Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(8):1067-1081. doi:10.7150/ijbs.20316 This issue Cite
Research Paper
Activation of Nrf2 Attenuates Pulmonary Vascular Remodeling via Inhibiting Endothelial-to-Mesenchymal Transition: an Insight from a Plant Polyphenol
Yucai Chen1, Tianyi Yuan1,2, Huifang Zhang1, Yu Yan1, Danshu Wang1, Lianhua Fang1 ![]() , Yang Lu3, Guanhua Du2
, Yang Lu3, Guanhua Du2 ![]()
1. State Key Laboratory of Bioactive Substances and Functions of Natural Medicines;
2. Beijing Key Laboratory of Drug Targets Identification and Drug Screening;
3. Beijing Key Laboratory of Polymorphic Drugs, Institute of Materia Medica Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China.
Received 2017-3-29; Accepted 2017-7-26; Published 2017-9-3
Abstract

The endothelial-to-mesenchymal transition (EndMT) has been demonstrated to be involved in pulmonary vascular remodeling. It is partly attributed to oxidative and inflammatory stresses in endothelial cells. In current study, we conducted a series of experiments to clarify the effect of salvianolic acid A (SAA), a kind of polyphenol compound, in the process of EndMT in human pulmonary arterial endothelial cells and in vivo therapeutic efficacy on vascular remodeling in monocrotaline (MCT)-induced EndMT. EndMT was induced by TGFβ1 in human pulmonary arterial endothelial cells (HPAECs). SAA significantly attenuated EndMT, simultaneously inhibited cell migration and reactive oxygen species (ROS) formation. In MCT-induced pulmonary arterial hypertension (PAH) model, SAA improved vascular function, decreased TGFβ1 level and inhibited inflammation. Mechanistically, SAA stimulated Nrf2 translocation and subsequent heme oxygenase-1 (HO-1) up-regulation. The effect of SAA on EndMT in vitro was abolished by ZnPP, a HO-1 inhibitor. In conclusion, this study indicates a deleterious impact of oxidative stress on EndMT. Polyphenol antioxidant treatment may provide an adjunctive action to alleviate pulmonary vascular remodeling via inhibiting EndMT.
Keywords: salvianolic acid A, endothelial-to-mesenchymal transition, TGFβ1, Nrf2, pulmonary arterial hypertension, antioxidant
Introduction
Pulmonary arterial hypertension (PAH) is a progressive syndrome, which is characterized by pulmonary arterial obstruction and subsequently increased pulmonary vascular resistance. Increased afterload causes right ventricular failure and ultimately death [1, 2]. The typical characteristic of PAH is structural remodeling of the small pulmonary arteries [3]. The pathobiology of pulmonary vascular remodeling has been poorly understood, and hyperplasia of pulmonary artery smooth muscle cells and endothelial dysfunction are considered to be the main causes.
There are various hypotheses for how pulmonary vascular remodeling develops. The epithelial-mesenchymal transition (EMT) has been widely considered as a vital cellular mechanism involved in embryonic tissue differentiation, tumor progression, and fibrosis [4]. Recently, endothelial-to-mesenchymal transition (EndMT), a special form of EMT, is deemed to participate in vascular remodeling in PAH [5-7]. During EndMT, endothelial cells (ECs) lose their endothelial phenotype, and gain mesenchymal phenotype, characterized by downregulation of endothelial markers like platelet endothelial cell adhesion molecule-1 (pecam-1, CD31) and upregulation of mesenchymal markers like α-smooth muscle actin (α-SMA) and vimentin [8-11]. In this process, stimulation of TGFβ results in loss of the cell-to-cell conjunction, transforming into a more spindle, fibroblast-like shape, migrating and invading into extracellular matrices of diverse composition. There are many signal conduction pathways and mechanisms that may participate in the endothelial-to-mesenchymal transition. The TGFβ/Smad pathway is confirmed to play a key role in the EndMT process [12], and be activated in animals with monocrotaline (MCT) - and hypoxia-induced PAH as well as patients with PAH [13-15]. Recent report indicated that there is an increase of TGF-β in MCT-induced EndMT and TGF-β1was also found to induced the EndMT by application of exogenous TGF-β1[7, 16]. Beyond that, non-Smad pathways are also activated during EndMT, including small GTPases such as RhoA, which implicated in actin and microtubule cytoskeleton organization [17].
It has been reported that reactive oxygen species (ROS) induces the EndMT during oxidative stress [18, 19]. For one thing, oxidative stress promotes TGFβ1 secretion, one of the main inducers of EndMT [20]. For another, it is reported that oxidants could activate latent TGF-β directly [21]. Furthermore, many oxidants could activate the NF-κB pathway [22], and the inflammatory cytokines produced subsequently are proved to be the direct stimulant of EndMT [8, 23].
NF-E2-related factor 2 (Nrf2), is the main regulatory factor of the antioxidant response through regulating expression of a series of antioxidant enzymes, including heme oxygenase-1 (HO-1) [24, 25]. In response to oxidative stress or electrophiles, Nrf2 separates from Kelch-like ECH-associated protein 1 (Keap1), a key Nrf2 inhibitory factor of Nrf2/HO-1 pathway, translocates into the nucleus and induces the expression of antioxidant proteins [26, 27].
It is demonstrated that polyphenol may be a class of Nrf2 activator. These compounds could bind with cysteine residue of Keap1 and promote de-methylation of Nrf2 promoter [24]. A great deal of researches indicates that polyphenols possess extensive pharmacological activities in cardiovascular system [28-31], including adjunctive treatment of microcirculation protection, endothelial protection, myocardial preservation and antioxidation. Especially, salvianolic acid A (SAA) has been gaining our attention due to its broad pharmacological effects on oxidative stress and fibrotic diseases [32-38], which are closely related with PAH [39-41]. SAA can exert antioxidant effect through many pathways. First of all, as a polyphenol compound, SAA has direct radical scavenging effect [42]. On the other hand, SAA suppresses expression of NADPH oxidase 4 (Nox4), the reactive oxygen species-generating enzyme. Moreover it has been shown that SAA could upregulate Nrf2/HO-1 signaling pathway to exert its antioxidant effect [43-45].
In recent years, there has been growing evidence in effect of oxidative stress on EndMT [20, 46] in different types of cells. It is not yet known whether suppressing oxidative stress in pulmonary artery endothelial cells can attenuate EndMT and subsequent pulmonary arterial remodeling. Therefore, in the present study, we used the polyphenolic natural antioxidant, SAA, to prove the hypothesis that antioxidant treatment may be an effective way to attenuate pulmonary vascular remodeling via inhibiting the EndMT.
Methods
Reagents
SAA with a purity of >98% by HPLC analysis was provided by the Institute of Materia Medica (Beijing, China). SB431542, Hoechst 33342, monocrotaline (MCT) and 2',7'-dichlorofluorescin diacetate (DCFH-DA) were purchased from Sigma-Aldrich (Shanghai, China). Antibodies against CD31, α-SMA, HO-1, Nrf2, 3-nitrotryosine, TGFβ1 and Nox4 were purchased from Abcam (Cambridge, UK). Antibodies against p-Smad2/3, CD68, RhoA, p-Cdc42, Cdc42, p-Cofilin, p-Smad1/5 and eNOS were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against GAPDH and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture
Human pulmonary arterial endothelial cells (HPAECs) was obtained from ScienCell (San Diego, US), maintained in Dulbecco's Modification of Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Grand Island, NY, USA). Cells were cultured at 37 °C in a 5% CO2 incubator. Cells between passages 3 and 9 were used for experiments.
Cell viability assay
Cell viability was evaluated using a Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) as previously described [47]. HPAECs were seeded into 96-well plates (3×103 cells per well) and cultured in DMEM containing 10% FBS for 24 h. The cells were then subjected to growth arrest without serum for 24 h and treated with various concentrations of SAA for 48 h. After 3 h incubation at 37 °C, the absorbance at 450 nm was measured using the ELISA reader.
Wound healing assay
Cells (105/ml) were seeded into 24-well culture plates (0.5 ml/well) until 90% confluence was reached. After starving in serum-free DMEM for one day, the cell monolayer was scraped using a standard 200-μL pipette tip. The medium was added with SB431542 (3 μM), a TGFβ receptor inhibitor, or various concentrations of SAA (0.3, 1 and 3 μM). At 0, 12 and 24 h after scratching, microphotographs were taken at the same location.
