Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(1):47-56. doi:10.7150/ijbs.22209 This issue Cite
Research Paper
Dioscin overcome TKI resistance in EGFR-mutated lung adenocarcinoma cells via down-regulation of tyrosine phosphatase SHP2 expression
Yao-Chen Wang1,2, De-Wei Wu5, Tzu-Chin Wu1,2, Lee Wang3, Chih-Yi Chen2,4, Huei Lee5 ![]()
1. Department of Internal Medicine, Chung Shan Medical University and Hospital, Taichung, Taiwan.
2. School of Medicine, Chung Shan Medical University and Hospital, Taichung, Taiwan.
3. Department of Public Health, Chung Shan Medical University, Taichung, Taiwan.
4. Department of Surgery, Chung Shan Medical University and Hospital, Taichung, Taiwan.
5. Graduate Institute of Cancer Biology and Drug Discovery, Taipei Medical University, Taipei, Taiwan.
Received 2017-8-3; Accepted 2017-12-14; Published 2018-1-11
Abstract

Resistance to tyrosine kinase inhibitors (TKIs) results in tumor relapse and poor prognosis in patients with lung adenocarcinoma. TKI resistance caused by epidermal growth factor receptor (EGFR) mutations at T790M and c-Met amplification occurs through persistent activation of the MEK/ERK and PI3K/AKT signaling pathways. We therefore expected that dual inhibitors of both signaling pathways could overcome TKI resistance in lung adenocarcinoma. Here, dioscin was selected from a product library of Chinese naturally occurring compounds and overcame TKI resistance in EGFR-mutated lung adenocarcinoma cells. Mechanistically, dioscin may down-regulate the expression of SH2 domain-containing phosphatase-2 (SHP2) at the transcription level by increasing p53 binding to the SHP2 promoter due to reactive oxygen species (ROS). Simultaneous inhibition of MEK/ERK and PI3K/AKT activation via decreased SHP2 expression and its interaction with GAB1 may be responsible for dioscin-mediated TKI sensitivity. A higher unfavorable response to TKI therapy occurred more commonly in patients with high SHP2 mRNA expression than in patients with low SHP2 mRNA expression. Therefore, we suggest that dioscin may act as a dual inhibitor of the MEK/ERK and PI3K/AKT signaling pathways to overcome TKI resistance via dysregulation of SHP2 expression in lung adenocarcinoma.
Keywords: Dioscin, EGFR, lung adenocarcinoma
Introduction
Tyrosine kinase inhibitors (TKIs) inhibit kinase activity of epidermal growth factor receptor (EGFR) and exhibit a better clinical benefit in the treatment of EGFR-mutated lung adenocarcinoma [1]. Unfortunately, nearly complete drug resistance and tumor relapse occur in patients who have received TKI-targeting therapy, even with third generation TKIs [2,3]. Therefore, new clinical approaches using precise medicine are needed to overcome TKI resistance and to suppress tumor recurrence for patients with lung adenocarcinoma.
We and other groups have indicated that the activation of MEK/ERK and PI3K/AKT signaling by EGFR and c-MET may induce TKI resistance in lung adenocarcinoma cells by modulating the epidermal-to-mesenchymal transition (EMT) and the apoptotic pathway [4-7]. Dual inhibition of both pathways may potentially result in more favorable efficacy when compared with inhibition of either pathway alone in patients with advanced cancers, but with the risk of greater toxicity [7]. However, a single agent that can simultaneously suppress both pathways is not yet available for overcoming TKI resistance.
The Src homology 2 (SH2) domain-containing PTP2 (SHP2) encoded by the PTPN11 gene is required for activation of the RAS/ERK signaling via regulation of Src family kinase activity [8]. The EGFR-activated Src family kinase was required for GAB1 phosphorylation and maintenance of the GAB1-SHP2 association, and consequently for the activation of PI3K/AKT signaling [9]. Therefore, we expected that inhibition of both the MEK/ERK and the PI3K/AKT signaling pathways by targeting SHP2 might be an effective strategy for overcoming TKI resistance in lung adenocarcinoma.
Dioscin a natural steroidal saponin which is abundant in some medicinal plants, such as Dioscorea nipponica Makino [10]. Dioscin exerts effects such as protection against acute chemically mediated liver injury, amelioration of cerebral ischemia/reperfusion injury, and anti-inflammatory activities [11-15]. Intriguingly, the anticancer potential of dioscin was effective in various cancer cells [15,16]. Doscin inhibits cancer cell survival via various mechanisms. For example, dioscin induces cancer cell apoptosis via the mitochondrial pathway in esophageal, laryngeal, and gastric cancer cells [17-23]. In non-small cell lung cancer (NSCLC) cells, dioscin inhibited the proliferation and induction of apoptosis in A549, H460, and H446 cells [20], indicated that dioscin-induce autophagy mitigates cell apoptosis through modulation of PI3K/AKT and ERK and JNK signaling pathways in NSCLC cells [19]. Treatment of A549 and H1299 cells with dioscin caused a dose-dependent increase in ERK1/2 and JNK1/2 activity, accompanied with a decreased PI3K expression and decreased phosphorylation of AKT and mTOR [19]. These results suggest dioscin as an anticancer agent in human cancers including NSCLC. Multidrug resistance is a serious obstacle encountered in cancer treatment. Dioscin restores the activity of the anticancer agent Adriamycin in multidrug-resistant human leukemia K562/Adriamycin cells by down-regulating MDR1 via inhibition of NF-κB signaling pathway [24]. Dioscin may decrease the resistance degree of HepG2/Adriamycin cells and significantly inhibit P-glycoprotein expression, and, in turn, increase the accumulation of Adriamycin in HepG2/Adriamycin cells [25]. These results suggest that dioscin may reverse multidrug resistance to act as a potential chemotherapy adjuvant. However, no report showed the effects of dioscin on TKI resistance in NSCLC.
