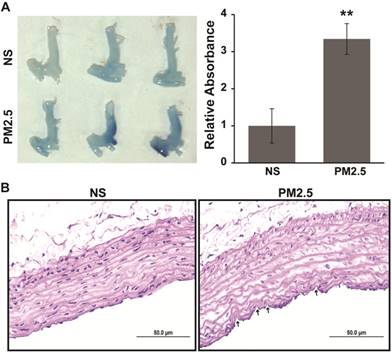
In our previous work, it has been demonstrated that the permeability of vascular endothelial cells significantly increased upon exposure to PM2.5 in vitro [14]. In this study, Evans Blue dye was injected via the tail vein to examine whether the vascular permeability could also increase upon exposure to PM2.5 in vivo. We showed that PM2.5 exposure accelerated the accumulation of Evans blue in the region of the aortic arch (Fig. 1A). This work further demonstrated that exposure to PM2.5 could significantly increase vascular permeability. In addition, to explore the effect of PM2.5 exposure on vascular structure, a detailed histological analysis of the aortic arches of NS- or PM2.5-treated rats was performed. As revealed in Fig. 1B, mild injury could be observed in the vascular endothelium. These results indicate that PM2.5 increased the vascular permeability and injured the vascular endothelial structure in vivo.
Aortic arch permeability alteration in SD rats exposed to PM2.5. PM2.5 or normal saline was injected PM2.5 intratracheally at 4 mg/kg body weight in each SD rat once every 3 days for 4 weeks. (A) 2% Evans Blue (solution in normal saline) was injected into the caudal vein of SD rats, and the aortic arch was lavaged using normal saline and photographed after 90 min, after which the Evans Blue was extracted with formamide at 60 ˚C for 24 h and quantified at 620 nm. (B) Hematoxylin and eosin staining analysis of the aortic arch from SD rats treated or untreated with PM2.5. Labels indicate areas of endothelial injury. **P< 0.01. Bar = 50 μm.
PM2.5 promotes vascular endothelial cell permeability by up-regulating the expression of miR-21
A previous report has indicated that miRNA plays a key role in regulating vascular endothelial barrier function [19]. Therefore, differential expression analysis of miRNAs was carried out with an Affymetrix GeneChip miRNA 1.0 Array to detect the potential miRNAs involved in the alteration of vascular endothelial cell permeability by PM2.5. As shown in Fig. 2A and 2B, the analysis results revealed that 11 miRNAs were significantly up-regulated and 4 miRNAs were down-regulated in PM2.5-treated compared with NS-treated HUVECs(>2-fold change, P<0.01). In addition, miR-21 was the most significantly changed miRNA during the PM2.5-induced increase of vascular endothelial cell permeability, and the expression of miR-21 was further verified by qRT-PCR (Fig. 2C). Moreover, the expression of miR-21 was significantly up-regulated in the aortic arches of PM2.5-treatedrats compared with the NS-treated group (Fig. 2D).
miR-21 is involved in the PM2.5-induced increase in vascular endothelial cell permeability. Total RNAs were extracted from NS- or PM2.5-treated HUVECs and aortic arches of SD rats. (A) Affymetrix GeneChip miRNA 1.0 Array analysis of the differential expression miRNAs in NS or PM2.5 treated HUVECs. Hierarchical clustering was performed using 84 miRNAs selected from among 2,536 miRNAs. Each row corresponds to one miRNA, and the two columns correspond to the NC and PM2.5 groups. (B) Volcano plot of miRNA analysis. Plots of log P value versus log2fold change for HUVECs treated and untreated with PM2.5. The two vertical lines in each panel mark twofold changes. Red spots indicate miRNAs with highly significant fold changes. Labels indicate miR-21. (C) miR-21 expression was verified using qRT-PCR from HUVECs treated or untreated with PM2.5 (80 µg/mL) for 24 h. (D) The expression of miR-21 in the aortic arches of rats treated with NS or PM2.5 was measured using qRT-PCR. (E) Effect of PM2.5 on HUVEC monolayer permeability. Cells were cultured until confluent in transwell inserts and incubated with or without the miR-21 inhibitor for 24 h and then stimulated with PM2.5 (80 µg/mL) or left untreated for 24 h, and then FITC-dextran was added to the top chamber. Samples were removed from the bottom chamber 24 h later and analyzed with a fluorometer.*P< 0.05, ***P< 0.001.
