Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Background
Material and Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(3):306-320. doi:10.7150/ijbs.23869 This issue Cite
Research Paper
Environmental adaptation of Acanthamoeba castellanii and Entamoeba histolytica at genome level as seen by comparative genomic analysis
Victoria Shabardina1, Tabea Kischka1, Hanna Kmita2, Yutaka Suzuki3, Wojciech Makałowski1 ![]()
1. Institute of Bioinformatics, University Münster, Niels-Stensen Strasse 14, Münster 48149, Germany
2. Laboratory of Bioenergetics, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University
3. Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8562, Japan
Received 2017-11-15; Accepted 2017-12-30; Published 2018-2-12
Abstract

Amoebozoans are in many aspects interesting research objects, as they combine features of single-cell organisms with complex signaling and defense systems, comparable to multicellular organisms. Acanthamoeba castellanii is a cosmopolitan species and developed diverged feeding abilities and strong anti-bacterial resistance; Entamoeba histolytica is a parasitic amoeba, who underwent massive gene loss and its genome is almost twice smaller than that of A. castellanii. Nevertheless, both species prosper, demonstrating fitness to their specific environments. Here we compare transcriptomes of A. castellanii and E. histolytica with application of orthologs' search and gene ontology to learn how different life strategies influence genome evolution and restructuring of physiology. A. castellanii demonstrates great metabolic activity and plasticity, while E. histolytica reveals several interesting features in its translational machinery, cytoskeleton, antioxidant protection, and nutritional behavior. In addition, we suggest new features in E. histolytica physiology that may explain its successful colonization of human colon and may facilitate medical research.
Keywords: Life strategy, genomic adaptation, comparative genomics, Acanthamoeba castellanii, Entamoeba histolytica
Background
Amoebozoa clade is the subject of interest for scientists from different fields of biology. These unicellular organisms are primitive eukaryotes and represent a convenient model for studying phagocytosis, cell migration, development of multi-cellularity and for evolutionary studies (1,2). Amoeba species inhabit wide variety of ecological niches and demonstrate amazing environmental adaptations: from slime molds that can form multicellular structure in response to specific stimuli, to Acanthamoeba castellanii that is able to feed on almost any natural source, to Entamoeba histolytica, an obligatory human parasite. At the same time the knowledge about amoebas' physiology and genetics is scarce and ambiguous due to difficulties in laboratory culturing and lack of fully sequenced genomes from different lineages (3-7). Thus, there are many poorly understood facts about amoebas and more studies are required for further understanding of these intriguing organisms.
A. castellanii (Ac) and E. histolytica (Eh) represent two extremes of cell functioning: diverse metabolic pathways in Ac versus parasitic reduction of biological functions in Eh. A. castellanii prospers in a range of environments, such as different water sources, wet surfaces, soil, including rhizosphere, and even human tissues, due to its ability to metabolite and synthetize a wide spectrum of substances (2). It can feed on plants and on soil bacteria due to presence of cellulase and PHB (poly(3-hydroxybutyrate)) depolymerase; the latter enzyme is very beneficial for the amoeba, as the majority of bacteria produce and store PHB in considerable amounts. Ac is also known as a symbiont for several soil and human microbes, and it is not surprising that cases of horizontal gene transfer (HGT) from bacteria is quite common for this amoeba (8-10). To be able to coordinate all the varieties of interactions Ac developed notably complex for unicellular organism signaling system, anti-microbial defense system and stress resistance systems.