Transwell invasion assay
The migration of HPAECs was also assessed by 24-well Transwell assay (Corning Inc., NY, USA) as previously described [48]. Briefly, cells with doses of SAA or SB431542 were added to the top of chamber. As the chemoattractant, a total of 600 μL of medium supplemented with or without TGFβ1 (5 ng/ml) and the indicated concentration of SAA was added to the lower chamber. After being incubated at 37 °C for 12 h, cells on the upper chamber was cleaned with a cotton swab, and the migrated cells on the bottom of the membrane was fixed with 90% ethanol, and stained by 0.5% crystal violet. Eight to ten fields per filter were taken randomly with a light microscope (Nikon, Japan).
Cell immunofluorescence Staining
The expression of CD31, α-SMA, HO-1 and nuclear translocation of Nrf2 were determined using immunofluorescence staining. After treatment, cells were washed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde. Then, cells were permeabilized with 0.5% Triton X-100 and blocked for 1 hours in 3% BSA at 37°C. Cells were immunostained with primary antibodies overnight at 4 °C, and then incubated with Alexa 488 or Alexa 555-conjugated secondary antibody (diluted 1:500) for 1 hour at room temperature. Nuclei were dyed with Hoechst 33342. Images were obtained and analyzed with a HCS Kinetic Scan (Cellomics ArrayScan VTI HCS) immediately.
Detection of ROS formation
The intracellular generation of ROS, as an index of oxidative stress, was determined using DCFH-DA. DCFH-DA is a cell-permeable probe. After diffusing into cells, it could be metabolized into the nonfluorescent DCFH, which could be oxidized by intracellular ROS into highly fluorescent DCF. After incubation with doses of SAA in the presence or absence of TGFβ1, HPAECs were rinsed with PBS and incubated with 5 µM DCFH-DA in serum-free medium in a light-free chamber for 30 min. Nuclei were dyed with Hoechst 33342. Images were obtained and analyzed with a HCS Kinetic Scan (Cellomics ArrayScan VTI HCS).
Animal experimental protocol
All of the animal experimental procedures were approved by the Institute of Animal Care and Use Committee at Chinese Academy of Medical Sciences. All the rats received a standard rodent chow and tap water, and were under a regular 12 h/12 h light/dark schedule in a temperature-controlled room at 22±3°C. For induction of pulmonary hypertension, rats received a single subcutaneous injection of 50 mg/kg monocrotaline (day 0). MCT was dissolved in 1 M HCl at a concentration of 40 mg/ml, adjusted to pH 7.4 with 1 M NaOH solution and diluted with distilled water. Adult male Wistar rats (purchased from Vital River Laboratory Animal Center (Beijing, China), 180 to 200g body weight) were randomly divided into five indicated groups: control group (injected with saline), model group (injected with MCT), SAA-treated groups (gavaged with SAA (0,3, 1 and 3 mg/kg) ten days after MCT injection when the vascular remodeling had already been established). At the end of the 28-days exposure period, rats were sacrificed for experimental measurements.
Morphologic, immunohistochemical and immunofluorescence analysis in lung tissue
Lungs were excised, washed with PBS solution, and fixed by 4% paraformaldehyde for 24 h. Then, specimens were embedded in paraffin and cut into sections 3-4 μm thick. Some lungs were frozen in liquid nitrogen immediately for immunofluorescence staining. A part of sections was processed for HE stainning, as described previously [49]. Immunohistochemical staining CD68 and 3-nitrotyrosine were performed to evaluate the quantity of macrophage and the oxidative stress status in lung. Elastic Van Gieson (EVG) staining and immunofluorescence staining for α-SMA were performed to calculate medial wall thickness of the pulmonary arterioles. Photomicrographs were obtained with microscope and analyzed using the software of Image Pro Plus 6.0 (Media Cybernetics Inc., Rockville, MD, USA).
Isometric tension studies
The vasodilation of isolated pulmonary artery rings was measured using multiwire myograph system, according to previous research [50]. After the animals were sacrificed, the lungs were isolated and bathed in a Krebs-Henseleit solution [NaCl, 120 mM; KCl, 4.8 mM; KH2PO4, 1.2 mM; NaHCO3, 25 mM; glucose, 11 mM; CaCl2, 2.5 mM; MgCl2, 1.4 mM; and ethylenediaminetetraacetic acid (EDTA), 0.01 mM]. The secondary branches of the pulmonary arteries were carefully dissected, cleaned up any connective tissue and cut into rings with an approximate length of 3 mm. Arterial segments were mounted in the myograph. During the course of the whole experiment, the solution was continuously bubbled with 95% O2 and 5% CO2 to maintain a pH of 7.4. After 30min equilibration period, the artery rings were precontracted by 10 nM endothelin-1 (ET-1). The cumulative dose-response curves were elicited by the cumulative application of acetylcholine (ACh: 10-8-10-4 M) or sodium nitroprusside (SNP: 10-8-10-5 M). The results were expressed as the percent relaxation of the maximum contractile tension to ET-1 (10 nM).
ELISA assay
Malondialdehyde (MDA; SenBeiJia Biotechnology, Nanjing, China) and glutathione peroxidase (GSH-Px; Cusabio, Wuhan, China) levels in serum were quantified using the commercially available rat ELISA kit according to manufacturer's instructions. A piece of lung tissue was cut out and grinded on ice with 1 mL PBS. Proinflammatory cytokines in lung, including TNF-α and IL-1β, were evaluated according to the instructions of detection kits (Cusabio, Wuhan, China). Lungs were dissected out and frozen in liquid nitrogen immediately.
Western blotting
The HPAECs or lungs in different groups were lysed with RIPA lysis buffer (Cell Signaling Technology, Danvers, USA) supplemented with protease inhibitors and subsequently centrifuged at 12,000 rpm for 10 min at 4°C. Protein concentration in the homogenates was estimated using the bicinchoninic acid protein assay. Equal amounts (50 μg) of protein were separated by 10% SDS-PAGE and electrotransferred to polyvinylidene difluoride membranes. The membranes were then blocked by 5% bovine serum albumin with 0.1% Tween-80 and incubated with appropriate primary antibodies overnight at 4°C. Then, the bands were immunoblotted with appropriate primary antibodies and were later incubated with anti-rabbit or anti-mouse horse radish peroxidase-conjugated secondary antibodies. Membranes were exposed to a mixture of enhanced chemiluminescence (ECL) solution and detected by ChemiDoc-It Imaging System (UVP, US). Quantitation of protein band was performed by Quantity One software (Bio-rad, US).
Statistical analysis
Statistical analysis was performed using SPSS software (version 21.0; SPSS Inc., Chicago, IL, USA). The results were expressed as the mean ± standard error of the mean (SEM). All data were submitted to one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparison test. Differences were considered statistically significant at p-values less than 0.05.
Results
SAA suppresses TGFβ1-induced EndMT in HPAECs and the canonical Smad2/3 signaling pathway
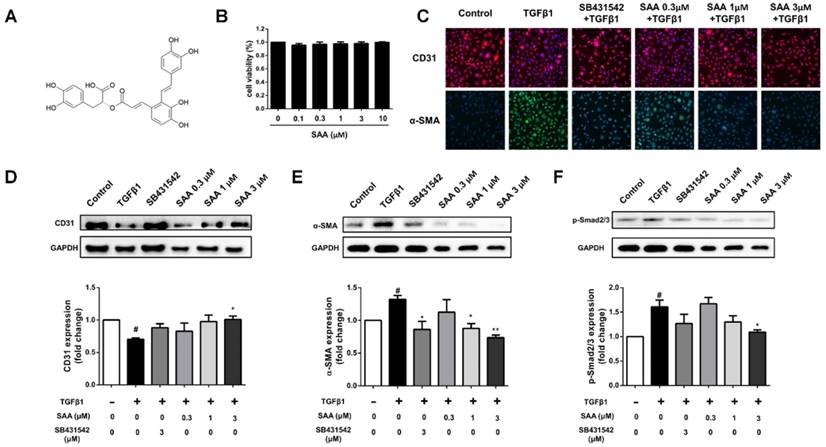
There was no significant impact on cell viability after exposure to SAA (0.1-10 μM) (Fig 1B). To examine the effect of SAA on TGFβ1-induced EndMT in HPAECs, expression of the endothelial cell marker CD31 and the mesenchymal marker α-SMA were detected. The reduction of CD31 and increase of α-SMA could be seen in TGFβ1-induced transition (Fig 1C-E). However, the phenomenon was attenuated after pre-incubation of doses of SAA and ALK5 inhibitor SB431542. Furthermore, as the canonical pathway, phosphorylation of Smad2/3 was increased after TGFβ1 treatment. Similarly, SAA treatment alleviated the change above (Fig 1F).