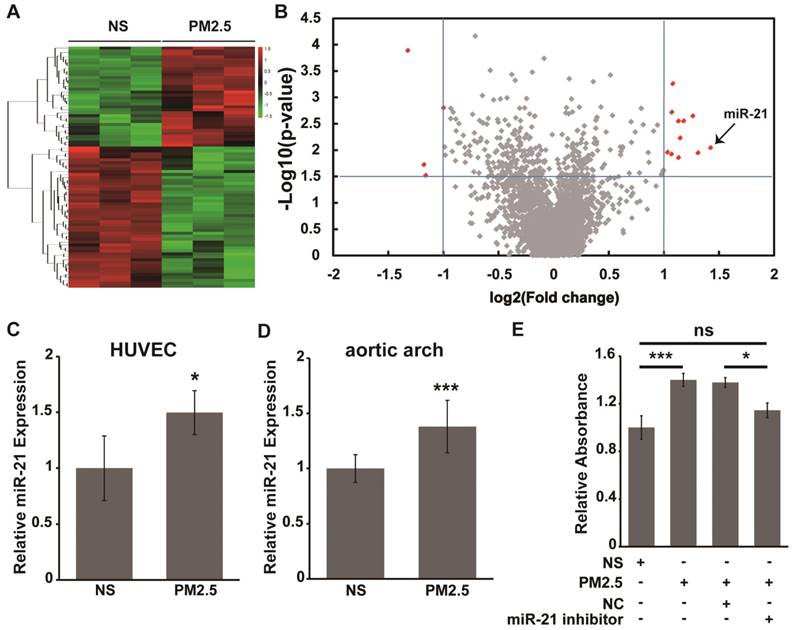
However, whether miR-21 is involved in the alteration of vascular endothelial cell permeability by PM2.5 remains to be further clarified. FITC-dextran was employed to confirm endothelial cell permeability after exposure with PM2.5. As shown in Fig. 2E, the diffusion of FITC-dextran across the HUVEC sheet was significantly after PM2.5 treatment compared with NS treatment. However, miR-21 inhibitor co-applied with PM2.5 could reverse the action of PM2.5 on the permeability of HUVECs. These data suggest that miR-21 is involve in the acceleration of vascular endothelial cell permeability induced by PM2.5.
TIMP3/MMP9 signaling is the direct target of miR-21 in PM2.5-induced increasing of vascular endothelial cell permeability
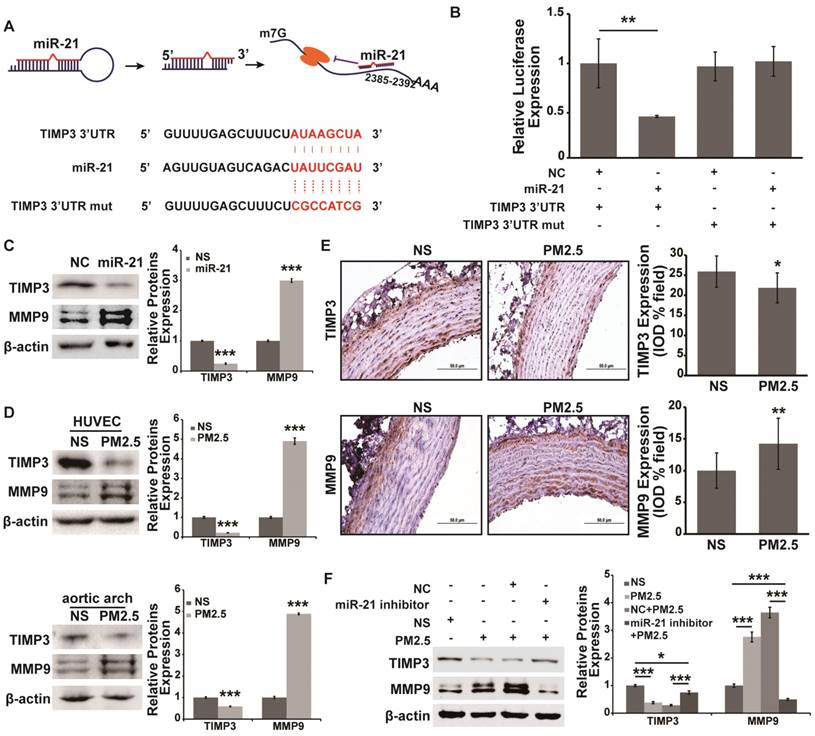
A previous report has indicated that the angiogenic effect of miR-21 acts by targeting TIMP3/MMP signaling [20]. Moreover, TIMP3/MMP9 signaling plays an important role in regulating vascular permeability [21, 22]. However, whether TIMP3/MMP9 signaling is involved in miR-21-mediated increasing of vascular endothelial cell permeability under PM2.5 treatment is unclear. The analysis result in RNA22 showed that TIMP3 was the potential direct target of miR-21 (Fig. 3A). A luciferase reporter assay was carried out to determine the direct binding of miR-21 to the TIMP3 3'UTR, and the assay showed that miR-21, compared with the control treatment, significantly suppressed the expression of Renilla luciferase carrying the wild-type target site of TIMP3 (Fig. 3B). In addition, the effect was reversed when the seed sequences complementary to the miR-21 were mutated (Fig. 3B). Furthermore, the protein expression of TIMP3 and MMP9 was measured and miR-21 significantly inhibited TIMP3 expression and up-regulated MMP9 expression (Fig. 3C). All the results indicated that