E. histolytica can transit from commensal life style in the human colon to tissue invasive behavior and consequently induce diarrhea and liver abscess, but the trigger to the transition from latent disease state to progressive amoebiasis is not yet understood. It was estimated that 40,000 to 100,000 people die from amoebiasis each year, mostly in developing countries (11,12). The genome of Eh is an example of the massive secondary gene loss, and most of the biosynthetic pathways are absent (amino acid synthesis, folate synthesis, most of the lipid synthesis), nucleotide metabolism and energy production are degenerate (13) and most metabolites are taken from the host. Mitochondria of the parasite are reduced to mitosomes, Golgi apparatus and rough ER are absent. At the same time Eh has some compensatory mechanisms, for example, it is able to produce energy from amino acids asparagine, aspartic acid, tryptophan, threonine, and methionine (Anderson & Loftus, 2005). Eh is an anaerobic organism, but it encounters micro-oxygenic environment during host tissue invasion, therefore Eh developed specific anti-oxidant protection engaging rubrerythrins and flavoproteins (14,15). The major way for E. histolytica cells to obtain nutrients and boost its energy metabolism is to phagocyte substances from the colon, in particular, lipids and carbohydrates, gut bacteria. Therefore, vesicular transport and signaling system are highly developed in Eh, and especially, regulation of kinase/phosphatase cycles and RhoGTPase activity (13,16,17). Moreover Eh is considered to be the only organism among protozoa that possesses genes for receptor serine/threonine kinases (13). During aggressive tissue colonization Eh actively uses protein and carbohydrates lysis enzymes, such as cysteine proteases and beta-amylase to break colon mucus and colon epithelium (18). These lysis enzymes plus amoebapores and Gal/GalNAc lectins (18,19) are the main pathogenic factors in Eh.
The both genomes of Ac and Eh have been sequenced (links to the database with the genome sequences are (20) and (21), respectively). The size of Ac's genome was estimated to be 42.02 megabases and 14974 protein-coding genes were annotated. Eh's genome is twice smaller - 20.84 megabases and with 8163 protein-coding genes being annotated. It is worth to note that only half of all genes in both, Ac and Eh, have assigned functions so far. Besides the extreme difference in gene number, the two amoebas differ also in nucleotide composition: A. castellanii's genome is characterized by 58% of GC content, the genome of E. histolytica by 24%. Moreover, Ac appears to contain genes with the high number of introns (~6,7 exons per gene) and demonstrates often HGT events: 2,7% of all Ac genes are expected to have a prokaryotic origin (8). Eh has approximately 1,2 exons per gene and 1,2% of all genes have been acquired via HGT (13). Several comparative studies of Amoebozoa genomes and transcriptomes were published, mostly highlighting differences and similarities between Dictyostelea and Entamoeba (22-24) or between different E. histolytica strains (18). Here we present the analysis of the RNAseq data for Ac and Eh in order to understand how life conditions shape genomes and to reveal distinctive features of Eh, as a parasite, contrasting with the greatly developed, “omnipotent” in the sense of environmental adjustments Ac. We have applied orthology relationship analysis and gene ontology (GO) characterization in combination with the gene expression estimation and for the first time compared expression patterns of different gene groups for A. castellanii and E. histolytica. The findings presented here improve our understanding of Ac and Eh physiology and evolution.
Material and Methods
E. histolytica and A. Castellanii culture and mRNA isolation
A. castellanii, strain Neff, was cultured as described before (25) in axenic media from 48 hours and yielded at the density of 4-5 x 106 cells/ml. E. histolytica, strain HM-1:IMSS, was grown in axenic conditions in TYI-S-33 medium (ATCC) at 37°C for 72 hours and was collected at the density of 2 x 105 cell/ml (26). Throphosoides of A. castellanii and E. histolytica were lysed with Trizol agent (Invitrogen) and RNA isolation procedure were performed according to the manufacturer's protocol. The mRNA-Seq Sample Preparation Kit (Illumina) was used according to the manufacturers protocol to generate necessary amount of cDNA. The quality and abundance of the samples were monitored with the Agilent Bioanalyzer (Agilent Technologies). Sequencing were done on HiSeq 2000 platform (Illumina) with 36 bp single-end reads for A. castellanii and 76 bp reads for E. histolytica.
RNAseq and data processing
The quality of the reads length was monitored using FastQC quality control tool (27). The reads for E. histolytica were trimmed to 36 bp (the reads length for A. castellanii) with the use of AWK programming language (28) to avoid bias during estimation of transcripts coverage. Reads mapping was performed with TopHat2 (29) with the option “--coverage-search” and segment length of 25. Reference genomes were downloaded from NCBI Genome database (30). 75.6% of reads from A. castellanii and 94.6% reads from E. histolytica were successfully mapped to their cognate genomes. Abundance of the reads per each transcript was counted in RPKM (Reads Per Kilobase of transcript per Million mapped reads) with the use of Cufflinks tool (31) and following options “-u” (--multi-read-correct) and “-b” (--frag-bias-correct) (32).