A natural product library of 124 naturally occurring compounds from Chinese herbs was used to determine which compound(s) could potentially overcome TKI resistance in EGFR-mutated lung adenocarcinoma cells. Our preliminary data identified dioscin as a candidate for overcoming TKI resistance (Supplementary Figure 1). In the present study, we provided molecular evidence that TKI resistance overcome by dioscin via simultaneous inhibition of both MEK/ERK and PI3K/AKT pathways through suppression of SHP2 expression at the transcription level.
Materials and Methods
Chemicals and antibodies
Gefitinib, Dioscin, Triptolide, and Nature product library were obtained from Selleckchem (Houston, TX). All other chemicals were acquired from Sigma Chemical (St. Louis, MO). Anti-EGFR, anti-total AKT, anti- phosphorylated (p) -AKT, anti-total ERK, and anti-(p)-ERK antibodies were purchased from Cell Signaling (Danvers, MA). Anti-SHP2 antibodies were obtained from Genetex (Irvine CA). All other antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX).
Cell lines
PC9, PC9GR and CL97 cells were kindly provided by Dr. P.-C. Yang and Dr. James C.-H. Yang (Department of Internal Medicine and Graduate Institute of Oncology, College of Medicine, National Taiwan University, Taiwan).H1650, H1975, and HCC827 cell lines were purchased from the American Type Culture Collection (ATCC) and cultured as described previously.
Human study subjects
The human study included 36 patients who underwent resection at China Medical University Hospital in Taichung, Taiwan, between October 2005 and December 2010. The tumor type and stage was determined according to the World Health Organization (WHO) classification system. The median follow-up time for this population was 1536 days (ranging from 151 to 3125 days) and the end of the follow-up period was December 2014. The IRB protocol (DMR101-IRB1-265) was approved by China Medical University Hospital.
Plasmid constructs and transfection
The SHP2 overexpression plasmid was purchased from Genscript (Piscatway, NJ). SHP2 (TRCN0000005003) and GAB1 (TRCN0000074283), were obtained from National RNAi Core Facility, Academia Sinica, Taiwan. The SHP2-Luciferase(Luc) plasmids were constructed with inserting a 1274, 902, 697, 380, and 108 bps fragment into pGL3-Basic vector (Promega Corp., Madison, WI). The P53-mutated binding site on the SHP2 promoter constructs containing multiple-point mutations were constructed by the QuickChange site-directed mutagenesis system (Stratagene, La Jolla, CA). The WT sequence (#1: GGGCGGG; #2: GGGCCGG; #3: GGGCAGC) for p53 binding sites on SHP2 promoter has been changed to the mutated sequence (#1: AGTCTGG; #2: AGTCCGG; #3: AGTCAGC). These plasmids were transiently transfected into lung cancer cells (1 × 106) using the Turbofect reagent (Formentas, Glen Burnie, MD). After 48 hours (hrs), cells were harvested to analyze in subsequent experiments.
Luciferase reporter assay
Cells were transfected with appropriate luciferase plasmid and indicated plasmid. Luciferase assays were performed using the luciferase reporter assay system (Promega) after 48 hrs. Normalized luciferase activity was used as luciferase activity/ β-galactosidase activity.
3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di-phenytetrazoliumromide (MTT) cytotoxicity assay
Before treatment, the cells in the exponential growth phase were pretreated with overexpression and knockdown plasmids for 24 hrs. The in vitro cytotoxic effects of these treatments were determined by MTT assay (at 570 nm) and the cell viability was expressed as a percentage of the vehicle control cells.
Annexin V- Propidium iodide (PI) staining analysis
The cells were collected by trypsinization and centrifugation at 1,000 g for 5 min. Following resuspension in binding buffer (10 mM HEPES-NaOH, 140 mM NaCl, 2.5 mM CaCl2) at a final cell density of 1-2 × 106 cells/ml, 500 μl of a single-cell suspension (1-2 × 105 cells) was incubated with 5 μl annexin V-FITC and 5 μl PI for 15 min at room temperature in the dark. After addition of 400 μl of binding buffer, the samples were analyzed with a BD FACS Calibur flow cytometer (BD Biosciences, San Jose, CA) within 1 hr. For each sample, 10,000 events were counted.
Colony formation assay
Cells were plated in 6-well plates in complement media overnight. After incubation, the culture media were replaced by fresh medium containing with Dioscin, DMSO as vehicle control for 48 hrs. These treated cells were cultured in the newly medium supplemented with 10 % FBS for another 10 days. Before the pictures of these colonies were taken, cells were stained with 0.01% crystal violet for 1 hr at room temperature.
Western blotting
Western blotting was performed as described previously [4]. Protein samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gels, transferred to poly-vinylidene difluoride membranes (Millipore, Billerica, MA), and finally immunoblotted with primary antibodies. Primer antibodies were used at 1:500 to 1:1000 dilution. Horseradish peroxidase-conjugated anti-mouse and rabbit secondary antibodies (Santa Cruz Biotechnology, Dallas, TX) were performed at 1:5000 dilution. Protein signals were detected by chemiluminescent reagents (Amersham Pharmacia, Piscataway, NJ).
Chromatin immunoprecipitation (ChIP) assay
ChIP analysis was performed as described in a previous report with the following modifications [26]. Immunoprecipitated DNA were re-suspended in 100 µl ddH2O and diluted 1:200 before PCR analysis. PCR amplification of immunoprecipitated DNA was carried out with diluted aliquots, using the primers consisting of the oligonucleotides that encompass the promoter region of SHP2. The forward primer, 5'- GTCGCGAGCGGTGACATCA-3' and the reverse primer, 5'- TCCAGGCCTGGGGATCCCGGAG -3' PCR products were separated on 2% agarose gels and analyzed using ethidium bromide staining. All ChIP assays were performed at least twice with similar results.