TIMP3/MMP9 signaling is the target pathway of miR-21. However, whether TIMP3/MMP9 signaling takes part in the PM2.5-induced increase of vascular endothelial permeability through miR-21 is not known. Therefore, we measured the expression of TIMP3 and MMP9 in PM2.5-treated HUVECs and rat aortic arches. It was shown that PM2.5 treatment could significantly inhibit TIMP3 expression and increase MMP9 expression both in HUVECs and in aortic arches (Fig. 3D and 3E). In addition, a miR-21 inhibitor co-applied with PM2.5 could reverse the alteration of TIMP3 and MMP9 expression in HUVECs (Fig. 3F). These data demonstrated that TIMP3/MMP9 signaling is the direct target of miR-21 in the PM2.5-induced increase in vascular endothelial permeability.
TIMP3/MMP9 is the direct target of miR-21 and is involved in miR-21-induced increases in vascular endothelial cell permeability underexposure to PM2.5. (A) The formation and regulation of miR-21 in inhibiting TIMP3 translation, and the putative binding site and mutation site of miR-21 in the TIMP3 3'UTR predicted by RNA22. (B) Luciferase assays indicated that miR-21 directly down-regulated the expression of TIMP3. psi-check2-TIMP3 3'UTR vector and the mutation vector were built, and then relative luciferase values were normalized to the group cotransfected with the NC. Renilla luciferase activity was normalized to firefly luciferase expression. (C) Western blotting shows the expression of TIMP3 and MMP9 in miR-21-overexpressing HUVECs. Western blotting (D) and IHC staining (E) show the expression of TIMP3 and MMP9 in NS- and PM2.5-treated HUVECs and aortic arches of SD rats. (F) Western blotting shows that the expression of TIMP3 was inhibited and MMP9 was up-regulated by PM2.5, and the effect was abolished by restraint of miR-21 in HUVECs. β-actin served as an internal control.*P< 0.05,**P< 0.01, ***P< 0.001. Bar = 50 μm.