InParanoid analysis and GO annotation
To compare transcriptomes of the two distinct species we analyzed a) the expression of their orthologs and b) what biological pathways are represented by the most expressed genes. To avoid artifacts and bias in our analysis, as we possessed only one replication of each experiment, we used the ranking approach: transcripts from each species were distributed in eight expression ranks, based on their RPKM counts (Table 1). RPKM values for each identified transcript can be found in Table S3 in Supplementary materials. Further analysis was performed referring to the expression rank of a transcript, but not to its “individual” expression value.
Division of the expressed transcripts into eight expression ranks
| Expression rank | RPKM interval (Cufflinks) | Ac proteins (% percent from all protein coding genes) | Eh proteins (% percent from all protein coding genes) |
|---|---|---|---|
| 1 | (0:1] | 1657 (11.06) | 1029 (12.61) |
| 2 | (1:5] | 2320 (15.49) | 1248 (15.29) |
| 3 | (5:10] | 2112 (14.10) | 1001 (12.26) |
| 4 | (10:20] | 2667 (17.80) | 1092 (13.38) |
| 5 | (20:40] | 2333 (15.58) | 1014 (12.42) |
| 6 | (40:100] | 1875 (15.02) | 924 (11.32) |
| 7 | (100:500] | 1361 (9.09) | 802 (10.82) |
| 8 | >500 | 386 (4.73) | 355 (4.34) |
945 genes from A. castellanii and 877 genes from E. histolytica have expression value zero (RPKM = 0). In the column “RPKM interval (Cufflinks)” parenthesis “(“ signifies exclusion of the number from the interval, square bracket “]” signifies inclusion of the number in the interval.
The search of orthologs between A. castellanii and E. histolytica were performed with InParanoid orthologs and in-paralogs finder (33) with Saccharomyces cerevisiae as the outgroup. Each revealed ortholog was assigned with its expression rank. Gene ontology analysis was performed with the use of BlastKOALA (KEGG Orthology And Links Annotation) online tool (34). Groups of proteins from each expression rank were run through BlastKOALA analysis separately. The results was structured in several pathway blocks, each block is divided into categories, each category contains one or several KEGG orthology groups with specific KO references; proteins in KO groups are assigned with K-numbers. The number of proteins in each biological pathway was counted and used for identification of over-represented pathways (see Table S2, Supplementary). It is worth to note that the protein count is redundant as the same protein can be included in several pathways, for example, Rap-1A protein (K-number K04353) is the part of MAPK signaling pathway (ko04010), Ras signaling pathway (ko04014), Rap1 signaling pathway (ko04015) and ten more. This phenomenon can also be the reason of non-specific annotation and attributing to Ac and Eh metabolisms of non-existing in Amoebozoa pathways. It will be later referred as “cross-annotating”. Example of BlastKOALA results structure for protein actin ACA_038460:
pathway block: Cellular Processes -> category: Cell Motility -> KO group: ko04810 Regulation of Actin Cytoskeleton -> K-number: K05692 actin beta/gamma 1.
Phylogenetic analysis
Proteins for the phylogenetic analysis were selected with the use of PSI-BLAST algorithm (35); multiple sequence alignment was performed with MAFFT software (36). Maximum Likelihood tree was built using RAxML 8.2.4 (37) with 200 bootstrap replicates. Interactive tree of life (iTOL) was used for the tree visualization (38) and Inkscape for the graphical editing (39).
Ribosomal proteins annotation
To search for missing ribosomal protein genes, the amino acid sequences of all annotated ribosomal proteins from the three amoeba species (A. castellanii, E. histolytica and D. discoideum) were selected and TBLASTN search with the default parameters was performed. The task was run three times: each time against one of the three amoebas' genomes. Genomic regions aligned with the known ribosomal proteins from another species but not annotated in the searched genome suggested that the gene is not missing from the genome but escaped the annotation of the given genome.
