Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(11):1398-1408. doi:10.7150/ijbs.22249 This issue Cite
Research Paper
Targeting the long noncoding RNA MALAT1 blocks the pro-angiogenic effects of osteosarcoma and suppresses tumour growth
Zhi-Chang Zhang1*, Chun Tang2*, Yang Dong1, Jing Zhang1, Ting Yuan1, Shi-Cong Tao1 ![]() , Xiao-Lin Li1
, Xiao-Lin Li1 ![]()
1. Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China;
2. Department of Nursing, Guangming Traditional Chinese Medicine Hospital, Pudong New Area, Shanghai 201300, China.
* Co-first authors: These authors contributed equally to this work.
Received 2017-8-6; Accepted 2017-9-22; Published 2017-11-1
Abstract

Osteosarcoma (OS), the commonest primary malignant tumour originating from bone, affects a substantial number of people, mostly during adolescent growth, and leads to a very poor prognosis as a result of the high rate of early metastases. Consequently, there is urgent demand for a novel treatment for this disease. There are growing concerns focused on OS-induced pro-angiogenic effects, but to date, the mechanism of OS-induced pro-angiogenesis is still insufficiently well-understood. Long noncoding RNAs (lncRNAs) have attracted increasing interest due to their strong correlation with a variety of diseases and their powerful capacity for epigenetic regulation. Recently, metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a lncRNA, has been discovered to be closely related to OS progression and hypoxia responses which are associated with angiogenesis. In this study, we confirm that MALAT1 induces pro-angiogenic effects, and demonstrate that the underlying mechanism involves a MALAT1/mechanistic target of rapamycin (mTOR)/hypoxia inducible factor-1α (HIF-1α) loop. With the help of chemically-modified small interfering RNAs targeting MALAT1 (siMALAT1), we confirm that siMALAT could provide a potential strategy to block the abnormally active OS-induced pro-angiogenic effect, and ultimately successfully suppress progression of OS tumours.
Keywords: osteosarcoma, MALAT1, siRNA, angiogenesis, oncotherapy.
Introduction
Osteosarcoma (OS) is the most common primary malignant tumour arising in bone, especially near the metaphyseal growth plates of long bones or around the knee, and has high morbidity with a major peak during adolescent growth, and a minor peak in patients in their 70s and 80s [1-3]. The early metastasis rate is high and, at the same time, patients with metastatic disease have a very poor prognosis which means that, to be specific, the five-year survival rate remains below 30% [4, 5]. To make matters worse, almost a quarter of OS patients have metastasized sarcoma cells at first presentation [6, 7], and hence, operative treatment including amputation surgery does not prove adequate. Consequently, novel treatments are in high demand.
Over the past few years, more and more studies have paid close attention to the role of angiogenesis, including the imbalance between anti-angiogenic and pro-angiogenic effects, in OS progression including proliferation, migration, and metastasis [8]. The foremost regulator of angiogenesis is vascular endothelial growth factor (VEGF), which participates in the pathogenesis and progression of several cancers [9], and has been reported to be closely related to the clinical stages of OS [10, 11]. However, a more detailed and essential understanding of the countless ties between OS-induced angiogenesis and OS progression still requires further investigation.
With improvements in knowledge of the human genome, it is now known that most genomic sequences are generally transcribed into RNAs including both protein-coding RNAs (or messenger RNA, mRNA) and non-coding RNAs (ncRNAs) [12]. Among the ncRNAs, long non-coding RNAs (lncRNAs), defined as ncRNAs with at least 200 nucleotides and without apparent protein-coding capability [13, 14] are attracting more and more interest due to the strong correlation with a variety of diseases and their powerful capacity for epigenetic regulation [15]. More interestingly, studies have shown that the expression profile of lncRNAs is shifted in OS [16, 17].
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was first identified as a member of the lncRNAs that is highly expressed in several types of cancer [18, 19]. The expression level of MALAT1 is closely related to OS progression, as, for instance, silence of MALAT1 would delay tumour growth of OS, indicating the potential of MALAT1 as a theranostic target of OS treatment [20]. From subsequent studies, the strong relationship between MALAT1 and OS progression has gradually emerged [21]. However, the detailed mechanism of MALAT1-induced OS progression still remains unknown and needs to be elucidated.
Expression of MALAT1 is dramatically enhanced in hypoxic conditions in which angiogenesis is promoted [22]. Further, the most recent studies have revealed the outline of the relationship between MALAT1 and pro-angiogenic ability [23-25]. In addition, it is reported that the vasculogenic mimicry or pro-angiogenic ability induced by MALAT1 is mediated by pro-angiogenic factors including VEGF and FGF2 [25, 26].
Based on the obvious similarity between the abnormally-high expression of MALAT1 in OS and the high expression of MALAT1induced by hypoxia, we surmised that the high level of expression of MALAT1 creates the illusion of a hypoxic environment, generating the abnormally strong angiogenic stimulus observed in OS, an illusion which might be mediated by hypoxia inducible factor-1α (HIF-1α). Although previous studies have implied the existence of a MALAT1/HIF-1α loop [27, 28], the detailed mechanism still remains unclear. Interestingly, MALAT1 has been shown to activate mechanistic target of rapamycin (mTOR) [29], which can further increase the level of HIF-1α [30, 31].
The present research confirmed that abnormally highly-expressed MALAT1 participates in a MALAT1/mTOR/HIF-1α positive feedback loop, playing a crucial role in OS-related pro-angiogenic ability in tumour progression. More specifically, abnormally high expression of MALAT1 in OS cells induces abnormally high expression of HIF-1α via the activation of mTOR, and this highly-expressed HIF-1α in turn induces abnormally high levels of angiogenesis which ultimately promote OS progression. Meanwhile, the high expression levels of HIF-1α in turn sustain the high expression of MALAT1 to a certain extent. Finally, we employed a chemically-modified siRNA of MALAT1, and proved its ability to retard or stop the progression of OS as well as to inhibit tumour-induced angiogenesis, suggesting it may play an important role in future anti-cancer strategies targeting OS.