Real-time quantitative RT-PCR analysis
DNase I-treated total RNA was subjected to polymerase chain reaction (PCR) analysis with the Reverse Transcription Kit (Life technologies, Foster city, CA) and a Real-Time Thermocycler 7500 (Life technologies, Foster city, CA). GAPDH was used as the RNA reference housekeeping gene. The following primer sequences were used for amplification of the SHP2 gene: the forward primer, 5'- GGAGTTGATGGCAGTTTTTTGG-3', and the reverse primer, 5'- TCTGAATCTTGATGTGGGTGACA-3'. The SHP2 mRNA levels in lung tumors that were higher than the median value were defined as “high”, while levels lower than the median value were defined as “low”.
Statistical analysis
Statistical analysis was performed with the SPSS statistical software (Version 13.0; Chicago, IL.). The association between tumor response and SHP2 mRNA expression were analyzed by the chi-square test. Survival plots were generated using the Kaplan-Meier method, and differences between patient groups were determined by the log-rank test. Cox regression analysis was performed to determine OS and RFS and stratified for all known variables (age, gender, smoking status and tumor stage).
Results
Dioscin suppresses colony formation efficacy and cell viability in TKI resistant lung adenocarcinoma cells via apoptotic pathway
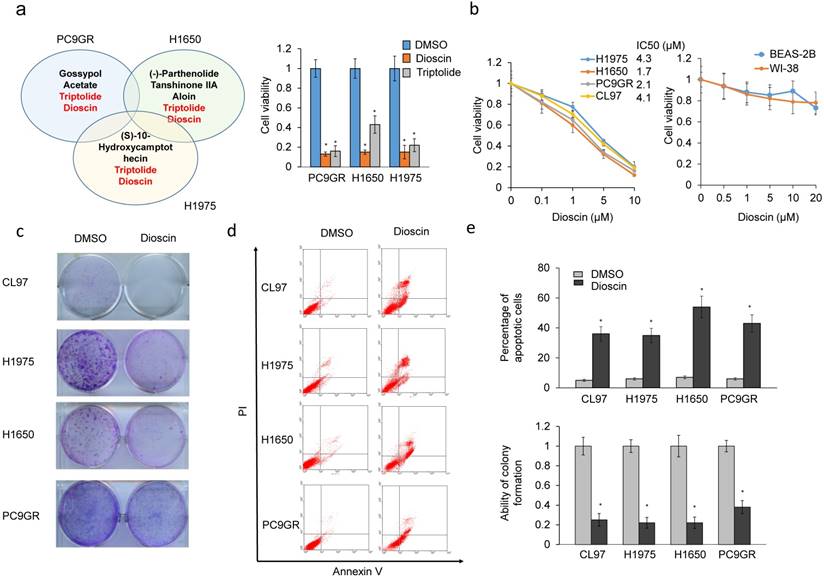
A natural product library was screened to identify compound(s) that could cause more than 50% cytotoxicity in three TKI resistant EGFR-mutated cell lines (PC9GR, H1650, and H1975). Among these compounds, triptolide and dioscin induced more than 50% cytotoxicity in these three cell types (Figure 1a left panel). The MTT assay indicated that dioscin was more effective than triptolide at inducing cytotoxicity in these three cell types (Figure 1a right panel). The MTT assay was performed to evaluate the inhibitory concentration of dioscin yielding 50% cell viability (IC50) based on dose-response curves. The lowest IC50 value of dioscin was 1.7μM for H1650 cells, followed by 2.1μM for PC9GR cells, 4.1μM for CL97 cells, and 4.3μM for H1975 cells (Figure 1b). Dioscin showed no cytotoxicity in normal WI38 and Beas-2B lung cell lines, as determined by MTT assays (Figure 1b). Figure 1c and 1d show the representative colony formation and annexin V-PI staining profiles in these four cell types. The colony formation efficacy was markedly decreased by dioscin treatment in all four cell types (Figure 1e lower panel). Annexin V-PI staining indicated that dioscin induced the highest percentage of apoptotic cells in the H1650 cell line, followed by PC9GR cells, CL97 cells, and H1975 cells (Figure 1e, upper panel). Therefore, dioscin may suppress colony formation and cell viability in TKI resistant EGFR-mutated lung adenocarcinoma cells via the apoptotic pathway.
Dioscin suppresses colony formation efficacy and cell viability in TKI-resistant lung adenocarcinoma cells via an apoptotic pathway. (a) PC9GR, H1650, and H1975 cells were treated with 124 compounds in a natural product library. These compounds showed as more than 50% cytotoxicity of these three cell types. (b) EGFR-mutated lung cancer cells (H1975, H1650, CL97, PC9GR, WI-38 and Beas-2B) were treated with dioscin at the indicated concentrations for 48 h and cell viability and IC50 were analyzed by the MTT assay. (c, d) Colony formation (c) and annexinV-PI staining assays (d) were performed in six EGFR-mutated lung cancer cell types subjected to dioscin treatment. (e) The relative colony formation ability and percentage of apoptotic cells modulated by dioscin are summarized in these cell types. All data were collected from three independent experiments. The mean value and standard deviation were indicated as the column with error bars. The significant differences in experimental groups were compared to vehicle (0.1% DMSO) treatment (*P < 0.05).