Results and Discussion
Orthology assignment to A. castellanii and E. histolytica proteins
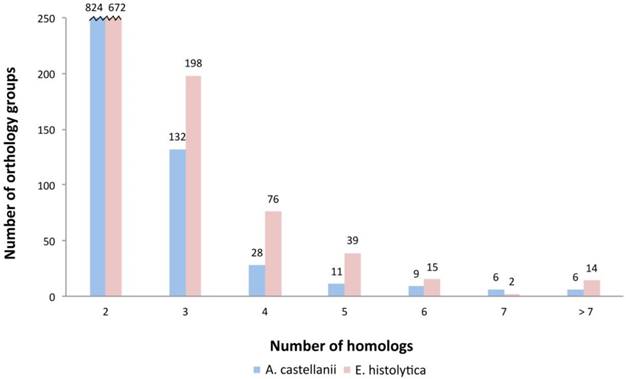
Orthology clustering between Ac and Eh with the use of InParanoid tool resulted in identification of 1016 orthology groups with 1357 homologs from Ac and 1657 homologs from Eh; Saccharomyces cerevisiae was used as an outgroup, what resulted in rejection of 303 putative homologs: 56 proteins from Eh and 221 from Ac. The rejected proteins might be out-paralogs with ancestor genes being lost during evolution or simply artifacts. We also performed InParanoid orthologs search between Eh and D. discoideum and have revealed 1499 orthology groups with 1904 homologs from D. discoideum and 3070 homologs from Eh. Song et al. demonstrated similar findings for Eh and D. discoideum with the use of reciprocal best hit (RBH) approach: 1510 orthology groups with 3216 homologs in D. discoideum and 3833 homologs in Eh (23). Entamoeba and Dictyostelea are expected to have closer relationship, as both belong to Conosa subclade, while Acanthamoeba is included in Lobosa. It is important to note that the phylogenetic analysis of Amoebazoa indicates high evolutionary rate of Eh, at least two times higher than of other lineages (7). Based on these data we conclude that the results obtained here for Ac and Eh are reliable. Interestingly, having almost twice lesser number of protein coding genes, Eh featured higher number of paralogs as compared to Ac. Number of orthology groups between the two amoebas with one-to-one relationship is 565 (55.6%); with one-to-many relationship: 259 (25.5%) groups with a single Ac ortholog and two or more Eh proteins and only 107 (10.5%) groups with a single Eh ortholog and two or more Ac proteins; and 85 groups of orthologs - with many-to-many relationship (8.4%) (Fig. 1). Poor correlation between number of paralogs for each group of genes in Ac and Eh (Pearson coefficient is 0.18; see Fig. S2 in Supplementary) suggests that the amoebas took different evolutional directions as different genes underwent duplication and conservation in the genome.
Distribution of homologs in orthology groups. This plot demonstrates that E. histolytica has more orthology groups with multiple paralogs.
An interesting example of InParanoid performance is the search for homologous proteins among thioredoxins. These enzymes are the major antioxidants in living organisms, and their main function is reducing disulfide bonds in proteins. Although their active site is conserved, thioredoxins are represented by several diverse forms. Ac expresses 21 thioredoxins or thioredoxin domain containing proteins at ranks 1 to 7; whereas Eh expresses 24 thioredoxins at ranks 2 to 8 (expression ranks are explained in Methods; shortly, RPKM expression values are grouped into eight ranks, rank 8 being the most expressed, rank 1 - the least expressed). However, InParanoid tool attributed only five Ac proteins and eight Eh proteins into five orthology groups. There are two reasons explaining this result. First, although all these proteins contain thioredoxin-like domain, they belong to different subclasses of Thioredoxin-like superfamily: Thioredoxins (XP_004335509.1 in Ac, XP_652635.1 and XP_656726.1 in Eh), DsbA_FrnE (XP_004333404.1 in Ac, XP_648689.1 in Eh), and P5 family (XP_004339861.1 in Ac and XP_650000.1 in Eh); as well as to different superfamilies: Pyridine nucleotide-disulphide oxidoreductase superfamily (XP_004351681.1 in Ac and XP_655748.1 in Eh) and YbbN superfamily (XP_004339861.1 in Ac and XP_650000.1 in Eh). As a consequence, all these thioredoxins participate in different biological processes. This is an example of how thorough orthology analysis can improve annotation of genomes, as function assignment to newly discovered genes is often based on their homology to already characterized genes from the related species. Second, InParanoid was designed to search similarity between whole proteins but not separate domains; this can explain why not all amoebic thioredoxins were included in the orthology groups. Actin proteins represent a similar case, as only part of the amoeba actins was clustered by InParanoid. As the subject of actin diversity seems to be complex in amoebas we describe the case in the separate division (see below).