SAA inhibits EndMT in HPAECs and the Smad2/3 phosphorylation. (A) The chemical structure of salvianolic acid A. (B) The cells were pretreated with different concentrations of salvianolic acid A (0.1 to 10 μM) for 48 hours. Cell viability was determined by CCK-8 assays (n=5). (C) Representative fluorescent microscopy images of CD31 and α-SMA staining in HPAECs. Red fluorescence represents CD31, green fluorescence represents α-SMA, and nuclei were visualized by Hoechst 33342 staining. (D, E) Relative density analysis of the CD31 and α-SMA protein bands by western blot with GAPDH as control (n=3). (F) Normalized band intensity quantification showing the phosphorylation of Smad2/3. (n=3). #p < 0.05 vs. HPAECs without SAA or TGFβ1 treatment, *p < 0.05 and **p < 0.01 vs. TGFβ1-induced HPAECs without SAA treatment. Data are expressed as X-fold induction compared to normal control.
SAA inhibits cell migration and activation of Rho-GTPases pathway
In the wound healing assay, SAA suppressed migration of HPAECs exposed to TGFβ1 at 12 and 24h (Fig 2A-C). The migration was further evaluated using transwell assay. As shown in Fig 2D and E, the migrated cell number was significantly higher after 12 h in TGFβ1-treated group than in controls (12.1 ± 0.6 versus 88.3 ± 10.6 cells per field), and SAA significantly inhibited this effect (62.6 ± 2.4, 47.3 ± 5.7 and 32.2 ± 2.4 versus 88.3 ± 10.6 cells per field for 0.3, 1 and 3 μM SAA).
Effect of SAA on cell migration and Rho-GTPases pathway. (A) Representative images show invading HPAECs after 2 h treatment with different SAA concentrations (0.3, 1, and 3 μM). Serum-starved HPAECs were wounded by a pointed tip. Confluent HPAEC monolayers were treated with TGFβ1 for 12 and 24 h, and then photographed by inverted microscope, the migration distance was calculated. Bar graph shows the migration distance of HPAECs in response to different doses of SAA after 12h (B) and 24 h (C). The migration was also analyzed by transwell assay with SAA (0.3, 1, and 3 μM) or SB431542 (3 μM). (D) Representative images of migrated HPAECs in transwell chamber assay after treatment with or without TGFβ1 in the presence or absence of SAA for 12 h. (E) The figure indicates the number of migrated cells. Cells were counted in 8-10 randomly chosen microscope fields per filter. (F-H) Western blot analysis of RhoA protein expression, phosphorylation of Cdc42 and Cofilin protein expression in HPAECs in the different groups. #p < 0.05 and ###p < 0.001 vs. HPAECs without SAA or TGFβ1 treatment, *p < 0.05, **p < 0.01 and ***p < 0.01 vs. TGFβ1-induced HPAECs without SAA treatment. Data are expressed as X-fold induction compared to normal control in western blot.
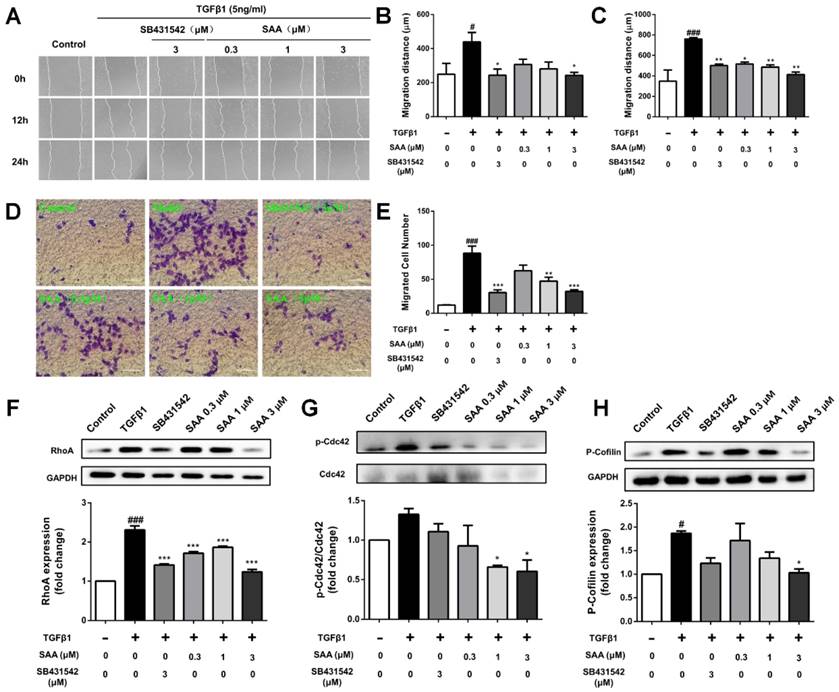
Compared with control group, p-Cdc42 and RhoA levels were significantly increased in the TGFβ1-treated HPAECs (Fig 2F and 2G). On the contrary, SAA (0.3, 1 and 3 μM) treatment prevented the TGFβ1-induced Cdc42 phosphorylation and RhoA expression. The phosphorylation of cofilin, as the main downstream signaling molecule of the above-mentioned two Rho-GTPases, increased in model group, whereas in SAA (0.3, 1 and 3 μM) treated HPAECs, the activation of cofilin was significantly reduced (Fig 2H).
SAA upregulates Nrf2/HO-1 pathway and reduces the intracellular ROS level
We next conducted an experiment to determine the effect of SAA on ROS formation induced by TGFβ1 in HPAECs. ROS formation in model group increased significantly compared to control group, whereas SAA reversed efficiency of TGFβ by reducing ROS production to nearly basal level (Fig 3B).
Effect of SAA on ROS formation and activation of Nrf2/HO-1 pathway. (A and B) Production of ROS in HPAECs was determined by DCFH-DA assay. The HPAECs were pretreated with different concentrations of SAA for 2h prior to treatment with TGFβ1. The bar graph shows the statistic results of fluorescence intensity. (C and D) Immunofluorescence analysis of Nrf2 nuclear translocation. HPAECs were treated with different doses of SAA (0.1, 0.3, 1 and 3 μM) for 6 hours. Anti-Nrf2 antibodiy was used as primary antibodies, followed by Alexa Fluor 488-conjugated secondary antibody. Nuclei were stained with Hoechst 33342, and then visualized by Cellomics array scan. (E and F) Immunofluorescence staining of HO-1 during TGFβ1-induced EndMT in HPAECs. (E) Representative images of HO-1, which were photoed by a high-content cytometer. The green fluorescence represents HO-1. (F) The statistic result shows the expression level of HO-1 with different doses of SAA in the absence or presence of TGFβ1 treatment. ##p < 0.01 and ###p < 0.001 vs. HPAECs without SAA or TGFβ1 treatment, *p < 0.05, **p < 0.01 and ***p < 0.01 vs. TGFβ1-induced HPAECs without SAA treatment. Data are expressed as X-fold induction compared to normal control.