Materials and Methods
Cell culture
In this study, we used a well-accepted cell line of human OS, MNNG/HOS [32, 33], which was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The MNNG/HOS cells were cultured in Dulbecco's modified eagle medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) with 10% foetal bovine serum (FBS; Gibco, Thermo Fisher Scientific) in a constant temperature humidified 37°C incubator with 5% carbon dioxide (CO2). We used a human microvascular endothelial cell line (HMEC-1), a widely-used cell model for the assessment of pro-angiogenic ability including proliferation, migration and tubule formation, to evaluate cell migration and tubule formation; the cell culture of HMEC-1 has been described previously [34, 35]. The use of HMEC-1 is generally accepted as an in vivo model for the study of cancer-related angiogenesis and the antiangiogenic properties of antitumor drugs [36-40].
Small interference RNA (siRNA)
To interfere with the function of MALAT1, we purchased a ready-to-use siRNA for MALAT1, the In Vivo Pre-designed siRNA for MALAT1 (siMALAT1; siRNA ID: n511399; Ambion, Thermo Fisher Scientific) as well as its negative control, In Vivo Negative Control siRNA (siCtrl; Ambion, Thermo Fisher Scientific). The siRNAs were transfected into cells with the help of Lipofectamine™ 2000 Transfection Reagent (Invitrogen, Thermo Fisher Scientific) according to the instructions supplied. The working solution for in vivo transfection was prepared using Invivofectamine™ 3.0 Reagent (Invitrogen) referring to the accessary supporting files.
Inhibitor and agonist
The inhibitors and agonist in this study (rapamycin, MHY1485 and BAY 87-2243) were purchased from Selleckchem (Houston, TX, USA). The working concentration of both rapamycin and BAY 87-2243 was 100 nM, while the working concentration of MHY1485 was 1 μM. Rapamycin is a well-recognized inhibitor of mTOR kinase [41, 42], MHY1485 is an effective agonist of mTOR [43, 44], and BAY 87-2243 is a highly selective inhibitor of HIF-1α [45, 46].
RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR)
After extraction of total RNA using TRIzol Reagent (Invitrogen), the complementary DNA (cDNA) was synthesised using a SuperScript™ IV First-Strand Synthesis System (Invitrogen) following the instructions provided by the manufacturer. Subsequent PCR amplification was performed with a Platinum™ Multiplex PCR Master Mix (Invitrogen) referring to the instructions provided by the manufacturer.
The PCR primers are as described below:
GAPDH-FOR: GGGAAGGTGAAGGTCGGAGT
GAPDH-REV: GGGGTCATTGATGGCAACA
MALAT1-FOR: GCATTTTGGGATGGTCTTAA
MALAT1-REV: CAGCGGTACACTCCTTCTCT
Polyacrylamide gel electrophoresis (PAGE)
After RT-PCR, differences in expression levels were analysed by PAGE. Briefly, the samples were mixed with SYBR™ Safe DNA Gel Stain (Invitrogen), BlueJuice™ Gel Loading Buffer (Invitrogen) and UltraPure™ DNase/RNase-Free Distilled Water (Invitrogen) according to the manufacturer's instructions. The prepared samples were loaded onto Novex™ TBE Gels (Invitrogen) and electrophoresed at a constant voltage of 150 V for 40 min, and finally the gel was observed using a Bio-Rad Gel Doc™ XR+ system (Bio-Rad, Hercules, CA, USA).
Cell Counting Kit-8 (CCK-8) assay
Cell counting kit-8 (CCK-8) was acquired from Dojindo Molecular Technologies (Kumamoto, Japan). Before CCK-8 assay, cells were treated with different samples of conditioned medium for 48 h. Then cells were plated into 96-well plates at 5 × 103 cells/well. On day 0, 1 and 3, a total of 10 μL of CCK-8 solution were added into each well before returning to the 37°C incubator for 1 h, after which the optical density (OD) values were detected by a Bio-Rad microplate reader. Wells without cells were used as blanks. The detailed procedures were as described previously[47].
Migration assay
After treating with conditioned medium from different cultures for 48 h, cells were inoculated onto the surface of the upper chamber of a Corning 8-μm transwell plate (Corning, NY, USA). The detailed procedures were as described previously [34, 35]. The method of quantifying the results of the migration assay involved counting the migrated cells as described previously [35].
Tubule formation assay
After treating the cells with different samples of conditioned medium for 48 h, angiogenic activity was evaluated using ECM gel (Sigma-Aldrich, St Louis, MO, USA). The detailed procedures were as described previously [34]. The quantitative method for evaluating tubule formation involved calculating the percentage of tube numbers relative to the positive control group (the CM-siCtrl group in this study) as described previously [48].
Immunoblotting and antibodies
The detailed procedures used for protein extraction and western blotting were as described previously [34, 49]. The primary antibodies used in this study, including p-mTOR, mTOR, HIF-1α, p-AKT, AKT, p-ERK, ERK, and β-actin were obtained from Cell Signaling Technology (Boston, MA, USA).
Enzyme-linked immunosorbent assay (ELISA)
The anti-VEGFA and anti-FGF2 primary antibodies for ELISA were purchased from Abcam (Cambridge, UK). The ELISA coating solution, TMB Substrate, and ELISA STOP solution were purchased from SolarBio (Beijing, China). The detailed procedures were as described previously [34].
Nude mouse transplantation tumour assay
To study therapeutic strategies for malignant tumours, the most well-recognized model is the nude mouse transplantation tumour assay [50]. We used 6-week-old BALB/c nude mice, which were raised in a specific pathogen free (SPF)-grade animal experimental centre which has a laminar flow device and a specific light-dark cycle (12 h light: 12 h dark).
All experimental and animal care procedures were approved by the Animal Research Ethics Committee of Shanghai Sixth People's Hospital, with reference to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines, the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education by the New York Academy of Sciences, and the National Institutes of Health (NIH) guide for the care and use of laboratory animals.