Further we will discuss the largest orthology groups in the studied amoebas, identified by InParanoid search (Table 2); to see expression rank for each of these proteins, refer to the Table S1 in Supplementary.
Most expanded orthology groups in Ac and Eh
| Orthology group | Number of Ac proteins | Number of Eh proteins |
|---|---|---|
| Major facilitator superfamily transporter | 1 | 14 |
| Hypothetical protein | 2 | 12 |
| Peroxiredoxin | 2 | 10 |
| Maltose acetyltransferase | 1 | 9 |
| Biotin protein ligase | 1 | 9 |
| Cysteine proteinase | 8 | 8 |
| START domain | 2 | 7 |
| Actin | 6 | 7 |
| Beta-amylase | 1 | 7 |
| Endonuclease V | 1 | 7 |
| Phosphatase 2C domain | 1 | 7 |
| (Ser/Thr phosphatases, family 2C) | ||
| Amino acid transporter | 1 | 7 |
| Lecithin:cholesterol Acyltransferase | 1 | 7 |
| Phospholipase, patatin family | 5 | 6 |
| RhoGEF | 2 | 6 |
| Pmp3 superfamily | 1 | 6 |
| Ubiquintin/ubiquintin domain | 6 | 5 |
| TolA like proteins | 18 | 4 |
| Von Willebrand factor domain | 8 | 4 |
| Rab32 | 8 | 3 |
| Ser/Thr phosphatase family | 9 | 1 |
| GATA zinc finger domain | 6 | 1 |
| Serpin | 6 | 1 |
Orthology groups expanded in A. castellanii
TolA like proteins have eighteen paralogs in Ac and four in Eh, but their functions have not been yet characterized in Amoebozoa. The majority of Ac paralogs are expressed in the ranks 4-6, while three Eh paralogs demonstrate higher expression levels - at the rank 7. In bacteria, TolA proteins are responsible for membrane integrity and prevent antibiotics from penetrating the membrane (40). Therefore, it is likely that TolA like proteins are responsible for resistance to bacteria toxins and aggressive chemical agents, especially in Ac, as it is constantly endures different environmental impacts. Another big orthology group is Rab32, represented by eight copies in Ac and three in Eh, all highly expressed. Rab32 is likely to be involved in amoebas resistance to parasites via facilitating phagocytosis as a response (41). Serpins are serine proteinase inhibitors and may play role in antibacterial protection, as serine proteases are known pathogenic factors in prokaryotes (42). Ac has six serpin genes, Eh only one but with the higher expression than any of the Ac's serpin genes. Other large orthology groups are von Willebrand factor type A domain containing proteins (facilitate adhesion), and GATA zinc finger domain containing proteins that mostly participate in DNA binding and are associated with several transcription factors. Again, in these two cases the only Eh ortholog is transcribed at higher levels than any of Ac paralogs. Interestingly, the tendency of multiple amoeba paralogs to exhibit lower expression than the only ortholog from another species has been noticed in our data also for thioredoxins, biotin protein ligases and endonucleases V. It might be that higher expression of a single gene in one species is compensated in another species by expression of several copies of this gene but at a moderate level.