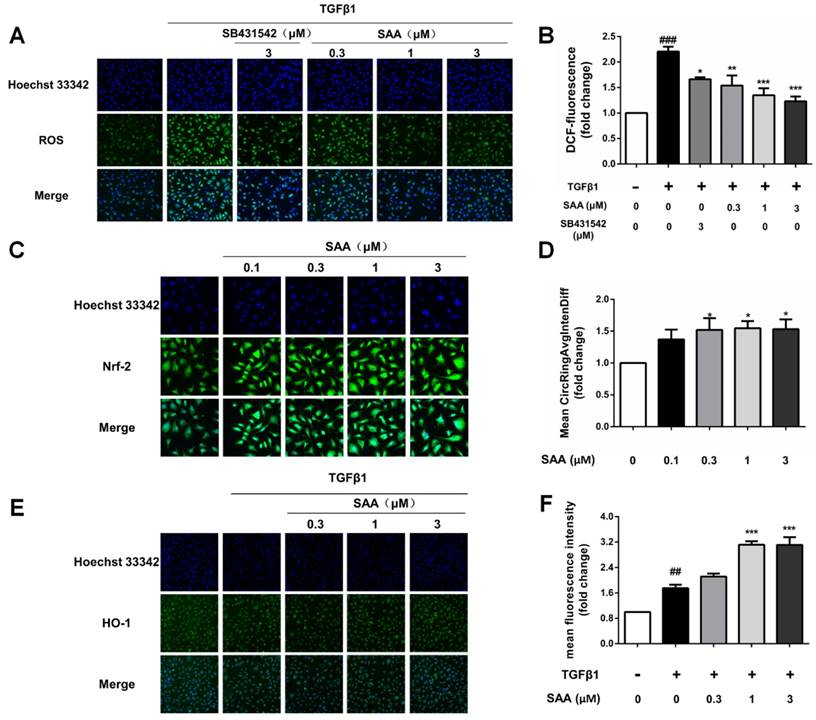
To explore the influence of SAA on Nrf2/HO-1 pathway, nuclear translocation of Nrf2 in HPAECs was determined. After SAA (0.1-3μM) treatment for 6h, nuclear translocation of Nrf2 was increased in HPAECs (Fig 3C and 3D). Consistent with the Nrf2 activation, SAA potently increased the expression of HO-1, even as HO-1 level increased after stimulation by TGFβ1 (Fig 3E and 3F).
SAA decreases TGFβ1 level and endothelial-mesenchymal transition in lung of PAH rats
As the main inducement of EndMT, the TGFβ1 level was determined in lung tissue of MCT-induced PAH rats. MCT significantly stimulated TGFβ1 level compared to the control group, which was accorded with previous studies about MCT-induced PAH model. Whereas SAA treatment, especially the 3mg/kg group, decreased TGFβ1 expression nearly to basal level (Fig 4A and B).
Effect of SAA on EndMT in lungs of PAH rats. (A) Photomicrographs of serial sections of rat lung from animals treated with different doses of SAA exposed to MCT for 4 weeks. Sections were stained for TGFβ1. (B-F) Relative density analysis of the TGFβ1, CD31, α-SMA, p-Smad2/3, and p-Smad1/5 protein bands by western blot (n=3). #p < 0.05, ###p < 0.001vs. lungs without SAA or MCT treatment, *p < 0.05 **p < 0.01 vs. lungs of PAH rats without SAA treatment. Data are expressed as X-fold induction compared to normal control.
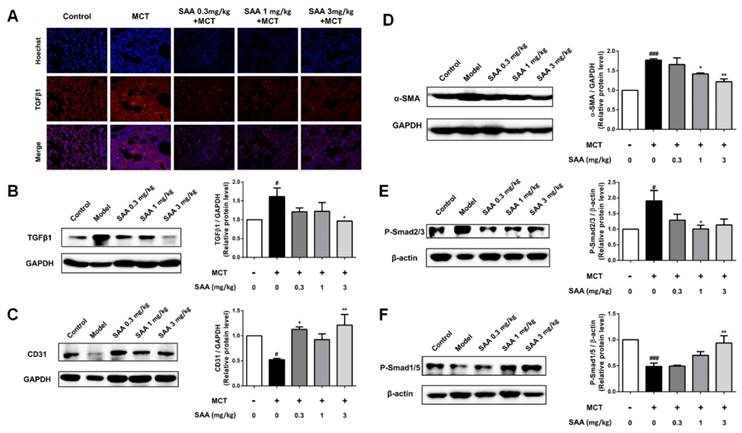
Compared with the control group, the fibroblast marker, α-SMA markedly increased and the endothelial markers CD31 decreased in lung of PAH rats. Accordance with the results of cell experiments, upon SAA treatment, the α-SMA level was significantly down-regulated and CD31 expression was up-regulated (Fig. 4C and D). As shown in Fig 4E, phosphorylation of Smad2/3 in lung was attenuated by SAA treatment. Meanwhile, the Smad1/5 activation was upregulated in SAA-treated groups in a dose-dependent manner (Fig 4F).
SAA restores pulmonary vascular remodeling and improves vascular relaxation in MCT-treated rats
In order to evaluate remodeling of different types of pulmonary arteries, the wall thickness of muscular artery and non-muscularized artery, classified according to their diameters in different range, was calculated. MCT treatment caused a significant pulmonary vascular remodeling as indicated by increased medial wall thickness, while the vascular remodeling was alleviated with SAA treatment (Fig 5 A-D), especially in arteries with diameter less than 150 µm as compared to the model group. Whereas for arteries with diameter lager than 150 µm, only the high-dose group had a mirror effect.
Effect of SAA on vascular remodeling and vascular relaxation. (A) EVG staining and immunofluorescence staining with antibodies against α-SMA were performed. Pulmonary arterioles were classified into three categories according to the diameter: (B) < 50 μm, (C) 50-150 μm and (D) >150 μm. The medial wall thickness of pulmonary arterioles was determined by calculating the ratio of the area of the media to that of the whole vessel. Concentration-response curves shows endothelium-dependent relaxation in response to Ach (E) and endothelium-independent relaxation in response to SNP (F) from different experimental groups at day 28 in pulmonary artery rings of MCT-treated rats treated with or without SAA. All rings were precontracted submaximally with ET-1 (10 nM) to obtain a stable plateau. ##p < 0.01, ###p < 0.001vs. lungs without SAA or MCT treatment, *p < 0.05, *p < 0.01 and ***p < 0.001 vs. lungs of PAH rats without SAA treatment. Data are expressed as X-fold induction compared to normal control.
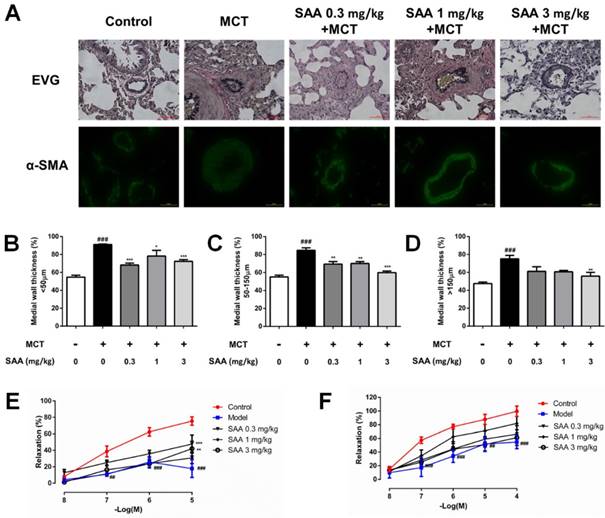
Impairment of relaxing function plays a key role in vascular remodeling. To evaluate MCT-induced impairment of endothelium-dependent relaxation in pulmonary artery rings isolated from different groups, the concentration-response to Ach (10-8-10-5 M) was recorded. MCT caused a significant decrease in the responsiveness to Ach, which was partially normalized by SAA (0.3 and 3 mg/kg). To elucidate MCT-induced impairment of endothelium-independent relaxation, the concentration-response to SNP (10-8-10-4 M) was recorded. The relaxant effect of SNP was significantly impaired in MCT-treated rats. But there was no obvious improvement in SAA-treated groups, as compared to the model group. Since the distinction, it was supposed that SAA improved vasodilatation function in a partially endothelium-dependent manner.