VE-cadherin is also involved in the miR-21-mediated increase of vascular endothelial cell permeability underPM2.5 treatment
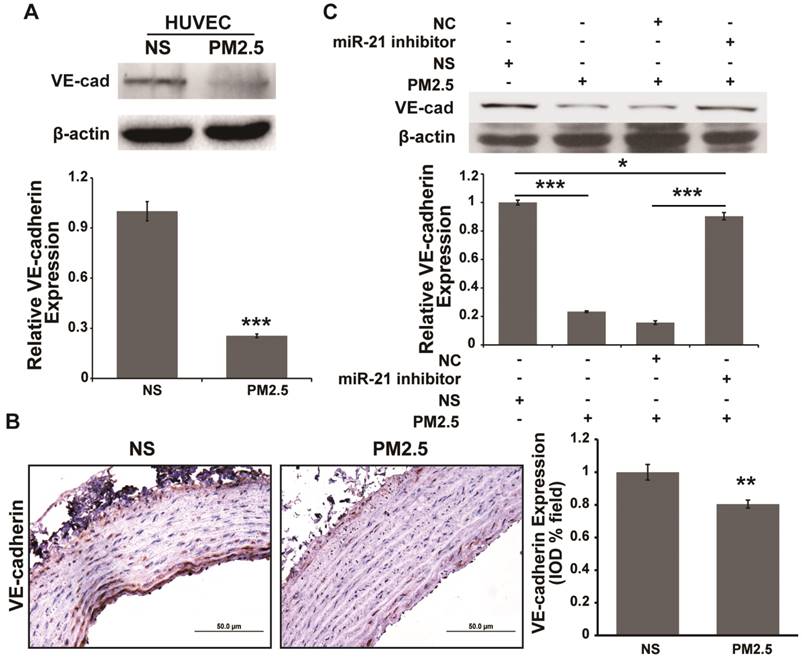
It has been reported that vascular endothelial cadherin (VE-cadherin) down-regulation could compromise barrier integrity in HUVECs [23, 24]. Reports indicated that miR-21 could indirectly regulate E-cadherin expression, which inhibits the epithelial to mesenchymal transition (EMT) [25, 26]. E-cadherin and VE-cadherin are both members of the cadherin family. Therefore, whether miR-21 regulates VE-cadherin expression in the PM2.5-induced increase of vascular endothelial permeability should be investigated. We found that PM2.5 treatment could significantly inhibit VE-cadherin expression in HUVECs and aortic arches (Fig. 4A and 4B). In addition, the inhibitory effect could be reversed by co-treatment with miR-21 inhibitor and PM2.5 in HUVECs (Fig. 4C). These data demonstrated that miR-21 could also target VE-cadherin to regulate vascular endothelial permeability under PM2.5 treatment.
VE-cadherin is regulated by miR-21 and is involved in PM2.5-induced increases in vascular endothelial cell permeability. Western blotting (A) and IHC staining (B) show the expression of VE-cadherin was inhibited in PM2.5-treated HUVECs and aortic arches of SD rats. (C) Western blotting shows that the expression of VE-cadherin was inhibited by PM2.5, and the effect was abolished by restraint of miR-21 in HUVECs. β-actin served as an internal control.*P< 0.05,**P< 0.01, ***P< 0.001. Bar = 50 μm.
STAT3 promotesmiR-21 transcription in response toPM2.5 treatment
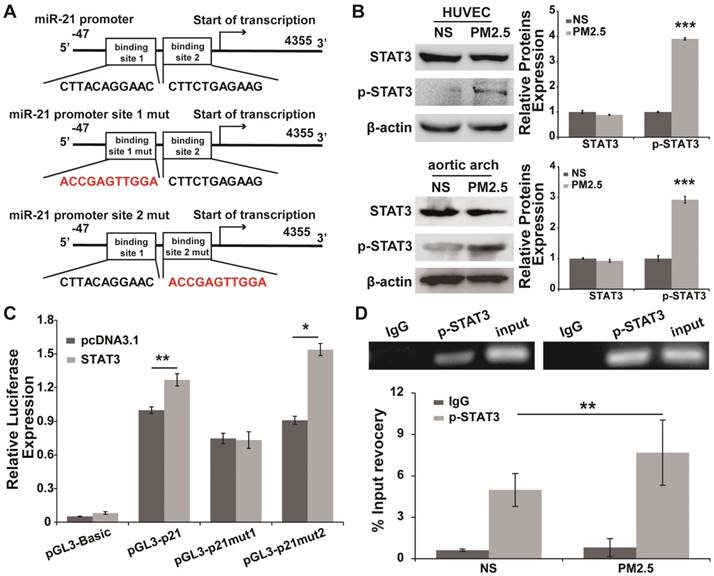
Putative transcription factor binding sites associated with the miR-21 promoter were predicted, and STAT3, a transcription factor may directly bind to the miR-21 promoter at two different sites (Fig. 5A). Additionally, STAT3 canactivatemiR-21 in cancer cells [25, 27]. Whether STAT3 promotes the transcription of miR-21 under PM2.5 treatment needs to be further confirmed. We found that the expression of STAT3 in HUVECs and aortic arches did not differ between the NS and PM2.5 groups, and the expression of p-STAT3 was significantly up-regulated in PM2.5-treated HUVECs and aortic arches compared with the NS group (Fig. 5B). The similar expression trend of miR-21 and p-STAT3 suggested that p-STAT3 might regulate the transcription of miR-21 in PM2.5-treated HUVECs compared with NS-treated ones. The luciferase reporter assay showed that STAT3 could significantly promote the expression of luciferase that carried the wild-type promoter of miR-21 compared with the control group (Fig. 5C). Remarkably, binding site 1, but not binding site 2, was a crucial