Dioscin overcomes TKI resistance via simultaneous inhibition of the MEK/ERK and PI3K/AKT signaling pathways by decreasing SHP2 expression via GAB1 signaling
CL97, H1650, H1975, and PC9GR cells were treated with dioscin to verify whether an increase in TKI sensitivity by dioscin could occur through inactivation of the PI3K/AKT and MEK/ERK signaling pathways. Western blotting indicated that dioscin treatment inactivated the actived form of AKT and ERK in a dose-dependent manner in these four cell types, but total protein levels were unchanged (Figure 2a). In addition, total EGFR and actived form of EGFR were unchanged by dioscin treatment (Figure 2a). We hypothesized that the inactivated in ctived form of AKT and ERK by dioscin could occur through dysregulation of SHP2. Western blotting indicated that SHP2 expression was markedly decreased by dioscin treatment in these four cell types (Figure 2b upper panel). Real-time PCR and luciferase reporter assays indicated a dose-dependent decrease in SHP2 promoter activity and its mRNA levels following dioscin treatment in these four cell types (Figure 2b lower panel).
Dioscin overcomes TKI resistance via simultaneous inhibition of the MEK/ERK and PI3K/AKT signaling pathways due to decreased SHP2 expression and its interaction with GAB1. (a) The effect of dioscin on AKT and ERK signaling. Five hours after dioscin (-5 μM) treatment, the cell lysates were harvested and analyzed for signaling alteration by weocistern blotting with the indicated antibodies. (b) The effect of dioscin on SHP2 transcriptional level. Five hours after dosing cells with dioscin (0-5 μM), the cell lysates were prepared and analyzed for mRNA and promoter activity by real-time PCR and luciferase assays. The relative luciferase activity and mRNA expression was shown as fold-activation relative to the cells with vehicle treatment. (c) The effect of SHP2 knockdown on changes in cell viability and apoptosis induced by dioscin treatment. H1975, CL97, H1650, and PC9GR cells transfected with SHP2 shRNA were treated with 5 μM dioscin for 48 h. The cell viability was measured by MTT assays. Cell apoptosis was measured by annexinV-PI staining assays and flow cytometry. (d) The effect of SHP2 overexpression or/and GAB1 knockdown on the apoptosis induced by dioscin treatment. H1975 and CL97 cells transfected with SHP2 overexpression plasmid or/and GAB1 shRNA were treated with 5 μM dioscin for 48 h. Cell apoptosis was measured by annexinV-PI staining assays and flow cytometry. All data were collected from three independent experiments. The mean value and standard deviation are indicated as the column with error bars. The significant differences (*P < 0.05) in experimental groups were compared to indicated groups. N.s., Non-significance.
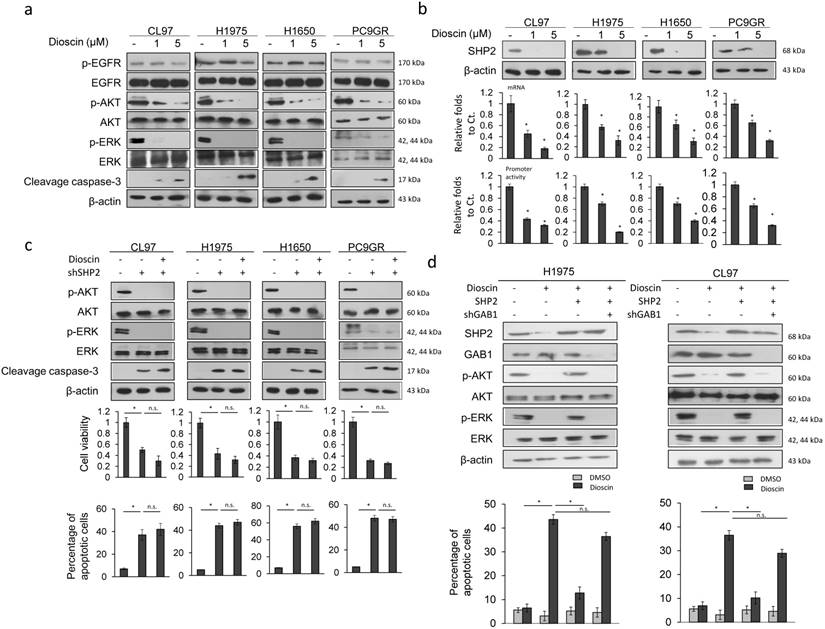
These four cell types were treated with dioscin and/or a small hairpin (sh)RNA for SHP2 to examine the possibility that dioscin inactivation of both pathways occurred through decreased SHP2 expression. Western blotting showed an almost complete inhibition of actived form of AKT and ERK in these four cell types by treatment with shSHP2, singly or in combination with dioscin (Figure 2c upper panel). Cell viability was markedly decreased and the percentage of apoptotic cells was markedly increased by shSHP2 transfection, singly or in combination with dioscin treatment, in these cell types (Figure 2c lower panel). The cleavage of caspase-3 expression was concomitantly increased in these four cell types with these treatments (Figure 2c upper panel). These results indicated that decreases in SHP2 expression by dioscin may block activation of MEK/ERK and PI3K/AKT signaling pathways and in turn kill TKI-resistant EGFR-mutated cells via apoptotic pathway.
The interaction between SHP2 and GAB1 simultaneously activates MEK/ERK and PI3K/AKT signaling pathways for endothelial cell proliferation [27]. We next examined the possibility that blockade of both signaling pathways by dioscin could occur through inhibition of SHP2 interaction with GAB1. H1975 and CL97 cells were treated with dioscin, an SHP2 expression plasmid, and/or shGAB. A nearly complete inhibition of actived form of AKT and ERK was observed in both cell types following dioscin treatment, but levels of both proteins were almost fully restored by the combination of dioscin with SHP2 expression plasmid treatment (Figure 2d upper panel). Further rescue of both protein expressions was achieved with a combination of dioscin, SHP2 expression plasmid, and shGAB1 treatment (Figure 2d upper panel). The percentage of apoptotic cells in both cell types was markedly increased by dioscin, but a combination of dioscin and SHP2 expression plasmid almost completely rescued this increase in apoptotic cells. The increase in the percentage of apoptotic cells was almost completely reversed by the combination of dioscin, SHP2 expression plasmid, and shGAB1 treatment (Figure 2d lower panel). These results suggest that dioscin kills TKI resistant cells through apoptotic pathway by inactivating the MEK/ERK and PI3K/AKT signaling pathways due to decreases in SHP2 expression via GAB1 signaling.