The nude mice were inoculated around the left side of the neck with 5 × 106 MNNG/HOS cells by subcutaneous injection. Seven days later, when the OS tumours had reached approximately 3 mm3, the mice were randomly allocated into the control group or the siMALAT1 group (n = 8 per group; five for long-term observation and three to obtain tissue specimens). In the groups for long-term observation, on day 7, 12, and 17 after inoculation, the siMALAT1 working solution was injected into the transplanted tumours. In the groups for obtaining tissue specimens, on day 7 after inoculation, the siMALAT1 working solution was injected into the transplanted tumours and the mice were euthanised with an overdose of anaesthetic, on day 9 after inoculation, before obtaining tissue specimens for western blotting or PCR assays. The siMALAT1 working solution was prepared by mixing siMALAT1 (10 mg/kg) with Invivofectamine™, and diluting using normal saline according to the manufacturer's instructions; in the control group, siMALAT1 was replaced by an equal quantity of siCtrl.
On day 21 after inoculation, the mice were euthanised with an overdose of anaesthetic. The tumours were carefully excised and prepared for paraffin embedding and sectioning, as described in a previous study [35].
Histological staining and immunofluorescence staining of paraffin sections
After sectioning, the sections were used for immunofluorescence staining, following the detailed procedures described in previous publications [35, 51]. The cleaved-caspase-3 primary antibodies were obtained from Cell Signaling Technology, while other primary antibodies including VEGF-A, Ki67 and CD31 were obtained from Abcam.
Statistical analysis
Numerical data are shown as means ( ) ± standard deviation (SD). One-way analysis of variance (ANOVA), which was performed using RStudio (Boston, MA, USA), was used to confirm the statistically-significant differences. When P values were < 0.05, the means were considered to be significantly different.
) ± standard deviation (SD). One-way analysis of variance (ANOVA), which was performed using RStudio (Boston, MA, USA), was used to confirm the statistically-significant differences. When P values were < 0.05, the means were considered to be significantly different.
Results
MALAT1/HIF-1α loop in OS cells
The expression level of MALAT1 in OS was measured by subjecting the RT-PCR products to PAGE (Fig. 1A). We found that expression of MALAT1 was dramatically increased in OS compared to bone marrow stromal cells (BMSCs), representative normal cells with osteogenic potential in the bone marrow. The siRNA of MALAT1, used in this study, was proven to be effective in suppressing the level of MALAT1 in OS cells (Fig. 1A). Analysis of ex vivo tissue specimens further confirmed the in vivo inhibiting ability of siMALAT1 (Fig. S1A).
MALAT1/HIF-1α loop in OS cells. (A) RT-PCR products detected by PAGE. (B) Phosphorylation level of mTOR detected by western blot. (C) HIF-1α detected by western blot. (D) RT-PCR products detected by PAGE.
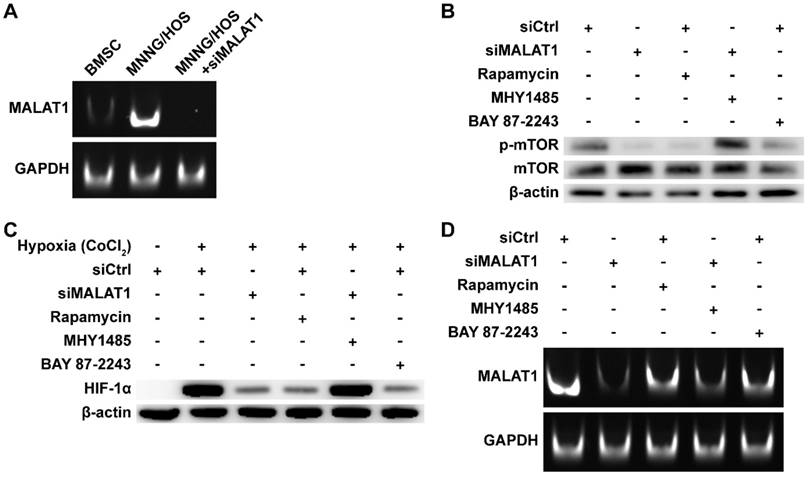
To confirm the existence of a MALAT1/HIF-1α loop [27, 28], we obtained inhibitors for the main nodes of this loop, including rapamycin, a classical inhibitor of mTOR [42, 52], and BAY 87-2243, a selective inhibitor of HIF-1α [45]. To further confirm that mTOR is a crucial node in the MALAT1/HIF-1α loop, we obtained MHY1485, an agonist of mTOR [43, 44].
We confirmed the efficiency of rapamycin and MHY1485 by western blotting (Fig. 1B). We observed that siMALAT1 markedly inhibited mTOR signalling (Fig. 1B), indicating that the abnormal activation of mTOR signalling in OS is induced by MALAT1. Rapamycin inhibited the phosphorylation of mTOR, simulating the effect caused by siMALAT1. MHY1485 rescued the siMALAT1-induced inhibition of mTOR phosphorylation. Confirmation of the HIF-1α/MALAT1/mTOR feedback mechanism was further confirmed using BAY 87-2243, the HIF-1α inhibitor. The result showed that inhibition of HIF-1α caused slight inhibition of mTOR via the feedback loop (Fig. 1B).
In consideration of the difficulty of detecting HIF-1α under normoxic conditions, the well-recognized solution is to use CoCl2 to simulate hypoxia and then detect HIF-1α [53]. Using this method we found that the expression of HIF-1α was not so hard to detect under simulated hypoxia, but addition of siMALAT1, rapamycin or BAY 87-2243 each inhibited the expression of HIF-1α (Fig. 1C). MHY1485, which rescued the siMALAT1-induced inhibition of mTOR phosphorylation, rescued the siMALAT1-induced inhibition of HIF-1α. These results show that mTOR is a critical player in the MALAT1/HIF-1α axis. The siMALAT1-induced inhibition of HIF-1α was further confirmed using ex vivo tumour tissue specimens (Fig. S1B).
We further used PAGE to investigate the HIF-1α/MALAT1 feedback to produce a complete picture (Fig. 1D). Our results showed that rapamycin-induced inhibition of mTOR and the inhibition of HIF-1α both slightly inhibited the expression of MALAT1, and this inhibition could be partially relieved by MHY1485.
Changes of the secretome mediated by the MALAT1/HIF-1α loop
Changes in the levels of growth factors including FGF2 and VEGF-A were measured using ELISA. The results showed that siMALAT1, rapamycin and BAY 87-2243 all inhibited the secretion of FGF2 and VEGF-A under both normoxia and hypoxia (Fig. 2A). Furthermore this effect of siMALAT1 was rescued by activation of mTOR using MHY1485 (Fig. 2A).