Orthology groups expanded in E. histolytica
The most numerous orthology group in Eh is major facilitator superfamily transporters, comprising 14 proteins. These membrane proteins are responsible for transport of small biomolecules (amino acids, sugar phosphates, nucleosides, drugs, ions) into the cell. Functionally related to them amino acid transporters are present in seven copies in Eh and one in Ac. The next highly enriched orthology group contains proteins with zinc-finger domain, also characterized in Pfam database as TLD-domain. Its function is unknown, although often zinc-finger domains are participants of DNA-binding or ubiquitinisation. Beta-amylases reflect ability of Eh to destruct colon mucus during tissue invasion. Beta-amylase depletion in cultured Eh resulted in amoeba's decreased ability to break mucus layer and, thus, eliminated colon invasion (18). Eh genome encodes eight beta-amylases, and mRNA of four of them is expressed in our data at ranks 1, 6, 6, 8. Ac uses mostly alpha-amylases and has only one beta-amylase gene (rank 4). Beta-amylase is characteristic to plants, fungi and bacteria and breaks polymeric carbohydrates into maltose molecules, those further can be directed into energy metabolism and biosynthesis. Maltose-acyltransferase, the enzyme participating in maltose and CoA metabolism, is represented with multiple gene copies in Eh and only one in Ac. During tissue invasion Eh encounters unusually high oxygen concentrations (18,43), and at that point antioxidant protection is crucial for the parasite survival. In agreement with that we observed relatively high number of transcribed peroxiredoxin genes in Eh, in particular, ten copies, while Ac possesses only two peroxiredoxins. Another stress protecting agent is endonuclease V that is involved in DNA repair and is represented by seven genes in Eh and one in Ac. Ac and Eh seem to expand different types of protein phosphatases: 2C domain containing phosphatases (PP2C domain) are more typical for Eh (seven gene copies, while only one gene in Ac). At the same time metallophosphatases (MPP domain) are expanded in Ac (nine paralogs, and only one is present in Eh). Rho GTPases are universal eukaryotic regulators of the cytoskeleton dynamics and the importance of Rho signaling system for the physiology of Eh is discussed by Loftus et al. (13). Our InParanoid analysis revealed several orthology groups for RhoGAP and RhoGEF. Biotin protein ligase (involved in biotinylation of different proteins) has one ortholog in Ac (rank 6) and nine copies in Eh, though all poorly expressed (ranks 1 and 2). It is not clear why E. histolytica “prefers” to use several gene copies for this enzyme and could be interesting to study whether it has any relation to parasitism or resistance to the host immune system.
Paralogs equally represented in A. castellanii and E. histolytica
Cysteine proteases, ubiquitins and actin proteins are equally well represented in both amoebas. Cysteine proteases are major enzymes in protein digestion. In particular, CP-A5 and CP-C1-A type peptidases are known as determining factors during host cells lysis by Eh (18). Our InParanoid search has identified one orthology group of C1-type cysteine proteases with equal number of paralogs for both species (eight), but those in Eh being expressed at higher levels (Fig. S1, Supplementary). Maximum likelihood phylogenetic analysis shows two distinct clusters with A. castellanii and with E. histolytica cysteine proteinases (see Fig. S1 in Supplementary). It is likely that these proteases perform different functions in the two amoebas, as it is thought that CP-C1-A type works extracellular in parasites for invading surrounding tissues and cells, while in free living eukaryotes it is localized mostly in lysosomes.
In general, we observe in Ac the tendency to have multiple copies of the genes responsible for antibacterial resistance and the genes coordinating response to changing external conditions. As well, our orthology analysis revealed specific genomic features in Eh. Namely, genes that are involved in the host tissue lysis and in uptake of nutrients from extracellular environment underwent extensive gene duplication. Gene duplication is the most common way for new genes to appear and is not rare in parasitic organisms, especially for those genes that contribute to parasite's pathogenicity and resistance to host immunity. This takes place, for example, in Giardia lamblia and several Apicomplexa (44,45). H. A. Lorenzi et al. conducted a rigorous study and revealed four different types of genomic duplications in Eh that are often accompanied by transposons (46). Another example is putative transcription regulator CudA; among all eukaryotes it is found only in Amoebozoa. It was shown that only parasitic Eh and Hartmannella vermiformis reveal CudA gene duplication, but not free-living amoebas (24). This observation supports the hypothesis that parasitic organisms are prompt to duplication events. Here, for the first time we point out that Eh uses gene duplication mechanism to improve its survival and adapt to its host; the whole genome structure and expression patterns can be influenced by this process.