Dioscin induces p53 expression via ROS generation to suppress SHP2 transcription
Five fragments of SHP2 promoters (-1040/+233, -669/+233, -464/+233, -147/+233, and +125/+233) were constructed by PCR and deletion mutations (Figure 3a left panel). Luciferase reporter assay indicated that the SHP2 promoter activity of these five fragments was nearly completely eliminated by dioscin in H1975 and CL97 cells (Figure 3a right panel). Dioscin induces ROS generation and causes oxidative DNA damage in cancer cells [28]. We therefore expected that the putative p53 and Sp1 binding sites located at the promoter (+125/+233) could play a role in the regulation of SHP2 transcription by dioscin. H1975 and CL97 cells were transfected with shSp1 and shp53 to examine whether both possible transcription factors could be involved in SHP2 transcription. The SHP2 promoter activity (+125/+233) was markedly elevated by shp53 transfection, but unchanged by shSp1 transfection in both cell types (Figure 3b). We constructed p53 binding site-mutated promoters (+125/+233) to verify an involvement of p53 in Dioscin-mediated SHP2 transcription. Luciferase reporter assays indicated that Dioscin-mediated SHP2 promoter activity in H1975 and CL97 cells depended on the number of p53 binding site mutations on the SHP2 promoter (Figure 3c). Western blotting showed that p53 expression was elevated by dioscin, whereas SHP2 expression was decreased by dioscin in H1975 and CL97 cells. However, the decrease in SHP2 expression by dioscin in both cell types was markedly elevated by shp53 treatment, singly or in combination with dioscin (Figure 3d upper panel). The elevation of SHP2 protein expression by shp53 and/or shp53+dioscin in both cell types was consistent with the SHP2 promoter activity (Figure 3d lower panel).
Dioscin induces p53 expression via ROS generation to suppress SHP2 transcription. (a) Schematic diagram of SHP2-promoter-driven luciferase reporters: SHP2 (-1041/+233)-Luc, SHP2 (-669/+233)-Luc, SHP2 (-464/+233)-Luc, SHP2 (-147/+233)-Luc and SHP2 (+125/+233)-Luc. SHP2 promoter activity in dioscin-treated H1975 and CL97 cells was evaluated by luciferase reporter activity assays. The relative luciferase activity was shown as fold-activation relative to activity of SHP2 (-1041/+233)-Luc in the cells with vehicle treatment. (b) The effect of Sp1 or p53 knockdown on the SHP2 promoter activity. SHP2 promoter activity in Sp1-knockdown or p53-knockdown H1975 and CL97 cells was evaluated by luciferase reporter activity assays. The relative luciferase activity was shown as fold-activation relative to activity of SHP2 (+125/+233)-Luc in the cells with vehicle treatment. (c) The four SHP2 promoter (WT, Mut1, Mut1/2, and Mut 1/2/3) constructs were transfected into the indicated cell types. Promoter activity in dioscin-treated H1975 and CL97 cells was evaluated by luciferase reporter activity assays. The relative luciferase activity was shown as fold-activation relative to activity of SHP2 (+125/+233)-Luc in the cells with vehicle treatment. (d) The effect of p53 knockdown on SHP2 promoter activity regulated by dioscin treatment. H1975 and CL97 cells transfected with p53 GAB1 shRNA were treated with 5 μM dioscin for 5 hrs. The cell lysates were harvested and analyzed for the promoter activity by luciferase assay. The relative luciferase activity was shown as fold-activation relative to activity of SHP2 (+125/+233)-Luc in the cells with vehicle treatment. (e) The effect of ROS on SHP2 promoter activity regulated by dioscin treatment. H1975 and CL97 cells were treated with 5 μM dioscin and/ or 1mM NAC for 5 hrs. The cell lysates were harvested and analyzed for the promoter activity by luciferase assay, protein expression by western blotting, and p53 binding ability on SHP2 promoter by ChIP assays. The relative luciferase activity was shown as fold-activation relative to activity of SHP2 (+125/+233)-Luc in the cells with vehicle treatment. The significant differences (*P < 0.05) in experimental groups were compared to indicated groups. N.s., Non-significance.