Changes of the secretome mediated by the MALAT1/HIF-1α loop. (A) Changes of FGF2 and VEGF-A detected by ELISA. Under normoxic conditions, *P < 0.05 compared to siCtrl and **P < 0.01 compared to siCtrl. Under hypoxic conditions, #P < 0.05 compared to siCtrl and ##P < 0.01 compared to siCtrl. (B) The phosphorylation level of AKT and ERK after vascular endothelial cells were treated by different conditioned medium (CM). The (C) proliferation rate, (D) migration rate and (E) tube formation ability of vascular endothelial cells (HMEC-1) after treated by different CM. *P < 0.05 compared to control.
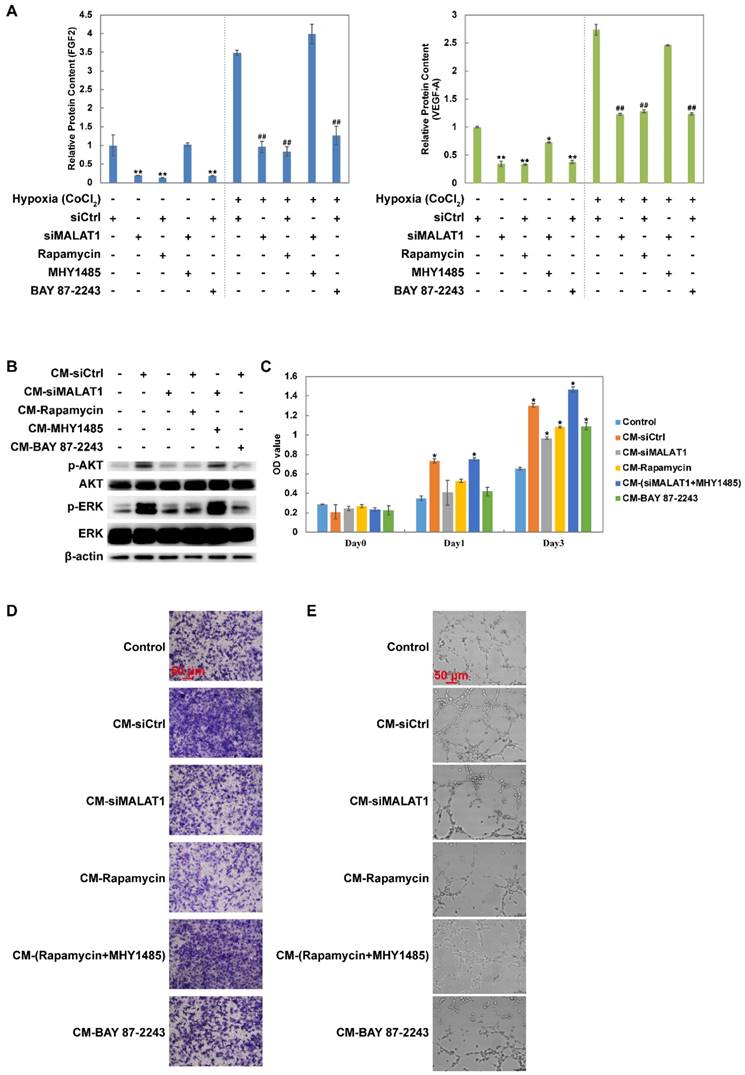
Conditioned medium (CM) from MNNG/HOS cells treated with siCtrl dramatically activated both AKT and ERK while CM from MNNG/HOS cells treated with siMALAT1, rapamycin or BAY 87-2243 had a weaker effect (Fig. 2B). The reduced effect of siMALAT1 could be rescued by combination treatment with MHY1485 (Fig. 2B).
The pro-angiogenic activity of CM was determined using CCK-8 assay (Fig. 2C), transwell assay (Fig. 2D and Fig. S1C) and tube formation assay (Fig. 2E and Fig. S1D). The angiogenic activity was in accordance with the activation level of AKT and ERK.
Suppression of tumour growth induced by chemically-modified siMALAT1 by blocking its pro-angiogenic ability
Tumour volume was significantly smaller in the siMALAT1 group than in the control group (Fig. 3A) indicating that chemically-modified siMALAT1 was effective in suppressing tumour growth.
(A) Tumour volume was significantly smaller after siMALAT1 treatment. The in vivo expression of (B) Ki67, (C) cleaved caspase-3, (D) VEGF-A and (E) CD31 were detected by immunofluorescence.
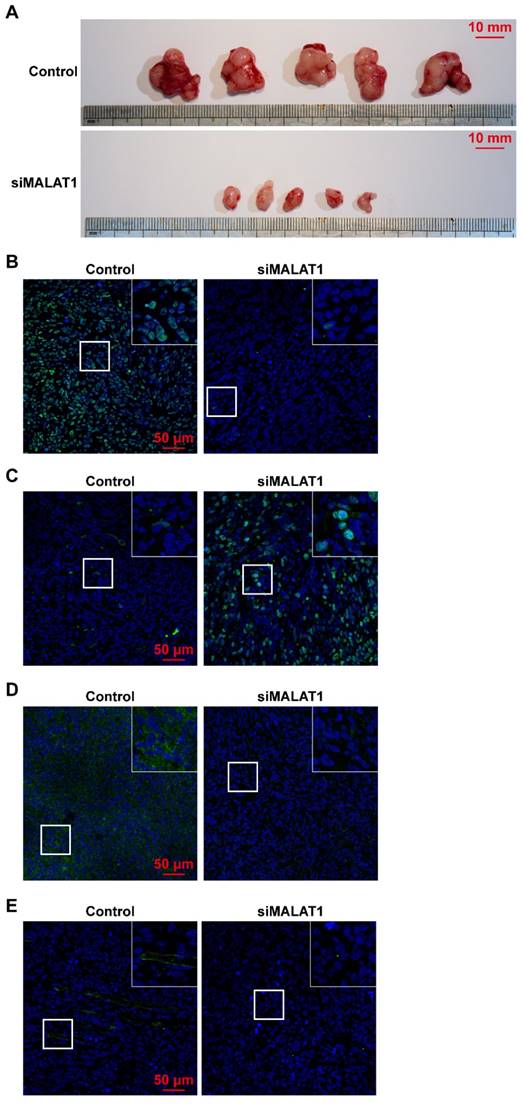
The proliferative ability of OS in vivo was measured by Ki67 staining, and the results showed that siMALAT significantly reduced the proliferative ability of OS cells (Fig. 3B). In addition, without an adequate blood supply provided by angiogenesis, numerous internal cells in the siMALAT1 group underwent apoptosis, measured by staining of cleaved caspase-3, but this was not observed in the control group (Fig. 3C).