Actin genes in A. castellanii and E. histolytica
Most of eukaryotes possess several actin genes, and often their protein products exhibit differentiated functions. This phenomena is well known in multicellular forms, but sometimes is also observed in unicellular organisms. For example, the study of amoeba class Arcellinida had distinguished two actin clusters with the differing sequence patterns, GC content and, possibly, functions (47). There are several phylogenetic studies of amoebas' actins (6,7,47) that demonstrate wide expansion of actin genes within Amoebozoa species. Based on our comparative analysis, we suggest that Ac and Eh went different paths in developing their cytoskeletal systems, as none of the homologous actins (seven in Eh and six in Ac) were detected in the RNAseq data (see Table S1 in the Supplementary), but rather those actins are expressed that were not defined by InParanoid as orthologs. In whole, the genome of A. castellanii encodes 12 actin genes and yet poorly annotated 17 actin subfamily genes; they are expressed at ranks 1 to 7. Eh genome contains 18 actin genes, nine of them are expressed in our experiment at ranks 3 to 7, and nine have zero expression. Further, we describe the example of three particular actin domain containing proteins in E. histolytica with the possibly specific functions acquired by fusing with new protein domains.
E. histolytica's RNAseq data revealed three highly expressed actin domain containing proteins that are poorly characterized so far. One of the proteins is XP_655533.1 with the length of 876 amino acids, expression rank 7. Using BLASTP with default settings against the non-redundant protein database we identified that its N-terminal 300 amino acids share sequence similarity with FtsY protein from Piscirickettsia salmonis (E value = 2e-44), the bacteria that parasite on salmon. FtsY protein is suggested to be the bacterial analog of eukaryotic SRP (signal recognition protein) receptor that participates in targeting nascent ribosome-mRNA-protein complexes to rough EPR (48). It is possible that XP_655533.1 protein in Eh participates in translocation of nascent proteins to lipid membrane structures within the cell. The sequence similarity to the bacterial protein also suggests HGT. Protein XP_652727.1/EAL47341.1, expression rank 7, exhibits similar characteristics. These two proteins have no homologs in other Amoebozoa, suggesting their specificity to Entamoeba. Another Eh protein with C-terminal actin domain is XP_652978.1, 763 amino acid long, expression rank 5. Its N-terminal part contains two filamin domains and one Ist1 domain. Filamin is used by the cell as a binding domain for actin and several other proteins; Ist1 regulates Vsp4 activity in vesicle formation (MVB pathway). Only six proteins with the domain structure “Ist1-filamin-filamin-actin” has been discovered so far and all from Amoebozoa: four in Entamoeba and two in Dictyostelea (Pfam database: http://pfam.xfam.org/family/actin#tabview=tab1). So far, none of these proteins were functionally characterized and may represent some unique Entamoeba features. Further molecular biology and biochemical experiments are needed to confirm biological meaning of these proteins and to reveal their functions.
Gene Ontology analysis
Ac and Eh proteins in each expression rank (ranks 1 to 8) were subjected to BlastKOALA annotation and classified into six pathway blocks: Metabolism, Genetic information processing, Environmental information processing, Cellular processes, Organismal systems, and Human diseases. To discuss the results we introduced two parameters: number of all proteins expressed in each pathway and expression pattern, i.e. distribution of proteins within expression ranks for each pathway (Fig. 3). Full analysis data can be seen in the Supplementary, Table S2. However, part of the Cellular processes pathways, such as oocyte meiosis, tight junctions, and the whole pathway block Organismal systems were excluded from the table and the discussion. Moreover, only category “amoebiasis” from Human diseases pathway was regarded. Obviously, the excluded categories are example of non-specific “cross-annotating” (see Methods). Further we summarize the most outstanding cases.
Expression pattern of A. castellanii and E. histolytica proteins for different pathway blocks.