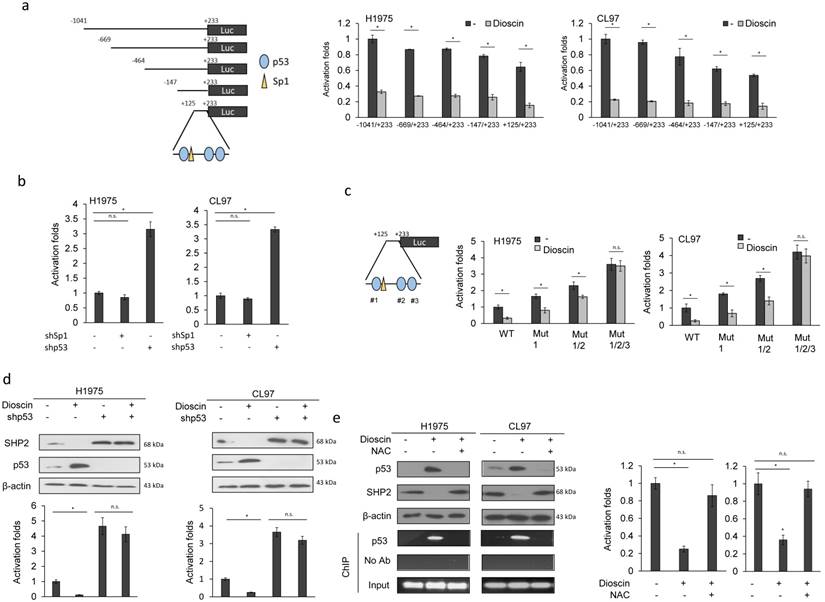
We next examined the possibility that ROS generation induced by dioscin could play a role in p53-regulating SHP2 expression. H1975 and CL97 cells were treated with dioscin singly or in combination with a ROS scavenger, N-acetylcysteine (NAC). Expression of p53 was markedly increased by dioscin treatment, but the elevation of p53 expression by dioscin was almost completely reversed by dioscin+NAC treatment in both cell types (Figure 3d). Chromatin immunoprecipitation indicated that p53 binding to the SHP2 promoter (+125/+233) was markedly increased by dioscin, but p53 binding to the SHP2 promoter was completely eliminated by the combination of dioscin and NAC in both cell types (Figure 3e left panel). Luciferase reporter assays further confirmed the change in SHP2 promoter activity by dioscin and by the combination of dioscin with NAC in both cell types (Figure 3e right panel). These results indicated that increased p53 expression due to dioscin-mediated ROS generation might suppress SHP2 transcription.
Low SHP2-expressing tumors may be associated with unfavorable response and poor overall survival in patients with lung adenocarcinoma who have received TKI therapy when compared to patients with high SHP2-expressing tumors
We enrolled 36 lung adenocarcinoma patients who had disease recurrence after surgical resection and receiving TKI therapy. Real-time PCR analysis was performed to evaluate SHP2 mRNA expression in tumor tissues from this study population. The median values of SHP2 mRNA levels were used as a cutoff point to divide patients into “high” and “low” subgroups. The high SHP2 subgroup had a higher frequency of patients >60 years old than <60 years old (65% vs. 31%, P = 0.044; Table 1). However, no correlation of SHP2 expression with other parameters was observed (Table 1). Patients in the high SHP2 subgroup were more likely to have an unfavorable response to TKI therapy when compared to patients in the low SHP2 subgroup (69% vs. 35%, P = 0.044; Table 2). The correlation of TKI response with SHP2 mRNA expression levels was also assessed. SHP2 mRNA expression levels were significantly higher in patients with unfavorable response than in patients with favorable response to TKI (P = 0.012; Figure 4a).
Relationships of SHP2 mRNA levels with clinico-pathological parameters in lung adenocarcinoma patients.
| SHP2 | ||||
|---|---|---|---|---|
| Variables | No. | Low | High | P |
| Age | ||||
| ≤60 | 16 | 5 (31) | 14 (69) | 0.044 |
| >60 | 20 | 13 (65) | 7 (35) | |
| Gender | ||||
| Female | 20 | 10 (50) | 10 (50) | 1.000 |
| Male | 16 | 8 (50) | 8 (50) | |
| Smoking status | ||||
| Nonsmokers | 29 | 14 (48) | 15 (52) | 1.000 |
| Smokers | 7 | 4 (57) | 3 (43) | |
| Stage | ||||
| I | 13 | 7 (54) | 6 (46) | 0.729 |
| II &III | 23 | 11 (48) | 12 (52) | |
The association of SHP2 mRNA level with the response to TKI therapy in lung adenocarcinoma patients.
| The response to TKI therapy | ||||
|---|---|---|---|---|
| Variables | Patient No. | Unfavorable (%) | Favorable (%) | P |
| SHP2 | ||||
| Low | 18 | 7 (39) | 11 (61) | 0.044 |
| High | 18 | 13 (72) | 5 (28) | |
The unfavorable response is defined at least 25% increase in size or number of the tumors; otherwise is defined as a favorable response. The median value of SHP2 mRNA levels was used as cutoff point to divide patients into “high” and “low” subgroups.
Low SHP2-expressing tumors may be associated with unfavorable response and poor overall survival in patients with lung adenocarcinoma who have received TKI therapy when compared to patients with high SHP2-expressing tumors. (a) SHP2 mRNA expression levels were significantly higher in patients with an unfavorable response than in patients with a favorable response to TKI. (b) Kaplan-Meier analysis was used to estimate prognosis in patients with high SHP2-expressing tumors to have poorer overall survival than in patients with low SHP2-expressing tumors.
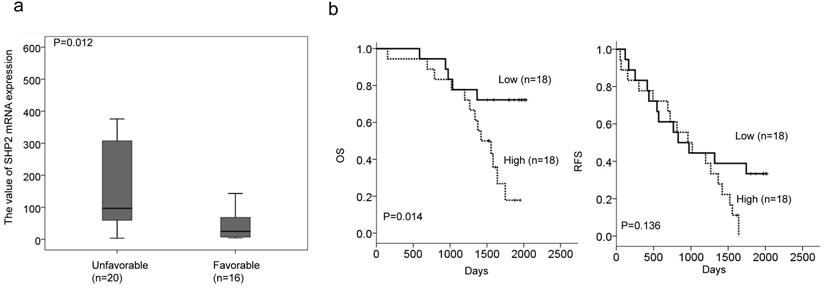
Kaplan-Meier analysis showed poorer overall survival in patients in the high SHP2 subgroup than in the in low SHP2 subgroup; however, SHP2 expression had no prognostic value for relapse-free survival in this study population (Figure 4b). We therefore suggest that high SHP2 expression may confer TKI resistance and an unfavorable prognosis in patients with lung adenocarcinoma.