We observed that in tumours treated with siMALAT1, the level of expression of VEGF-A was much lower compared to the control group (Fig. 3D). Virtually no newly-formed vessels (observed by CD31 staining) were observed in the siMALAT1 group, while there were plenty of newly-formed vessels in the control group (Fig. 3E).
Discussion
Malignant osteoblasts, which constitute OS, produce immature and infantile osseous or osteoid tissue [54]. OS tends to occur in children and adolescents who are undergoing a growth spurt [55]. The main therapeutic method for OS is surgical treatment, but the survival rate remains low at approximately 15-17% without the help of chemotherapy [56, 57]. BMSCs give rise to pre-osteoblastic cells, which express osteoblast-specific proteins including alkaline phosphatase (ALP) and type I collagen [58]. We used BMSCs as the normal control and found that MNNG/HOS, an OS cell line, expressed abnormally high levels of MALAT1. We suspected that this abnormal expression might be the 'chief culprit' of OS progression.
The growth and metastasis of a tumour is largely controlled by the microenvironment [59]. Angiogenesis is one of the hallmarks of tumour progression and plays an important role as a permissive action [60]. Angio-prevention, targeting angiogenesis induced by tumour cells, could retain the tumour cell in an indolent state and restrain tumour progression [61].
For the past few years, an increasing number of studies have revealed that angiogenesis plays a crucial part in OS progression including in proliferation, migration and metastasis [62]. In the present study, the results showed that conditioned medium from MNNG/HOS cells had powerfully pro-angiogenic abilities. After blocking this ability, OS tumour progression in vivo was dramatically inhibited.
With gradual deepening of the understanding of the crucial role of lncRNAs in tumour progression, increasing evidence has been found connecting lncRNA to the pathogenesis of several malignant tumours including OS [63]. We found that MALAT1, which has a high expression level in a variety of malignant tumours [18, 19], was closely related to OS progression [20], suggesting that MALAT1 could be a potential therapeutic target. However, the detailed mechanism of MALAT1-induced OS progression is still unknown. In this study, we confirmed that expression of MALAT1, which is correlated to hypoxia-induced angiogenesis [22], is closely related to the pro-angiogenic effect induced by OS [23-25], by enhancing the expression level of pro-angiogenic factors including VEGF-A and FGF2.
In the process of tumour-induced angiogenesis, VEGF-A is regarded as a key participant [64]. In this study, our results indicated that the abnormally-high MALAT1 expression caused up-regulation of pro-angiogenic factors including VEGF-A. In particular, MALAT1 promotes the expression of VEGF-A and consequently, angiogenesis, through the MALAT1/mTOR/HIF-1α pathway, and the increased level of HIF-1α has a positive-feedback effect causing increased expression of MALAT1.
As the number of tumour cells increased, an oxygen-deficient environment gradually formed, leading to the development of an environment that the tumour cells could not survive without strong angiogenic ability [65, 66]. Consequently, abnormally-active pro-angiogenic ability is one of the hallmarks of malignancy [66]. HIF-1α, which accumulates in hypoxia and is degraded in normoxia [67], participates in the regulation of angiogenesis by influencing the expression level of several pro-angiogenic factors including VEGF-A [68, 69]. In the present study, we inhibited HIF-1α using BAY 87-2243, and found that HIF-1α-inhibition could suppress the OS-induced pro-angiogenic effect. However, HIF-1α is not a specific target and is present in a variety of cell types. The use of a HIF-1α inhibitor has risks including ischemic necrosis of normal tissue. In contrast, MALAT1 is relatively specific to OS rather than normal tissue. Therapeutic strategies targeting MALAT1 could thus inhibit the abnormal OS-induced angiogenesis, without affecting normal tissue-induced angiogenesis.
The kinase mTOR participates in integration and transmission of signals in a variety of signal pathways involved in cell growth and survival, by regulating mRNA expression, metabolism and autophagy [70]. The activation of mTOR phosphorylates several proteins including ribosomal protein S6 kinase (p70S6K) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1) [71]. The synthesis of HIF-1α is influenced by the mTOR signal pathway [72]. Previous studies found that rapamycin, an inhibitor of mTOR which acts by inhibiting the mTOR/p70S6K/4E-BP1 pathway, could reduce the metastasis of malignant osteosarcoma [73, 74], in which the inhibition of neo-angiogenesis may play an important role [74]. However, mTOR participates in multifarious physiological functions of normal cells [75-77]. Hence, inhibition of mTOR is not a treatment strategy which specifically targets OS. In the present study, our data showed that inhibition of MALAT1, a relatively specific target of OS, could inhibit mTOR and its downstream function, having a similar effect to that of rapamycin. Consequently we believe that MALAT1 will be a better potential target for the treatment of OS.
Current research has indicated the possibility of the existence of a MALAT1/HIF-1α loop. The positive correlation between MALAT1 and HIF-1α was recently revealed [78] and, further, increased HIF-1α levels were found to significantly increase the expression level of MALAT1 [27]. Moreover, the intimate relationship between MALAT1 and the mTOR pathway was also elucidated recently [29, 79], and, in particular, the fact that MALAT1 can activate mTOR via modulation of S6K1 alternative splicing [29]. More interestingly, mTOR is a widely-recognized upstream modulator of HIF-1α [72]. Thus, our work, inspired by these previous studies, verified the existence of a MALAT1/HIF-1α loop, and further identified the important role of mTOR in the MALAT1/HIF-1α loop. However, certain inadequacies persist in current knowledge, and more in-depth studies are still needed.