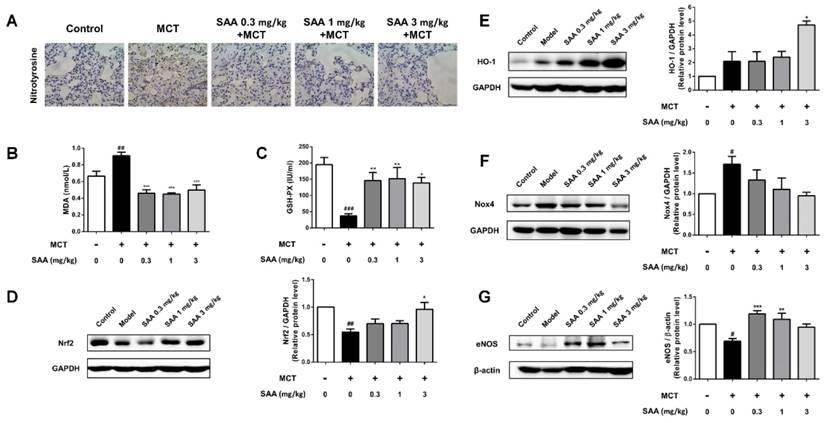
SAA attenuates oxidative stress level and upregulated Nrf2/HO-1 pathway in lung of PAH rats
Superoxide free radicals could react with nitric oxide to generate peroxynitrite. The expression of nitrotyrosine in lung tissue of MCT-induced PAH rats partly reflected oxidative damage in PAH rats. The change could be alleviated in SAA treated groups. The administration of SAA caused a significant decrease in serum MDA level and an increase in serum GSH-PX level as compared to MCT-treated rats. Protein level of Nox4, the major source of ROS production, and HO-1, were significantly increased in model group compared to the control group, whereas the Nrf2 expression decreased. MCT-induced Nox4 protein expression was slightly attenuated by SAA-treatment, but the difference between model group and SAA-treated groups was not statistically significant. Furthermore, the expression of Nrf2 and HO-1 was elevated after SAA supplementation.
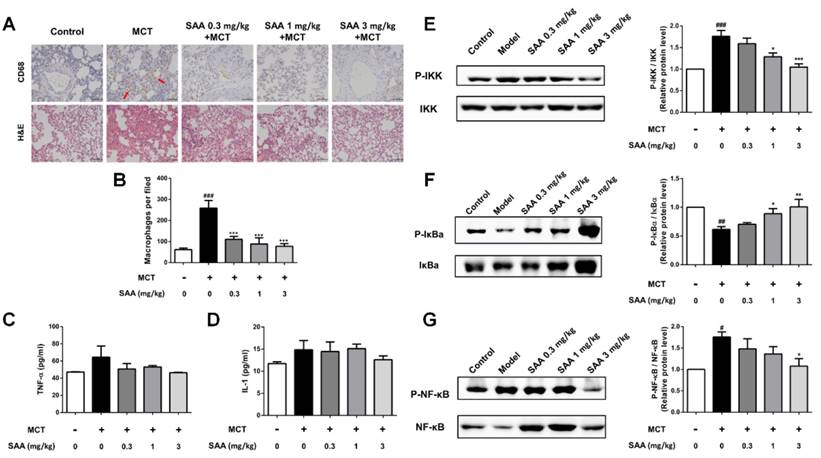
SAA attenuates inflammation in lung tissue of MCT-induced PAH rats
In the lungs of MCT-treated rats, the number of CD68-positive cells was significantly increased (Fig 7A). SAA treatment prevented infiltration of CD68-positive cells. ELISA results show that the TNF-α and IL-1β levels in lung tissues were higher in the model group than in the control group. Notably, the SAA-treated groups showed a decrease after 18 days of administration. Furthermore, we also examined the effects of SAA on NF-κB signaling pathway. As shown in Fig 7B, MCT increased the phosphorylation of NF-κB and IKK and decreased that of IκBa in the lung tissues of rats as compared to control rats. However, SAA alleviated this trend, demonstrating that SAA can suppress the activation of NF-κB signaling pathway in lungs of PAH rats.
SAA prevents MCT-induced oxidative stress in vivo. (A) Typical immunohistochemical analysis using lung tissue from the different groups was conducted to show changes of nitrotyrosine. (B)Lipid peroxide accumulation was quantified using MDA assay. (C) Measurements of GSH-PX activities in serum. (D-G) Relative density analysis of the Nrf2, HO-1, Nox4 and eNOS protein bands by western blot (n=3). #p < 0.05, ##p < 0.01, ###p < 0.001vs. lungs without SAA or MCT treatment, *p < 0.05, **p < 0.01, ***p < 0.001 vs. lungs of PAH rats without SAA treatment. Data are expressed as X-fold induction compared to normal control.
Effects of SAA on inflammation in the lung. Inflammation in lung was evaluated using ELISA and immunohistochemistry of CD68 in the formalin-fixed lung tissues. (A) Representative images for immunohistochemical staining for macrophage marker CD68 and H&E staining. (B) Quantity of CD68 positive cells was calculated from 9 separate images of different fields. (C and D) The level of the inflammation markers TNF-α and IL-1β in the PAH lungs. (E-G) Western blot analysis of phosphorylation of NF-κB, IκBα and IKK in different groups. #p < 0.05, ##p < 0.01, ###p < 0.001vs. lungs without SAA or MCT treatment, *p < 0.05, **p < 0.01, ***p < 0.001 vs. lungs of PAH rats without SAA treatment. Data are expressed as X-fold induction compared to normal control.
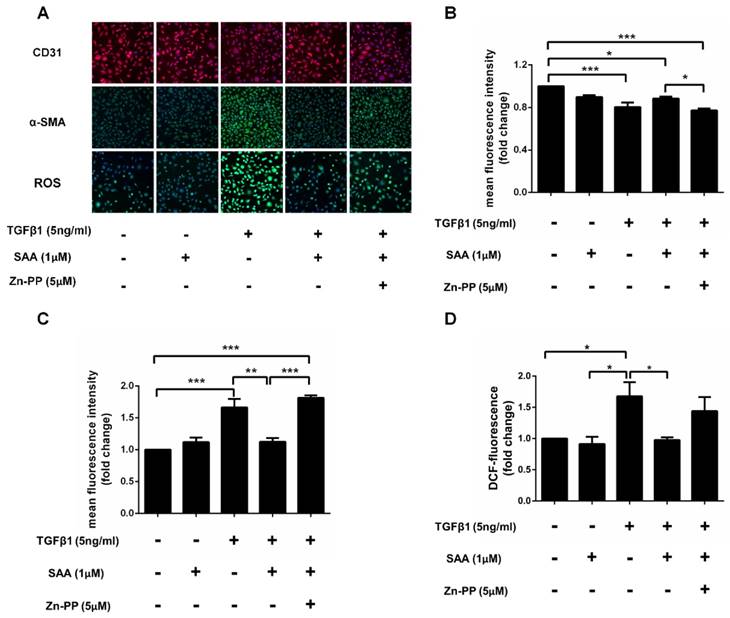
Effect of SAA on EndMT is depended on HO-1 induced by Nrf2
After TGFβ1 treatment, the expression of α-SMA was dramatically increased in HPAECs exposed to TGFβ1 than those without it, and SAA treatment ameliorated this change. However, the protective effects of SAA were abolished by HO-1 inhibitor ZnPP. Similarly, SAA treatment significantly increased CD31 level compared to cells with TGFβ1 treatment, while ZnPP reversed the increase of the CD31 caused by SAA. Beyond that TGFβ1 treatment significantly increased ROS levels in HPAECs and the change was prevented by SAA. The antioxidant effect of SAA was partially alleviated by Zn-PP (Fig 8C). However, there was no significant difference between these two groups.
HO-1 is involved in the cytoprotection of SAA against TGFβ1-induced EndMT. (A) Representative images show that pharmacological inhibition of heme oxygenase-1 with ZnPP eliminated the protective effect of SAA against TGFβ1-induced EndMT. Cells were pretreated with SAA alone or SAA combination with ZnPP for 2 h followed by TGFβ1 for a further 48 h. The expression of CD31 (B) and α-SMA (C) was measured by immunofluorescent staining. The ROS level (D) in HPAECs was determined by DCFH-DA assay. *p < 0.05, **p < 0.01 and ***p < 0.01. Data are expressed as X-fold induction compared to normal control.
Discussion
One of the main goals of this experiment was to verify the important role of oxidative stress in pulmonary endothelial-mesenchymal transition, and evaluate the protective effect of SAA, a phenolic antioxidant, as complementary therapeutics in pulmonary vascular remodeling.
In this study, we proved that SAA, a polyphenol Nrf2 activator, suppresses TGFβ1-induced EndMT via activating Nrf2/HO-1 pathway and reducing intracellular ROS generation. Furthermore, in monocrotaline-induced PAH model, SAA attenuates EndMT in lung and alleviates pulmonary vascular remodeling in treatment strategy, which may be related with activation of Nrf2/HO-1 signaling pathway and inhibition of oxidative stress.