site, because its mutation completely abolished the effects of p-STAT3 on the miR-21 promoter reporter (Fig. 5C). This conclusion was further confirmed by a chromatin immunoprecipitation assay. As shown in Fig. 5D, p-STAT3 directly binds to binding site 1 of the miR-21 promoter in HUVECs, and PM2.5 treatment could promote the binding effect of p-STAT3 in miR-21 promoter. Together, these results showed that miR-21 functions as an effector of p-STAT3 in PM2.5-treated HUVECs.
p-STAT3 is involved in the promotion of transcriptional regulation of miR-21 in PM2.5-treated HUVECs. (A) Schematic representation of predicted binding sites of STAT3 in the human miR-21 gene and information on the STAT3 binding site mutations in the miR-21 promoter region. (B) Western blotting shows that p-STAT3 was up-regulated by PM2.5, but there was no differential expression of STAT3 in NS- and PM2.5-treated HUVECs or aortic arches of SD rats. β-Actin served as an internal control. (C) Luciferase assays show the effects of STAT3 on the reporter constructs containing the wild-type and mutant miR-21 promoter. pGL3-miR-21 promoter and the mutation vectors were built, and then the constructs were co-transfected into HEK-293T cells with vectors expressing STAT3 as well as the pRL-TK vector as a transfection control. A vector expressing red fluorescence protein (dsRed) serves as a negative control for STAT3. The relative luciferase activities are the ratio of Firefly/Renilla luciferase normalized to the negative control. The data of the luciferase assays are presented as the mean ± SD from three separate experiments. (D) Chromatin immunoprecipitation assays show the in vivo interaction between p-STAT3 and miR-21 promoter binding site 1 as well as the increasing binding induced by PM2.5. HUVEC chromatin fragments were immunoprecipitated with antibodies against p-STAT3 or a negative control antibody (normal human immunoglobulin). HUVECs were treated or untreated with PM2.5 (80 µg/mL) for 24 h. The ChIP-enriched DNA levels were measured by qPCR and then normalized to the quantity of input DNA, followed by subtraction of nonspecific binding as determined by control IgG. All results represent three independent experiments, and the data are expressed as the mean±SD. *P< 0.05,**P< 0.01, ***P< 0.001.
Discussion
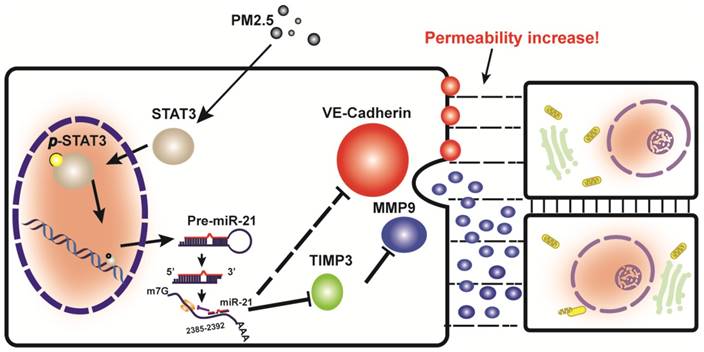
This study provided support for a model in which exposure to PM2.5 can up-regulate the expression of p-STAT3, which is involved in promoting the transcriptional regulation of miR-21. It directly inhibits TIMP3/MMP9 signaling and indirectly inhibits VE-cadherin expression, both of which are involved in the control of vascular endothelial cell permeability (Fig. 6).