Although siRNAs are commonly-used tools in gene regulation, they have powerful potential for expansion. When combined with a variety of smart nanovesicle technology [80-82], especially with exosome technology [83, 84], the delivery of siRNA could be targeted to specific cells [85], and exosome-encapsulated siRNA could provide a more stable and effective source with less influence on normal cells [35, 86]. Thus, in the future, siMALAT1 could not only be administered via intra-tumour injection but there is also the possibility of systemic delivery when combined with smart nanovesicles such as exosomes which target OS cells.
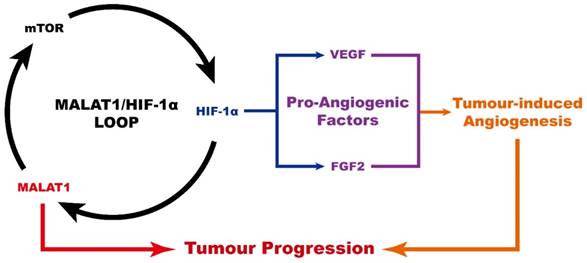
In conclusion (Fig. 4), MALAT1 causes an abnormally high pro-angiogenic effect by enhancing the expression level of pro-angiogenic factors including VEGF-A and FGF2, acting via the MALAT1/mTOR/HIF-1α pathway as well as through positive feedback from HIF-1α to MALAT1. As a consequence, MALAT1 may be considered as a new promising therapeutic target to prevent OS progression.
Schematic diagram showing the role of MALAT1 in the progression of osteosarcoma.
Supplementary Material
Figure S1.
Acknowledgements
This study was supported by the National Natural Science Foundation of China [Grant No.81572239].
Author Contributions
Conception and design: Shi-Cong Tao, Xiao-Lin Li
Development of methodology: Ting Yuan
Acquisition of data: Zhi-Chang Zhang, Chun Tang
Analysis and interpretation of data: Jing Zhang, Yang Dong
Writing of the manuscript: Shi-Cong Tao, Zhi-Chang Zhang
Study supervision: Xiao-Lin Li
All authors reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mirabello L, Troisi R, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009;115:1531-43
2. Mirabello L, Troisi R, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. International Journal of Cancer. 2009;125:229-34
3. Savage SA, Mirabello L. Using Epidemiology and Genomics to Understand Osteosarcoma Etiology. Sarcoma. 2011;2011:548151 -
4. Kager L, Zoubek A, Potschger U, Kastner U, Flege S, Kempfbielack B. et al. Primary Metastatic Osteosarcoma: Presentation and Outcome of Patients Treated on Neoadjuvant Cooperative Osteosarcoma Study Group Protocols. Journal of Clinical Oncology. 2016;21:2011-8
5. Mialou V, Philip T, Kalifa C, Perol D, Gentet J, Marecberard P. et al. Metastatic osteosarcoma at diagnosis: prognostic factors and long-term outcome-the French pediatric experience. Cancer. 2005;104:1100-9
6. Gorlick R, Khanna C. Osteosarcoma. J Bone Miner Res. 2010;25:683-91
7. Miller BJ, Cram P, Lynch CF, Buckwalter JA. Risk factors for metastatic disease at presentation with osteosarcoma: an analysis of the SEER database. J Bone Joint Surg Am. 2013;95:e89
8. Chang LC, Yu YL. Dietary components as epigenetic-regulating agents against cancer. Biomedicine (Taipei). 2016;6:2
9. Matsumoto K, Ema M. Roles of VEGF-A signalling in development, regeneration, and tumours. J Biochem. 2014;156:1-10
10. Padma VV. An overview of targeted cancer therapy. Biomedicine (Taipei). 2015;5:19
11. Liao YY, Tsai HC, Chou PY, Wang SW, Chen HT, Lin YM. et al. CCL3 promotes angiogenesis by dysregulation of miR-374b/ VEGF-A axis in human osteosarcoma cells. Oncotarget. 2016;7:4310-25
12. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A. et al. Landscape of transcription in human cells. Nature. 2012;489:101-8
13. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H. et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Research. 2012;22:1775-89
14. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47-62
15. Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73:2491-509
16. Wang B, Su Y, Yang Q, Lv D, Zhang W, Tang K. et al. Overexpression of Long Non-Coding RNA HOTAIR Promotes Tumor Growth and Metastasis in Human Osteosarcoma. Mol Cells. 2015;38:432-40
17. Cong M, Li J, Jing R, Li Z. Long non-coding RNA tumor suppressor candidate 7 functions as a tumor suppressor and inhibits proliferation in osteosarcoma. Tumour Biol. 2016;37:9441-50
18. Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM. et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031-41
19. Lin R, Maeda S, Liu C, Karin M, Edgington TS. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007;26:851-8
20. Cai X, Liu Y, Yang W, Xia Y, Yang C, Yang S. et al. Long noncoding RNA MALAT1 as a potential therapeutic target in osteosarcoma. J Orthop Res. 2016;34:932-41
21. Dong Y, Liang G, Yuan B, Yang C, Gao R, Zhou X. MALAT1 promotes the proliferation and metastasis of osteosarcoma cells by activating the PI3K/Akt pathway. Tumour Biol. 2015;36:1477-86
22. Thum T, Fiedler J. LINCing MALAT1 and angiogenesis. Circ Res. 2014;114:1366-8
23. Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T. et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389-97
24. Sun JY, Zhao ZW, Li WM, Yang G, Jing PY, Li P. et al. Knockdown of MALAT1 expression inhibits HUVEC proliferation by upregulation of miR-320a and downregulation of FOXM1 expression. Oncotarget. 2017
25. Huang JK, Ma L, Song WH, Lu BY, Huang YB, Dong HM. et al. LncRNA-MALAT1 Promotes Angiogenesis of Thyroid Cancer by Modulating Tumor-Associated Macrophage FGF2 Protein Secretion. J Cell Biochem. 2017
26. Li X, Song Y, Liu F, Liu D, Miao H, Ren J. et al. Long Non-Coding RNA MALAT1 Promotes Proliferation, Angiogenesis, and Immunosuppressive Properties of Mesenchymal Stem Cells by Inducing VEGF and IDO. J Cell Biochem. 2017