In previous studies we have shown that prophylactic administration of SAA slowed the progression of pulmonary vascular remodeling, and may be associated with the Smad signaling pathway [32]. More recent evidence also reveals that SAA alleviates hypertension through inhibiting oxidative stress and TGF-β signal transduction pathway [34].
In MCT-induced PAH model, experimental design can be divided into prevention strategy and treatment strategy. The former starts medication before or on the same day of MCT injection. The latter doesn't start treatment until pathological features of PAH appear. Despite more obvious effect of prevention strategy in animal model, it does not apply to clinical practice. To evaluate the therapeutical effects, the administration of SAA was started on day 10 after MCT injection. SAA could inhibit remodeling in peribronchial or intra-alveolar vessels. For studies in wire myograph, acetylcholine is employed to investigate endothelium-dependent relaxation effect, whose mechanism was to activate the M receptor on the endothelial cells. SNP, as a nitric oxide donor, is used for investigating endothelium-independent relaxation effect [51]. SAA treatment significantly normalized endothelium-dependent relaxation of main pulmonary arteries, while the endothelium-independent relaxation response was not affected. These results indicate that SAA may improve diastolic function of pulmonary arteries by protecting vascular endothelium.
EndMT is a phenotypic change during which endothelial cells lose endothelial phenotype and gain more invasive and migratory properties. Ranchoux et al [7] demonstrated that EndMT is involved in pulmonary vascular remodeling. Therefore, researching the underlying cellular and molecular mechanisms inhibiting EndMT is crucial to enhance the effectiveness of the prevention and treatment against pulmonary arterial hypertension. The PAH model could be well established via a single injection of MCT, a plant-derived alkaloid. Recent studies indicate that EndMT is a major cause of pulmonary artery remodeling in MCT-induced PAH in rats [16]. Using double immunofluorescence staining and semi-quantitative analysis, Nikitopoulou et al found that endothelial markers are downregulated and mesenchymal markers are upregulated in MCT-induced pulmonary hypertension [52]. Ghosh et al found that treatment with SB431542, the TGFβ receptor inhibitor, could completely block TGFβ-induced EndMT [53]. Consistent with these previous reports, EndMT phenomena can be obviously observed in lung tissues of rats injected with MCT in our study. SAA treatment significantly increased the expression of endothelial cell marker, CD31, and decreased the level of mesenchymal cell marker, α-SMA. In order to further prove our in vivo observations, we conducted in vitro experiments in HPAECs incubated with TGFβ1, a classical inducer of EndMT. Similar to our in vivo study, treatment with TGFβ1 resulted in down-regulation of CD31 and up-regulation of the mesenchymal marker α-SMA. These changes could be effectively attenuated by SAA treatment.
In the process of EndMT, upregulation of transcription factor including Slug and Snail due to stimulation of TGFβ results in increased migration and invasion into extracellular matrices of diverse composition. TGFβ could induce cytoskeleton reorganization through activating Small Rho GTPases. Cytoskeleton reorganization is the prerequisite for cell migration and adhesion. It is also found that inhibition of Rho/ROCK pathway remarkably inhibits EndMT and decreases the endothelial permeability [54]. The migration is based on losing cell-cell contact mediated by membrane proteins such as CD31 and VE-cadherin [19], which is in keeping with the above results. Firstly, classical wound healing assay was employed to analyze cell migration. The results showed that SAA could decrease the mean migration distance induced by TGFβ1. To clarify this result further, free from the influence by cell proliferation, we chose transwell invasion assay to evaluate cell migration. In accordance with scratch assay, SAA treatment reduced the number of migrated cell compared with the model group. Furthermore, studies indicate that TGFβ could induce reorganization of actin cytoskeleton, which is the foundation of morphological change and cell migration, requires activation of small GTPases Cdc42 and RhoA [17, 55].
Based on the observation that SAA inhibits cell migration, we took an in-depth study on the expression of small GTPases Cdc42 and RhoA, and the phosphorylation of cofilin, their downstream effector. In accordance with pharmacodynamic results, SAA could inhibit RhoA expression, phosphorylation of Cdc42 and cofilin in a dose-dependent manner
Imbalance in redox signaling is associated with pathological process including hypertension and pulmonary vascular remodeling [56, 57]. It has been reported that ROS plays a key role in promoting the TGF-β1-induced EMT genetic changes [58].
Toshio Suzuki found that EndMT of pulmonary vascular endothelial cells is involved in the development of acute lung injury, partly mediated by ROS formation, and the process is closely affected by NAD(P)H oxidase-dependent ROS production [59]. The possible molecular mechanism, at least in part, is that oxidative stress increases the expression and secretion of TGFβ1 and TGFβ2 [20]. Furthermore, TGFβ-induced EndMT is partly influenced by ROS formation via stimulating miR-21 synthesis [60], and oxidative stress results in fibrosis in various kinds of cells. Thus, ROS is identified as the major mediator of EndMT, EndMT-related fibrosis and vascular remodeling. In HPAECs with TGFβ1 treatment, the increase of ROS formation could be attenuated by different doses of SAA, as well as SB431542.
In terms of the mechanism underlying the protective effects of SAA on oxidative stress and EndMT, we subsequently turn our attention to Nrf2/ARE pathway. Nrf2/ARE pathway plays a key role in cellular redox homeostasis. Nrf2 dissociates from Keap1 due to different stressors, including endogenous substances, radiation and ROS, and subsequently translocate into nucleus. The accumulation of Nrf2 in nucleus promotes transcription of a wide variety of cytoprotective genes. Recent studies reported that SAA protects from oxidative stress via activating the Nrf2/HO-1 axis in epithelial cells [61]. Furthermore, SAA could improve the recovery of neurological function after SCI via the miR-101/Nrf2/HO-1 signaling pathway [62]. In the present study, the translocation of Nrf2 into nucleus was increased after SAA treatment in HPAECs and the Nrf2 expression was up-regulated in lung tissues of SAA treated rats compared with the model group, implying the activation of Nrf2 function.
HO-1 is a stress-inducible antioxidant enzyme at down-stream signal transduction of Nrf2. HO-1 catalyzes the degradation of heme to bilirubin, carbon monoxide (CO), and ferrous iron. The antioxidative effect of HO-1 is related to the production of these enzymatic products [63]. Similarly, in the current study, we found that SAA application increased the HO-1 protein expression both in vivo and in vitro. More importantly, inhibition of HO-1 by ZnPP significantly abolished the inhibitory effects of SAA on TGFβ1-induced EndMT in HPAECs, indicating that these protective effects of SAA on EndMT were mediated by Nrf2/HO-1 signaling pathway.
In terms of the regulation of EndMT, it is reported that the Nrf2/HO-1 signaling pathway may also interact with TGFβ/Smad signaling. In a major advance in 2007, Yan et al. found that HO-1 induced through a Smad-dependent signaling pathway protect HK-2 (human proximal tubule cell line) from H2O2-induced oxidative damage [64]. Moreover, a recent literature on this area [65] found that there may be a kind of protein-protein interaction between Nrf2 and Smad, through which the TGFβ1-related gene expression was suppressed. Although the precise mechanisms remain to be explored, the present research may provide insights into the role of Nrf2/HO-1 pathway in EndMT and suggest a new way for application of polyphenols.
NAPDH oxidases (Noxs) are a group of heme-containing transmembrane proteins and important ROS producers for both phagocytic and non-phagocytic cells. Not only has that, accumulating evidence indicated that Nox4 plays a pivotal role in mediating TGFβ signaling pathway [66]. After MCT injection for 4 weeks, the Nox4 level was increased in rat lungs, and treatment with SAA prevented MCT-induced Nox4 expression. As the primary source of NO formation, there was a reduction of eNOS formation in lung tissues of rats in model group compared with control group, which was in line with previous study [17]. In addition to vasodilative effect, eNOS-derived NO is also an antioxidant due to its inhibitory effect on NADPH oxidase activity [67]. Furthermore, our results show that the level of 3-nitrotyrosine (a product of tyrosine nitration by peroxynitrite) decreased with SAA treatment. This may be the result of SAA eliminating the overproduction of superoxide anions and subsequently reducing the peroxynitrite level in vivo.