A schematic illustration of how PM2.5 might increase endothelial permeability.
It has been demonstrated that the small-diameter particulate matter known as PM2.5 can enter the respiratory system and diffuse into the blood circulation, after which it induces endothelial dysfunction in pulmonary and systemic arteries [28-30]. Vascular permeability refers the capacity of a blood vessel wall to allow the passage of small molecules or whole cells in and out of the vessel. A single layer of endothelial cells line the inner layer of blood vessel walls. Therefore, the endothelial cell is the first barrier that maintains vascular permeability. Our previous study clarified that PM2.5 disrupts vascular endothelial cell barrier function [14]. Therefore, the totality of the evidence suggested that PM2.5 may induce endothelial dysfunction through direct damage to vascular endothelial cells. In this work, we observed that the inner layer of the blood vessel wall was injured and the permeability of the vascular endothelium was increased by PM2.5. The dysfunction of the vascular endothelium as a barrier can contribute to the development of atherosclerosis, hypertension, heart failure and cancer [31-34]. Our in vivo work is consistent with previously reported effects of PM2.5 on endothelial dysfunction and further demonstrate that the barrier function of vascular endothelial was abolished by PM2.5, suggesting that PM2.5 might induce cardiovascular diseases by altering vascular endothelial cell permeability.
MicroRNAs are small non-coding RNAs that bind to the 3'UTR of target genes and leading to translational repression. Recent research has shown that miR-147b regulates endothelial permeability [19]. However, whether there are more miRNAs involved in the regulation of endothelial permeability remains to be further explored. Our in silico analyses have identified 15 miRNAs whose expression were markedly changed in PM2.5-treated HUVECs. Since miR-21 is the most significantly changed miRNA among them, it became the focus of this study. It has been demonstrated that miR-21 plays an important role in tumors and heart disease [35-40]. In addition, miR-21 is involved in the regulation of intestinal permeability, angiogenesis and retinal blood vessel caliber upon exposure to PM10 [20, 41-43].Although miR-21 has been identified as an important regulator of blood vessel and intestinal permeability, there is no information available concerning its function in vascular endothelial cell permeability. Thus, the purpose of our study is to clarify this novel role of miR-21 in vascular endothelial cells with regard to permeability regulation. In vivo and in vitro studies have indicated that changes in miR-21 expression are associated with exposure to PM2.5. We were the first to confirm associations between expression of miR-21 and vascular endothelial cell permeability.
The mechanisms by which miR-21 up-regulation alters endothelial barrier function in response to PM2.5 exposure are not well understood. TIMP3/MMP9 signaling is the direct target of miR-21 in angiogenesis [20]. Recently, studies have indicated that TIMP3/MMP9 signaling and VE-cadherin are known to be dominant pathways for maintaining the endothelial barrier [44]. In this study, we clarified that miR-21 promotes vascular endothelial cell permeability by not only inhibiting TIMP3/MMP9 signaling but also suppressing VE-cadherin.
In summary, the current study reports miR-21 as a novel target of PM2.5 through which it alters vascular endothelial cell permeability to disrupt vascular barrier function. Our data suggest that targeting of TIMP3/MMP9 signaling and VE-cadherin by miR-21 promotes vascular endothelial cell permeability.
Supplementary Material
Table S1.
Acknowledgements
The authors thank Dr Xiaodong Fu for providing excellent technical assistance.
Funding
This research was supported by the National Natural Science Foundation of China (81670040), the Natural Science Foundation of Guangdong Province, China (2014A030313582 and 2016A030313601), the Science and Technology Planning Project of Guangzhou (2015A030302083), the Science and Technology Foundation of Guangzhou, China (201510010076, 201504010018), and the Pearl River S&T Nova Program of Guangzhou, China (201610010045).
Competing Interests
The authors have declared that no competing interest exists.