Discussion
We provided evidence that decreases in SHP2 expression at the transcription level and blockade of SHP2-GAB1 complex formation due to dioscin may overcome TKI resistance and lead apoptosis in lung adenocarcinoma cells by simultaneous inhibition of MEK/ERK and PI3K/AKT activation. Western blotting analysis indicated that dioscin suppressed the expression of Bcl-2, Bcl-w, Bcl-xL, but elevated the expression of Bim and Bax in CL97 and H1975 cells when compared both cells without dioscin treatment (Supplementary Figure 2). These results were partially consistent with previous studies to indicate that dioscin decreased Bcl-2 and increased Bad expression to lead cell apoptosis in human gastric cancer and esophageal cancer cells [17, 20]. In addition, dioscin inhibited colon tumor growth and tumor angiogenesis through regulation of VEGFR2 and AKT/MAPK signaling pathways [16]. Inhibition of tumor progression by dioscin by deregulation of AKT/MAPK signaling pathway seemed to support the possibility that dioscin not only suppressed tumor progression and metastasis but also overcome TKI resistance in lung adenocarcinoma cells via simultaneous inhibition of PI3K/AKT and MEK/ERK signaling pathways. The possible mechanism of dioscin in overcoming TKI resistance of EGFR-mutated-lung adenocarcinoma cells has been proposed in Supplementary Figure 3.
Dioscin suppresses SHP2 transcription through decreased Sp1 binding to the SHP2 promoter due to dioscin-induced p53 expression in response to ROS generation. Therefore, wild-type p53 may act as a repressor to suppress SHP2 promoter, but this was only observed in EGFR-mutated lung adenocarcinoma cells that harbored wild-type p53. However, targeting SHP2 by its inhibitor II-B08 may overcome TKI resistance in H1975 cells through inhibition of MEK/ERK activation, but not through the PI3K/AKT signaling [29]. Accumulating evidence shows that SHP2 synergistically promotes RAS-mediated MEK/ERK activation [8,9,29-33]. Therefore, to the best our knowledge, this is the first report to show that dioscin may act as a dual inhibitor of MEK/ERK and PI3K/AKT by decreased SHP2 expression to overcome TKI resistance in lung adenocarcinoma cells.
SHP2 is an essential transducer of mitogenic and cell survival signaling in the EGFR signaling pathway [9,29,32,34]. SHP2 promoted RAS activation by blocking the RAS GTPase-activating protein-induced downregulation of RAS [8]. EGFR-activated Src family kinases maintained a longer persistence of GAB1-SHP2 complexes than EGFR phosphorylation in response to EGF in EGFR-mutated H1666 lung adenocarcinoma cells [9]. Therefore, SHP2 activated survival signaling in an EGFR-dependent pathway. However, we and other groups reported that persistent activation of MEK/ERK and PI3K/AKT signaling pathways in TKI resistant lung adenocarcinoma cells occurred through an EGFR-independent pathway. For example, paxillin treatment promoted MEK/ERK activation to confer TKI resistance [4]. PAK1 may persistently activate PI3K/AKT signaling pathway to confer TKI resistance in lung adenocarcinoma cells [6]. In the present study, the involvement of both signaling pathways in TKI resistance can be simultaneously dysregulated by dioscin and consequently overcome TKI resistance in EGFR-mutated lung adenocarcinoma cells.
In summary, we provided evidence that dioscin may act as a dual inhibitor of MEK/ERK and PI3K/AKT signaling pathways to overcome TKI resistance via down-regulation of SHP2 expression. In addition, high SHP2 expression was associated with unfavorable response to TKI and poor overall survival in patients with lung adenocarcinoma. We therefore suggest that dioscin may potentially improve TKI resistance and outcome in patients with lung adenocarcinoma.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was jointly supported by grants from This work was jointly supported by grants from Tung's Taichung Metro-Harbor Hospital (TTM-TMU-106-02), lung cancer research fund, Taipei Medical University (104-6602-001-400), Taiwan, ROC.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nature reviews Cancer. 2010;10(11):760-774
2. Kim TM, Song A, Kim DW. et al. Mechanisms of Acquired Resistance to AZD9291: A Mutation-Selective, Irreversible EGFR Inhibitor. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2015;10(12):1736-1744
3. Morgillo F, Della Corte CM, Fasano M, Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open. 2016;1(3e):000060
4. Wu DW, Chen CY, Chu CL, Lee H. Paxillin confers resistance to tyrosine kinase inhibitors in EGFR-mutant lung cancers via modulating BIM and Mcl-1 protein stability. Oncogene. 2016;35(5):621-630
5. Wu DW, Chen TC, Huang HS, Lee H. TC-N19, a novel dual inhibitor of EGFR and cMET, efficiently overcomes EGFR-TKI resistance in non-small-cell lung cancer cells. Cell death & disease. 2016;7(6e):2290
6. Wu DW, Wu TC, Chen CY, Lee H. PAK1 Is a Novel Therapeutic Target in Tyrosine Kinase Inhibitor-Resistant Lung Adenocarcinoma Activated by the PI3K/AKT Signaling Regardless of EGFR Mutation. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22(21):5370-5382
7. Shimizu T, Tolcher AW, Papadopoulos KP. et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18(8):2316-2325
8. Zhang SQ, Yang W, Kontaridis MI. et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell. 2004;13(3):341-355