EndMT could be induced by inflammatory cytokines including TNFα and IL-6. Recent evidence suggests that NF-κB signaling pathway regulates this critical phenotype switch in the pathogenesis of PAH. Transfection of IκBα (AA) mutant plasmid could reverse the EndMT process [68]. What's more, there is an interactive relationship between inflammation and oxidative stress. An example in that some inflammatory cytokines can cause oxidative stress via inducing NADPH oxidases enzymes that lead to ROS production [69]. As well, there was evidence that oxidative stress might also activate NF-κB pathway [70]. In accordance with the previous evidence, activation of NF-κB signal pathway was shown in MCT-induced PAH model. SAA decreased NF-κB p65 and IKK phosphorylation in lung.
Recently, the molecular mechanism of phenolic Nrf2 activators has been unraveled. Due to the electrophilicity and reactivity to sulfhydryl group, they could directly disrupt Protein-Protein interactions between Keap1 and Nrf2 or promote the de-methylation of Nrf2 promoter [71]. Even though the present study demonstrates the protective effect of SAA on EndMT in vivo and in vitro, and its association with Nrf2/HO-1 signaling pathway. Further studies are needed to determine exactly how SAA affects the Nrf2/HO-1 pathway.
In summary, SAA could attenuate EndMT, showing as CD31 up-regulation and α-SMA down-regulation, suppress oxidative stress and alleviate pulmonary vascular remodeling. The Nrf2/HO-1 signaling pathway may be involved in the underlying mechanism.
Abbreviations
EndMT: endothelial-to-mesenchymal transition; SAA: salvianolic acid A; MCT: monocrotaline; HPAECs: human pulmonary arterial endothelial cells; ROS: reactive oxygen species; PAH: pulmonary arterial hypertension; EMT: epithelial-mesenchymal transition; Nrf2: NF-E2-related factor 2; Keap1: Kelch-like ECH-associated protein 1; Nox4: NADPH oxidase 4; DCFH-DA: 2',7'-dichlorofluorescin diacetate; DMEM: Dulbecco's Modification of Eagle's Medium; FBS: fetal bovine serum; CCK-8: Cell Counting Kit-8; PBS: phosphate buffered saline; EVG: Elastic Van Gieson; ET-1: endothelin-1; ACh: acetylcholine; SNP: sodium nitroprusside; MDA: malondialdehyde; GSH-Px: glutathione peroxidase.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81573645, 81603101) and the Natural Science Foundation of Beijing (7174322) and CAMS Innovation Fund for Medical Sciences (2016-I2M-1-010).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zamanian RT, Kudelko KT, Sung YK, de Jesus Perez V, Liu J, Spiekerkoetter E. Current clinical management of pulmonary arterial hypertension. Circulation research. 2014;115:131-47
2. Michelakis ED. Pulmonary arterial hypertension: yesterday, today, tomorrow. Circulation research. 2014;115:109-14
3. Song D, Wang HL, Wang S, Zhang XH. 5-Hydroxytryptamine-induced proliferation of pulmonary artery smooth muscle cells are extracellular signal-regulated kinase pathway dependent. Acta Pharmacol Sin. 2005;26:563-7
4. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Science signaling. 2014;7:re8
5. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. The Journal of clinical investigation. 2012;122:4306-13
6. Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. American journal of physiology Lung cellular and molecular physiology. 2007;293:L1-8
7. Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ. et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006-18
8. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ. et al. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Artery Hypertension. The American journal of pathology. 2015
9. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E. et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature medicine. 2007;13:952-61
10. Zhu K, Pan Q, Jia LQ, Dai Z, Ke AW, Zeng HY. et al. MiR-302c inhibits tumor growth of hepatocellular carcinoma by suppressing the endothelial-mesenchymal transition of endothelial cells. Sci Rep. 2014;4:5524
11. Yoshida M, Okubo N, Chosa N, Hasegawa T, Ibi M, Kamo M. et al. TGF-beta-operated growth inhibition and translineage commitment into smooth muscle cells of periodontal ligament-derived endothelial progenitor cells through Smad- and p38 MAPK-dependent signals. International journal of biological sciences. 2012;8:1062-74
12. Saito A. EMT and EndMT: regulated in similar ways? Journal of biochemistry. 2013;153:493-5
13. Arcot SS, Lipke DW, Gillespie MN, Olson JW. Alterations of growth factor transcripts in rat lungs during development of monocrotaline-induced pulmonary hypertension. Biochemical pharmacology. 1993;46:1086-91
14. Long L, Crosby A, Yang X, Southwood M, Upton PD, Kim DK. et al. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119:566-76
15. Thomas M, Docx C, Holmes AM, Beach S, Duggan N, England K. et al. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. The American journal of pathology. 2009;174:380-9
16. Song S, Zhang M, Yi Z, Zhang H, Shen T, Yu X. et al. The role of PDGF-B/TGF-beta1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cellular signalling. 2016;28:1489-501
17. Mihira H, Suzuki HI, Akatsu Y, Yoshimatsu Y, Igarashi T, Miyazono K. et al. TGF-beta-induced mesenchymal transition of MS-1 endothelial cells requires Smad-dependent cooperative activation of Rho signals and MRTF-A. Journal of biochemistry. 2012;151:145-56
18. Guo Y, Li P, Bledsoe G, Yang ZR, Chao L, Chao J. Kallistatin inhibits TGF-beta-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Experimental cell research. 2015;337:103-10
19. Yan F, Zhang GH, Feng M, Zhang W, Zhang JN, Dong WQ. et al. Glucagon-Like Peptide 1 Protects against Hyperglycemic-Induced Endothelial-to-Mesenchymal Transition and Improves Myocardial Dysfunction by Suppressing Poly(ADP-Ribose) Polymerase 1 Activity. Molecular medicine (Cambridge, Mass). 2015;21:15-25
20. Montorfano I, Becerra A, Cerro R, Echeverria C, Saez E, Morales MG. et al. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-beta1 and TGF-beta2-dependent pathway. Laboratory investigation; a journal of technical methods and pathology. 2014;94:1068-82
21. Pociask DA, Sime PJ, Brody AR. Asbestos-derived reactive oxygen species activate TGF-beta1. Laboratory investigation; a journal of technical methods and pathology. 2004;84:1013-23
22. Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochimica et biophysica acta. 2016
23. Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM. et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. The American journal of pathology. 2011;179:2660-73
24. Huang Y, Li W, Su ZY, Kong AN. The complexity of the Nrf2 pathway: beyond the antioxidant response. The Journal of nutritional biochemistry. 2015;26:1401-13
25. Cominacini L, Mozzini C, Garbin U, Pasini A, Stranieri C, Solani E. et al. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free radical biology & medicine. 2015;88:233-42
26. Chen B, Lu Y, Chen Y, Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. The Journal of endocrinology. 2015;225:R83-99
27. Mimura J, Itoh K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free radical biology & medicine. 2015;88:221-32
28. Grootaert C, Kamiloglu S, Capanoglu E, Van Camp J. Cell Systems to Investigate the Impact of Polyphenols on Cardiovascular Health. Nutrients. 2015;7:9229-55
29. Rangel-Huerta OD, Pastor-Villaescusa B, Aguilera CM, Gil A. A Systematic Review of the Efficacy of Bioactive Compounds in Cardiovascular Disease: Phenolic Compounds. Nutrients. 2015;7:5177-216
30. Bjorklund G, Chirumbolo S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition (Burbank, Los Angeles County, Calif). 2017;33:311-21
31. Hugel HM, Jackson N, May B, Zhang AL, Xue CC. Polyphenol protection and treatment of hypertension. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2016;23:220-31
32. Chen YC, Yuan TY, Zhang HF, Wang DS, Yan Y, Niu ZR. et al. Salvianolic acid A attenuates vascular remodeling in a pulmonary arterial hypertension rat model. Acta Pharmacol Sin. 2016;37:772-82