References
1. M. D, McDonald, Baluk P. Significance of Blood Vessel Leakiness in Cancer. CANCER RESEARCH. 2002;62:5381-5
2. van Hinsbergh VWM, van Nieuw Amerongen GP. Endothelial hyperpermeability in vascular leakage. Vascular Pharmacology. 2002;39:171-2
3. Vincent J D Krouwer, Liesbeth H P Hekking, Miriam Langelaar-Makkinje, Elsa Regan-Klapisz, Post JA. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Krouwer et al Vascular Cell. 2012;4:12
4. Aslan A, van Meurs M, Moser J. et al. Organ-Specific Differences in Endothelial Permeability-Regulating Molecular Responses in Mouse and Human Sepsis. Shock. 2017
5. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circulation research. 2003;92:827-39
6. Murphy G, Nguyenl Q, Cockettll MI. et al. Assessment of the Role of the Fibronectin-like Domain of Gelatinase A by Analysis of a Deletion Muta. B IO-ICACLHE MIFIXY. 1994;269:6632-6
7. Shipley JM, Doyle GAR, Fliszar CJ. et al. The Structural Basis for the Elastolytic Activity of the 92-kDa and 72-kDa Gelatinases. BIOLOGICAL CHEMISTRY. 1996;271:4335-41
8. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
9. Mishra A, Paul S, Swarnakar S. Downregulation of matrix metalloproteinase-9 by melatonin during prevention of alcohol-induced liver injury in mice. Biochimie. 2011;93:854-66
10. Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:1323-33
11. Cho WC. OncomiRs: the discovery and progress of microRNAs in cancers. Molecular cancer. 2007;6:60
12. McGuinn LA, Ward-Caviness CK, Neas LM3 SA. et al. MiRNAs and their potential for use against cancer and other diseases. 2007.
13. Chen SZ, Ning LF, Xu X. et al. The miR-181d-regulated metalloproteinase Adamts1 enzymatically impairs adipogenesis via ECM remodeling. Cell Death and Differentiation. 2016;23:1778-91
14. Dai J, Sun C, Yao Z, Chen W, Yu L, Long M. Exposure to concentrated ambient fine particulate matter disrupts vascular endothelial cell barrier function via the IL-6/HIF-1alpha signaling pathway. FEBS open bio. 2016;6:720-8
15. Marin V, Kaplanski G, Gres S, Farnarier C, Bongrand P. Endothelial cell culture: protocol to obtain and cultivate human umbilical endothelial cells. Journal of immunological methods. 2001;254:183-90
16. Menge T, Zhao Y, Zhao J. et al. Mesenchymal stem cells regulate blood-brain barrier integrity through TIMP3 release after traumatic brain injury. Science translational medicine. 2012;4:161ra50
17. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101-8
18. Xu H, He JH, Xiao ZD. et al. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52:1431-42
19. Chatterjee V, Beard RS Jr, Reynolds JJ. et al. MicroRNA-147b regulates vascular endothelial barrier function by targeting ADAM15 expression. PloS one. 2014;9:e110286
20. Hu J, Ni S, Cao Y. et al. The Angiogenic Effect of microRNA-21 Targeting TIMP3 through the Regulation of MMP2 and MMP9. PloS one. 2016;11:e0149537
21. Arpino V, Mehta S, Wang L. et al. Tissue inhibitor of metalloproteinases 3-dependent microvascular endothelial cell barrier function is disrupted under septic conditions. American journal of physiology Heart and circulatory physiology. 2016;310:H1455-67
22. Munjal C, Tyagi N, Lominadze D, Tyagi SC. Matrix metalloproteinase-9 in homocysteine-induced intestinal microvascular endothelial paracellular and transcellular permeability. Journal of cellular biochemistry. 2012;113:1159-69
23. Fainaru O, Adini I, Benny O. et al. Doxycycline induces membrane expression of VE-cadherin on endothelial cells and prevents vascular hyperpermeability. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:3728-35
24. Hebda JK, Leclair HM, Azzi S. et al. The C-terminus region of beta-arrestin1 modulates VE-cadherin expression and endothelial cell permeability. Cell communication and signaling: CCS. 2013;11:37
25. Sun SS, Zhou X, Huang YY. et al. Targeting STAT3/miR-21 axis inhibits epithelial-mesenchymal transition via regulating CDK5 in head and neck squamous cell carcinoma. Molecular cancer. 2015;14:213