9. Furcht CM, Buonato JM, Lazzara MJ. EGFR-activated Src family kinases maintain GAB1-SHP2 complexes distal from EGFR. Sci Signal. 2015;8(376ra):46
10. Brautbar N, Williams J 2nd. Industrial solvents and liver toxicity: risk assessment, risk factors and mechanisms. International journal of hygiene and environmental health. 2002;205(6):479-491
11. Xu T, Zheng L, Xu L. et al. Protective effects of dioscin against alcohol-induced liver injury. Arch Toxicol. 2014;88(3):739-753
12. Yao H, Hu C, Yin L. et al. Dioscin reduces lipopolysaccharide-induced inflammatory liver injury via regulating TLR4/MyD88 signal pathway. Int Immunopharmacol. 2016;36:132-141
13. Lu B, Xu Y, Xu L. et al. Mechanism investigation of dioscin against CCl4-induced acute liver damage in mice. Environ Toxicol Pharmacol. 2012;34(2):127-135
14. Zhang X, Han X, Yin L. et al. Potent effects of dioscin against liver fibrosis. Sci Rep. 2015;5:9713
15. Tao X, Sun X, Yin L. et al. Dioscin ameliorates cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via HMGB-1 inhibition. Free radical biology & medicine. 2015;84:103-115
16. Wu S, Xu H, Peng J. et al. Potent anti-inflammatory effect of dioscin mediated by suppression of TNF-alpha-induced VCAM-1, ICAM-1and EL expression via the NF-kappaB pathway. Biochimie. 2015;110:62-72
17. Liu MJ, Wang Z, Ju Y, Zhou JB, Wang Y, Wong RN. The mitotic-arresting and apoptosis-inducing effects of diosgenyl saponins on human leukemia cell lines. Biological & pharmaceutical bulletin. 2004;27(7):1059-1065
18. Wang Z, Zhou J, Ju Y, Zhang H, Liu M, Li X. Effects of two saponins extracted from the polygonatum Zanlanscianense pamp on the human leukemia (HL-60) cells. Biological & pharmaceutical bulletin. 2001;24(2):159-162
19. Hsieh MJ, Tsai TL, Hsieh YS, Wang CJ, Chiou HL. Dioscin-induced autophagy mitigates cell apoptosis through modulation of PI3K/Akt and ERK and JNK signaling pathways in human lung cancer cell lines. Archives of toxicology. 2013;87(11):1927-1937
20. Wei Y, Xu Y, Han X. et al. Anti-cancer effects of dioscin on three kinds of human lung cancer cell lines through inducing DNA damage and activating mitochondrial signal pathway. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2013;59:118-128
21. He X, Qiao A, Wang X. et al. Structural identification of methyl protodioscin metabolites in rats' urine and their antiproliferative activities against human tumor cell lines. Steroids. 2006;71(9):828-833
22. Wang Z, Cheng Y, Wang N. et al. Dioscin induces cancer cell apoptosis through elevated oxidative stress mediated by downregulation of peroxiredoxins. Cancer biology & therapy. 2012;13(3):138-147
23. Hsieh MJ, Yang SF, Hsieh YS, Chen TY, Chiou HL. Autophagy inhibition enhances apoptosis induced by dioscin in huh7 cells. Evidence-based complementary and alternative medicine: eCAM. 2012;2012:134512
24. Wang L, Meng Q, Wang C. et al. Dioscin restores the activity of the anticancer agent adriamycin in multidrug-resistant human leukemia K562/adriamycin cells by down-regulating MDR1 via a mechanism involving NF-kappaB signaling inhibition. Journal of natural products. 2013;76(5):909-914
25. Sun BT, Zheng LH, Bao YL. et al. Reversal effect of Dioscin on multidrug resistance in human hepatoma HepG2/adriamycin cells. European journal of pharmacology. 2011;654(2):129-134
26. Wu DW, Lin PL, Wang L, Huang CC, Lee H. The YAP1/SIX2 axis is required for DDX3-mediated tumor aggressiveness and cetuximab resistance in KRAS-wild-type colorectal cancer. Theranostics. 2017;7(5):1114-1132
27. Lauriol J, Jaffre F, Kontaridis MI. The role of the protein tyrosine phosphatase SHP2 in cardiac development and disease. Semin Cell Dev Biol. 2015;37:73-81
28. Lv L, Zheng L, Dong D. et al. Dioscin, a natural steroid saponin, induces apoptosis and DNA damage through reactive oxygen species: a potential new drug for treatment of glioblastoma multiforme. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2013;59:657-669
29. Xu J, Zeng LF, Shen W, Turchi JJ, Zhang ZY. Targeting SHP2 for EGFR inhibitor resistant non-small cell lung carcinoma. Biochemical and biophysical research communications. 2013;439(4):586-590
30. Tien SC, Chang ZF. Oncogenic Shp2 disturbs microtubule regulation to cause HDAC6-dependent ERK hyperactivation. Oncogene. 2014;33(22):2938-2946
31. Chen L, Sung SS, Yip ML. et al. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70(2):562-570
32. Lazzara MJ, Lane K, Chan R. et al. Impaired SHP2-mediated extracellular signal-regulated kinase activation contributes to gefitinib sensitivity of lung cancer cells with epidermal growth factor receptor-activating mutations. Cancer research. 2010;70(9):3843-3850
33. Zeng LF, Zhang RY, Yu ZH. et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J Med Chem. 2014;57(15):6594-6609
34. Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. 2003;23(21):7875-7886
Author contact
![]() Corresponding author: Huei Lee, Ph. D., Professor, Room 5, 12th floor, F building, Park Street, Nangang District, Taipei, Taiwan 115. Tel: 886-2-27361661 ext. 7616; Fax: 886-2-26558562; E-mail: hledu.tw
Corresponding author: Huei Lee, Ph. D., Professor, Room 5, 12th floor, F building, Park Street, Nangang District, Taipei, Taiwan 115. Tel: 886-2-27361661 ext. 7616; Fax: 886-2-26558562; E-mail: hledu.tw