27. Salle-Lefort S, Miard S, Nolin MA, Boivin L, Pare ME, Debigare R. et al. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1alpha axis. Int J Oncol. 2016;49:1731-6
28. Luo F, Liu X, Ling M, Lu L, Shi L, Lu X. et al. The lncRNA MALAT1, acting through HIF-1alpha stabilization, enhances arsenite-induced glycolysis in human hepatic L-02 cells. Biochim Biophys Acta. 2016;1862:1685-95
29. Malakar P, Shilo A, Mogilevsky A, Stein I, Pikarsky E, Nevo Y. et al. Long Noncoding RNA MALAT1 Promotes Hepatocellular Carcinoma Development by SRSF1 Upregulation and mTOR Activation. Cancer Res. 2017;77:1155-67
30. Sudhagar S, Sathya S, Lakshmi BS. Rapid non-genomic signalling by 17beta-oestradiol through c-Src involves mTOR-dependent expression of HIF-1alpha in breast cancer cells. Br J Cancer. 2011;105:953-60
31. Lin CH, Li CH, Liao PL, Tse LS, Huang WK, Cheng HW. et al. Silibinin inhibits VEGF secretion and age-related macular degeneration in a hypoxia-dependent manner through the PI-3 kinase/Akt/mTOR pathway. Br J Pharmacol. 2013;168:920-31
32. Zhang Y, Sun S, Chen J, Ren P, Hu Y, Cao Z. et al. Oxymatrine induces mitochondria dependent apoptosis in human osteosarcoma MNNG/HOS cells through inhibition of PI3K/Akt pathway. Tumour Biol. 2014;35:1619-25
33. Pinski J, Schally AV, Groot K, Halmos G, Szepeshazi K, Zarandi M. et al. Inhibition of Growth of Human Osteosarcomas by Antagonists of Growth Hormone-Releasing Hormone. Journal of the National Cancer Institute. 1995;87:1787-94
34. Tao SC, Yuan T, Rui BY, Zhu ZZ, Guo SC, Zhang CQ. Exosomes derived from human platelet-rich plasma prevent apoptosis induced by glucocorticoid-associated endoplasmic reticulum stress in rat osteonecrosis of the femoral head via the Akt/Bad/Bcl-2 signal pathway. Theranostics. 2017;7:733-50
35. Guo SC, Tao SC, Yin WJ, Qi X, Yuan T, Zhang CQ. Exosomes derived from platelet-rich plasma promote the re-epithelization of chronic cutaneous wounds via activation of YAP in a diabetic rat model. Theranostics. 2017;7:81-96
36. Pontillo C, Espanol A, Chiappini F, Miret N, Cocca C, Alvarez L. et al. Hexachlorobenzene promotes angiogenesis in vivo, in a breast cancer model and neovasculogenesis in vitro, in the human microvascular endothelial cell line HMEC-1. Toxicology letters. 2015;239:53-64
37. van der Schaft DW, Seftor RE, Seftor EA, Hess AR, Gruman LM, Kirschmann DA. et al. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J Natl Cancer Inst. 2004;96:1473-7
38. Zhang C, Yang F, Zhang XW, Wang SC, Li MH, Lin LP. et al. Grateloupia longifolia polysaccharide inhibits angiogenesis by downregulating tissue factor expression in HMEC-1 endothelial cells. Br J Pharmacol. 2006;148:741-51
39. Huang L, Perrault C, Coelho-Martins J, Hu C, Dulong C, Varna M. et al. Induction of acquired drug resistance in endothelial cells and its involvement in anticancer therapy. Journal of hematology & oncology. 2013;6:49
40. Clark JC, Akiyama T, Thomas DM, Labrinidis A, Evdokiou A, Galloway SJ. et al. RECK in osteosarcoma: a novel role in tumour vasculature and inhibition of tumorigenesis in an orthotopic model. Cancer. 2011;117:3517-28
41. Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB. et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336-46
42. Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell. 1996;7:25-42
43. Cheng Y, Kim J, Li XX, Hsueh AJ. Promotion of ovarian follicle growth following mTOR activation: synergistic effects of AKT stimulators. PLoS One. 2015;10:e0117769
44. Choi YJ, Park YJ, Park JY, Jeong HO, Kim DH, Ha YM. et al. Inhibitory effect of mTOR activator MHY1485 on autophagy: suppression of lysosomal fusion. PLoS One. 2012;7:e43418
45. Ellinghaus P, Heisler I, Unterschemmann K, Haerter M, Beck H, Greschat S. et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013;2:611-24
46. Helbig L, Koi L, Bruchner K, Gurtner K, Hess-Stumpp H, Unterschemmann K. et al. BAY 87-2243, a novel inhibitor of hypoxia-induced gene activation, improves local tumor control after fractionated irradiation in a schedule-dependent manner in head and neck human xenografts. Radiat Oncol. 2014;9:207
47. Tao SC, Yuan T, Zhang YL, Yin WJ, Guo SC, Zhang CQ. Exosomes derived from miR-140-5p-overexpressing human synovial mesenchymal stem cells enhance cartilage tissue regeneration and prevent osteoarthritis of the knee in a rat model. Theranostics. 2017;7:180-95
48. Amin MA, Rabquer BJ, Mansfield PJ, Ruth JH, Marotte H, Haas CS. et al. Interleukin 18 induces angiogenesis in vitro and in vivo via Src and Jnk kinases. Annals of the rheumatic diseases. 2010;69:2204-12
49. Tao SC, Gao YS, Zhu HY, Yin JH, Chen YX, Zhang YL. et al. Decreased extracellular pH inhibits osteogenesis through proton-sensing GPR4-mediated suppression of yes-associated protein. Sci Rep. 2016;6:26835
50. Hasei J, Sasaki T, Tazawa H, Osaki S, Yamakawa Y, Kunisada T. et al. Dual programmed cell death pathways induced by p53 transactivation overcome resistance to oncolytic adenovirus in human osteosarcoma cells. Mol Cancer Ther. 2013;12:314-25
51. Guo SC, Tao SC, Yin WJ, Qi X, Sheng JG, Zhang CQ. Exosomes from Human Synovial-Derived Mesenchymal Stem Cells Prevent Glucocorticoid-Induced Osteonecrosis of the Femoral Head in the Rat. Int J Biol Sci. 2016;12:1262-72
52. Edwards SR, Wandless TJ. The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J Biol Chem. 2007;282:13395-401
53. Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S. et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691-703
54. Doyle LA. Sarcoma classification: An update based on the 2013 World Health Organization Classification of Tumors of Soft Tissue and Bone. Cancer. 2014;120:1763-74
55. Gill J, Ahluwalia MK, Geller D, Gorlick R. New targets and approaches in osteosarcoma. Pharmacol Ther. 2013;137:89-99
56. Bernthal NM, Federman N, Eilber FR, Nelson SD, Eckardt JJ, Eilber FC. et al. Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer. 2012;118:5888-93
57. Link MP, Goorin AM, Miser AW, Green AA, Pratt CB, Belasco JB. et al. The Effect of Adjuvant Chemotherapy on Relapse-Free Survival in Patients with Osteosarcoma of the Extremity. The New England Journal of Medicine. 1986;314:1600-6
58. Wang D, Christensen KN, Chawla K, Xiao G, Krebsbach PH, Franceschi RT. Isolation and Characterization of MC3T3-E1 Preosteoblast Subclones with Distinct In Vitro and In Vivo Differentiation/Mineralization Potential†. Journal of Bone and Mineral Research. 1999;14:893-903
59. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T. et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130-3
60. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
61. Albini A, Tosetti F, Li VW, Noonan DM, Li WW. Cancer prevention by targeting angiogenesis. Nat Rev Clin Oncol. 2012;9:498-509
62. Geller DS, Gorlick R. Osteosarcoma: a review of diagnosis, management, and treatment strategies. Clin Adv Hematol Oncol. 2010;8:705-18
63. Zhou Q, Chen F, Fei Z, Zhao J, Liang Y, Pan W. et al. Genetic variants of lncRNA HOTAIR contribute to the risk of osteosarcoma. Oncotarget. 2016;7:19928-34
64. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273:114-27
65. Wang LH, Tsai HC, Cheng YC, Lin CY, Huang YL, Tsai CH. et al. CTGF promotes osteosarcoma angiogenesis by regulating miR-543/angiopoietin 2 signaling. Cancer Lett. 2017;391:28-37
66. Bid HK, Roberts RD, Cam M, Audino A, Kurmasheva RT, Lin J. et al. DeltaNp63 promotes pediatric neuroblastoma and osteosarcoma by regulating tumor angiogenesis. Cancer Res. 2014;74:320-9
67. Wan J, Chai H, Yu Z, Ge W, Kang N, Xia W. et al. HIF-1alpha effects on angiogenic potential in human small cell lung carcinoma. J Exp Clin Cancer Res. 2011;30:77
68. Rankin EB, Rha J, Unger TL, Wu CH, Shutt HP, Johnson RS. et al. Hypoxia-inducible factor-2 regulates vascular tumorigenesis in mice. Oncogene. 2008;27:5354-8
69. Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet. 2016;388:518-29
70. Guertin DA, Sabatini DM. Defining the Role of mTOR in Cancer. Cancer Cell. 2007;12:9-22
71. Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Molecular and Cellular Biology. 2004;24:2986-97
72. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851-64
73. Wan X, Mendoza A, Khanna C, Helman LJ. Rapamycin inhibits ezrin-mediated metastatic behavior in a murine model of osteosarcoma. Cancer Res. 2005;65:2406-11
74. Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J. et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779-85
75. Bockaert J, Marin P. mTOR in Brain Physiology and Pathologies. Physiol Rev. 2015;95:1157-87
76. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V. et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684
77. Hu JK, Du W, Shelton SJ, Oldham MC, DiPersio CM, Klein OD. An FAK-YAP-mTOR Signaling Axis Regulates Stem Cell-Based Tissue Renewal in Mice. Cell Stem Cell. 2017;21:91-106 e6
78. Lelli A, Nolan KA, Santambrogio S, Goncalves AF, Schonenberger MJ, Guinot A. et al. Induction of long noncoding RNA MALAT1 in hypoxic mice. Hypoxia (Auckland, NZ). 2015;3:45-52
79. Covach A, Patel S, Hardin H, Lloyd RV. Phosphorylated Mechanistic Target of Rapamycin (p-mTOR) and Noncoding RNA Expression in Follicular and Hurthle Cell Thyroid Neoplasm. Endocrine pathology. 2017
80. Guo P, Yang J, Jia D, Moses MA, Auguste DT. ICAM-1-Targeted, Lcn2 siRNA-Encapsulating Liposomes are Potent Anti-angiogenic Agents for Triple Negative Breast Cancer. Theranostics. 2016;6:1-13
81. Yu H, Guo C, Feng B, Liu J, Chen X, Wang D. et al. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics. 2016;6:14-27
82. Yang T, Zhao P, Rong Z, Li B, Xue H, You J. et al. Anti-tumor Efficiency of Lipid-coated Cisplatin Nanoparticles Co-loaded with MicroRNA-375. Theranostics. 2016;6:142-54
83. Tao SC, Guo SC, Li M, Ke QF, Guo YP, Zhang CQ. Chitosan Wound Dressings Incorporating Exosomes Derived from MicroRNA-126-Overexpressing Synovium Mesenchymal Stem Cells Provide Sustained Release of Exosomes and Heal Full-Thickness Skin Defects in a Diabetic Rat Model. Stem Cells Transl Med. 2017;6:736-47
84. Tao SC, Guo SC, Zhang CQ. Platelet-derived Extracellular Vesicles: An Emerging Therapeutic Approach. Int J Biol Sci. 2017;13:828-34
85. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-5
86. Liu D, Yang F, Xiong F, Gu N. The Smart Drug Delivery System and Its Clinical Potential. Theranostics. 2016;6:1306-23
Author contact
![]() Corresponding authors: Xiao-Lin Li, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China E-mail: lixiaolinedu.cn Shi-Cong Tao, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China E-mail: jerrytao1990com
Corresponding authors: Xiao-Lin Li, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China E-mail: lixiaolinedu.cn Shi-Cong Tao, Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China E-mail: jerrytao1990com