26. Huo W, Zhao G, Yin J. et al. Lentiviral CRISPR/Cas9 vector mediated miR-21 gene editing inhibits the epithelial to mesenchymal transition in ovarian cancer cells. Journal of Cancer. 2017;8:57-64
27. Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Molecular cell. 2010;39:493-506
28. Davel AP, Lemos M, Pastro LM. et al. Endothelial dysfunction in the pulmonary artery induced by concentrated fine particulate matter exposure is associated with local but not systemic inflammation. Toxicology. 2012;295:39-46
29. Nurkiewicz TR, Porter DW, Barger M. et al. Systemic microvascular dysfunction and inflammation after pulmonary particulate matter exposure. Environmental health perspectives. 2006;114:412-9
30. Kampfrath T, Maiseyeu A, Ying Z. et al. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circulation research. 2011;108:716-26
31. Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899-906
32. Brunner H, Cockcroft JR, Deanfield J. et al. Endothelial function and dysfunction. Part II: Association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. Journal of hypertension. 2005;23:233-46
33. Christensen HM, Schou M, Goetze JP. et al. Body mass index in chronic heart failure: association with biomarkers of neurohormonal activation, inflammation and endothelial dysfunction. BMC cardiovascular disorders. 2013;13:80
34. Sima AV, Stancu CS, Simionescu M. Vascular endothelium in atherosclerosis. Cell and tissue research. 2009;335:191-203
35. Liu ZL, Wang H, Liu J, Wang ZX. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radioresistance in non-small cell lung cancer cells by targeting PTEN. Molecular and cellular biochemistry. 2013;372:35-45
36. Qi M, Liu D, Zhang S. MicroRNA-21 contributes to the discrimination of chemoresistance in metastatic gastric cancer. Cancer biomarkers: section A of Disease markers. 2017
37. Yang Y, Guo JX, Shao ZQ. miR-21 targets and inhibits tumor suppressor gene PTEN to promote prostate cancer cell proliferation and invasion: An experimental study. Asian Pacific journal of tropical medicine. 2017;10:87-91
38. Thum T, Gross C, Fiedler J. et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980-4
39. Patrick DM, Montgomery RL, Qi X. et al. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. The Journal of clinical investigation. 2010;120:3912-6
40. Dong S, Cheng Y, Yang J. et al. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. The Journal of biological chemistry. 2009;284:29514-25
41. Liu LZ, Li C, Chen Q. et al. MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1alpha expression. PloS one. 2011;6:e19139
42. Louwies T, Vuegen C, Panis LI. et al. miRNA expression profiles and retinal blood vessel calibers are associated with short-term particulate matter air pollution exposure. Environmental research. 2016;147:24-31
43. Zhang L, Shen J, Cheng J, Fan X. MicroRNA-21 regulates intestinal epithelial tight junction permeability. Cell biochemistry and function. 2015;33:235-40
44. Corada M, Liao F, Lindgren M. et al. Monoclonal antibodies directed to different regions of vascular endothelial cadherin extracellular domain affect adhesion and clustering of the protein and modulate endothelial permeability. Blood. 2001;97:1679-84
Author contact
![]() Corresponding author: Jianwei Dai (Guangzhou Medical University, Xinzao, Panyu District, Guangzhou, 511436, P.R. China. Tel: 86-20-37103208, E-mail: daijwedu.cn); Qian-Qian Zhang (Guangdong Pharmaceutical University, Waihuan Road East #280, Guangzhou Higher Education Mega Center, Guangzhou, Guangdong 510006, P.R. China. Tel: 86-20-39352397, E-mail: zhangqianqianedu.cn).
Corresponding author: Jianwei Dai (Guangzhou Medical University, Xinzao, Panyu District, Guangzhou, 511436, P.R. China. Tel: 86-20-37103208, E-mail: daijwedu.cn); Qian-Qian Zhang (Guangdong Pharmaceutical University, Waihuan Road East #280, Guangzhou Higher Education Mega Center, Guangzhou, Guangdong 510006, P.R. China. Tel: 86-20-39352397, E-mail: zhangqianqianedu.cn).