33. Sun L, Zhao R, Zhang L, Zhang W, He G, Yang S. et al. Prevention of vascular smooth muscle cell proliferation and injury-induced neointimal hyperplasia by CREB-mediated p21 induction: An insight from a plant polyphenol. Biochemical pharmacology. 2016;103:40-52
34. Zhang J, An SJ, Fu JQ, Liu P, Shao TM, Li M. et al. Mixed Aqueous Extract of Salvia Miltiorrhiza Reduces Blood Pressure through Inhibition of Vascular Remodelling and Oxidative Stress in Spontaneously Hypertensive Rats. Cell Physiol Biochem. 2016;40:347-60
35. Yu XY, Zhang L, Yang XY, Li XT, Du GH. Salvianolic acid A improves intestinal motility in diabetic rats through antioxidant capacity and upregulation of nNOS. Journal of digestive diseases. 2016;17:441-7
36. Pan Y, Fu H, Kong Q, Xiao Y, Shou Q, Chen H. et al. Prevention of pulmonary fibrosis with salvianolic acid a by inducing fibroblast cell cycle arrest and promoting apoptosis. J Ethnopharmacol. 2014;155:1589-96
37. Teng F, Yin Y, Cui Y, Deng Y, Li D, Cho K. et al. Salvianolic acid A inhibits endothelial dysfunction and vascular remodeling in spontaneously hypertensive rats. Life sciences. 2016;144:86-93
38. Jin H, Liu M, Zhang X, Pan J, Han J, Wang Y. et al. Grape seed procyanidin extract attenuates hypoxic pulmonary hypertension by inhibiting oxidative stress and pulmonary arterial smooth muscle cells proliferation. The Journal of nutritional biochemistry. 2016;36:81-8
39. Bryant AJ, Carrick RP, McConaha ME, Jones BR, Shay SD, Moore CS. et al. Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. American journal of physiology Lung cellular and molecular physiology. 2016;310:L249-62
40. Jimenez SA, Piera-Velazquez S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix biology: journal of the International Society for Matrix Biology. 2016;51:26-36
41. Funke M, Geiser T, Schoch OD. Pulmonary hypertension associated with chronic lung diseases. Swiss medical weekly. 2016;146:w14363
42. Fang YZ, Yang S, Wu G. Free radicals, antioxidants, and nutrition. Nutrition (Burbank, Los Angeles County, Calif). 2002;18:872-9
43. Yang LL, Li DY, Zhang YB, Zhu MY, Chen D, Xu TD. Salvianolic acid A inhibits angiotensin II-induced proliferation of human umbilical vein endothelial cells by attenuating the production of ROS. Acta Pharmacol Sin. 2012;33:41-8
44. Tang H, Pan CS, Mao XW, Liu YY, Yan L, Zhou CM. et al. Role of NADPH oxidase in total salvianolic acid injection attenuating ischemia-reperfusion impaired cerebral microcirculation and neurons: implication of AMPK/Akt/PKC. Microcirculation (New York, NY: 1994). 2014;21:615-27
45. Tsai MK, Lin YL, Huang YT. Effects of salvianolic acids on oxidative stress and hepatic fibrosis in rats. Toxicology and applied pharmacology. 2010;242:155-64
46. Shu Y, Liu Y, Li X, Cao L, Yuan X, Li W. et al. Aspirin-Triggered Resolvin D1 Inhibits TGF-beta1-Induced EndMT through Increasing the Expression of Smad7 and Is Closely Related to Oxidative Stress. Biomolecules & therapeutics. 2016;24:132-9
47. Yin Q, Lu H, Bai Y, Tian A, Yang Q, Wu J. et al. A metabolite of Danshen formulae attenuates cardiac fibrosis induced by isoprenaline, via a NOX2/ROS/p38 pathway. British journal of pharmacology. 2015;172:5573-85
48. Guo J, Li L, Wu YJ, Yan Y, Xu XN, Wang SB. et al. Inhibitory effects of Brazilin on the vascular smooth muscle cell proliferation and migration induced by PDGF-BB. The American journal of Chinese medicine. 2013;41:1283-96
49. Derrett-Smith EC, Dooley A, Gilbane AJ, Trinder SL, Khan K, Baliga R. et al. Endothelial injury in a transforming growth factor beta-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis and rheumatism. 2013;65:2928-39
50. Chen YC, Yuan TY, Zhang HF, Wang DS, Niu ZR, Li L. et al. Fasudil evokes vasodilatation of rat mesenteric vascular bed via Ca(2+) channels and Rho/ROCK pathway. European journal of pharmacology. 2016;788:226-33
51. Mokhtar SS, Vanhoutte PM, Leung SW, Suppian R, Yusof MI, Rasool AH. Reduced nitric oxide-mediated relaxation and endothelial nitric oxide synthase expression in the tail arteries of streptozotocin-induced diabetic rats. Eur J Pharmacol. 2016;773:78-84
52. Nikitopoulou I, Orfanos SE, Kotanidou A, Maltabe V, Manitsopoulos N, Karras P. et al. Vascular endothelial-cadherin downregulation as a feature of endothelial transdifferentiation in monocrotaline-induced pulmonary hypertension. American journal of physiology Lung cellular and molecular physiology. 2016;311:L352-63
53. Ghosh AK, Nagpal V, Covington JW, Michaels MA, Vaughan DE. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cellular signalling. 2012;24:1031-6
54. Peng H, Li Y, Wang C, Zhang J, Chen Y, Chen W. et al. ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Scientific reports. 2016;6:20304
55. Edlund S, Landstrom M, Heldin CH, Aspenstrom P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Molecular biology of the cell. 2002;13:902-14
56. Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Comprehensive Physiology. 2013;3:1011-34
57. Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovascular research. 2006;71:247-58
58. Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Current medicinal chemistry. 2010;17:4256-69
59. Suzuki T, Tada Y, Nishimura R, Kawasaki T, Sekine A, Urushibara T. et al. Endothelial-to-mesenchymal transition in lipopolysaccharide-induced acute lung injury drives a progenitor cell-like phenotype. American journal of physiology Lung cellular and molecular physiology. 2016;310:L1185-98
60. Ling M, Li Y, Xu Y, Pang Y, Shen L, Jiang R. et al. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-kappaB in arsenite-induced cell transformation. Free radical biology & medicine. 2012;52:1508-18
61. Zhang H, Liu YY, Jiang Q, Li KR, Zhao YX, Cao C. et al. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free radical biology & medicine. 2014;69:219-28
62. Chien MY, Chuang CH, Chern CM, Liou KT, Liu DZ, Hou YC. et al. Salvianolic acid A alleviates ischemic brain injury through the inhibition of inflammation and apoptosis and the promotion of neurogenesis in mice. Free radical biology & medicine. 2016;99:508-19
63. Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. American journal of respiratory cell and molecular biology. 2007;36:175-82
64. Yan J, Yang S, Zhang J, Zhai C, Zhu T. BMP6 attenuates oxidant injury in HK-2 cells via Smad-dependent HO-1 induction. Free radical biology & medicine. 2009;46:1275-82
65. Oh CJ, Kim JY, Min AK, Park KG, Harris RA, Kim HJ. et al. Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-beta/Smad signaling. Free radical biology & medicine. 2012;52:671-82
66. Samarakoon R, Overstreet JM, Higgins PJ. TGF-beta signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cellular signalling. 2013;25:264-8
67. Fujii H, Ichimori K, Hoshiai K, Nakazawa H. Nitric oxide inactivates NADPH oxidase in pig neutrophils by inhibiting its assembling process. The Journal of biological chemistry. 1997;272:32773-8
68. Li L, Wei C, Kim IK, Janssen-Heininger Y, Gupta S. Inhibition of nuclear factor-kappaB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension. 2014;63:1260-9
69. Crosswhite P, Sun Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. Journal of hypertension. 2010;28:201-12
70. Dorfmuller P, Chaumais MC, Giannakouli M, Durand-Gasselin I, Raymond N, Fadel E. et al. Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension. Respir Res. 2011;12:119
71. Kim S, Lee HG, Park SA, Kundu JK, Keum YS, Cha YN. et al. Keap1 cysteine 288 as a potential target for diallyl trisulfide-induced Nrf2 activation. PloS one. 2014;9:e85984
Author contact
![]() Corresponding authors: Professor Lianhua Fang and Professor Guanhua Du, National center for pharmaceutical screening, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, 2 Nan Wei Road, Beijing 100050, China; Tel: 86-10-63165184; E-mails: fanglhac.cn (LH Fang), dughac.cn (GH Du).
Corresponding authors: Professor Lianhua Fang and Professor Guanhua Du, National center for pharmaceutical screening, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, 2 Nan Wei Road, Beijing 100050, China; Tel: 86-10-63165184; E-mails: fanglhac.cn (LH Fang), dughac.cn (GH Du).