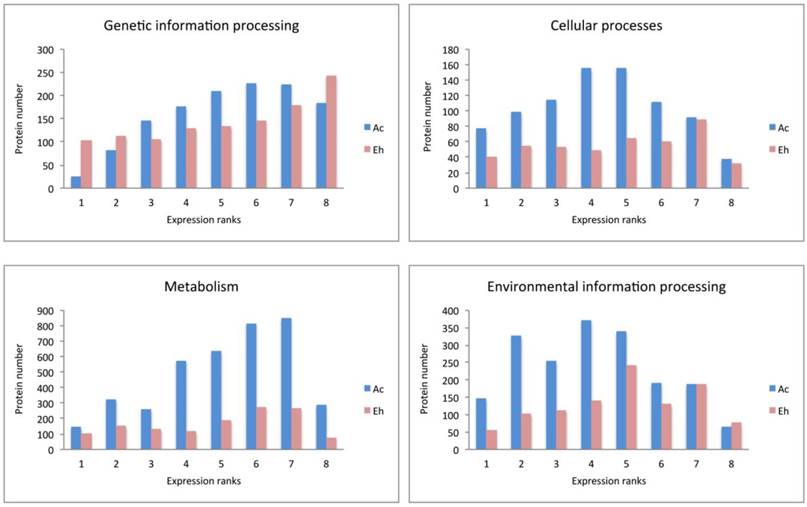
Expression patterns in A. castellanii and E. histolytica
Expression patterns in Ac and Eh are distinct (Fig. 2 and Fig. 3), except Genetic information processing pathway block. In general, Eh genes seem to be evenly distributed between all expression ranks, with some preference for the top expressed ranks (6-8). Such phenomena of intense expression at high levels can be related to the overall gene deficiency in E. histolytica and represent the compensatory effect. Ac demonstrates enrichment in the ranks with moderately expressed genes (ranks 3-6) for Cellular processes and Environmental information processing, and shift to higher expression levels for Metabolism and Genetic information processing. This can be an evidence of active growth of the amoeba; at the same time nutrient rich growth medium and the constant laboratory environment may result in the lesser need of the amoeba for mobility and adjustment to external conditions.
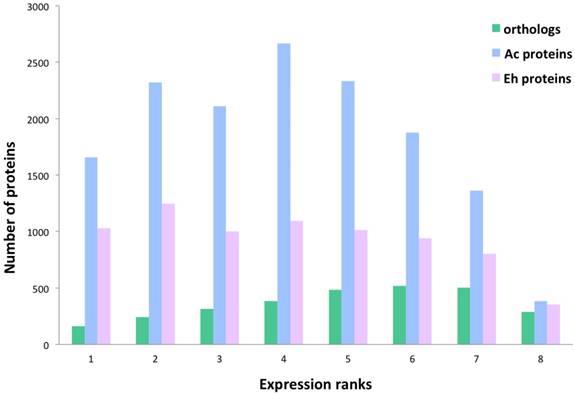
Overall protein distribution within the expression ranks. Number of Ac and Eh proteins that are present in orthology groups show correlation with the expression levels, thus there is a tendency to have more homologous proteins expressed in the ranks 5-7 than in the ranks 1-4. Spearman correlation coefficient between the overall orthologs number and expression rank is 0.73.
Genetic information processing
This block describes transcription, translation, folding-sorting-degradation of proteins, replication and repair processes. In general, all the pathways are similarly well represented in both amoebas, though there are some differences. In particular, protein number in Eh is smaller for ubiquitin mediated proteolysis and SNARE interaction in vesicular transport. As well, repair processes, such as homologous recombination, DNA replication and base excision/scission repair is the least represented in Eh.
Interesting observation concerns ribosomal proteins and aminoacyl-tRNA synthetases. The Eh genome harbors more than twice as many copies for ribosomal protein genes than the Ac genome: 213 versus 86 genes (Table S2, Supplementary). Consistently, InParanoid tool identified 64 orthology groups including 62 ribosomal proteins from Ac, and 141 from Eh. Table 3 compares the number of the multi-copy ribosomal protein genes between Ac and Eh. We have performed TBLASTN search for possibly missing ribosomal genes in Ac and Eh genomes and could discover new candidates, not annotated yet in the latest genome assembly versions: 37 genes for Ac and 2 genes for Eh (Table S4, Supplementary).
Copy-number of genes coding for ribosomal proteins
| Gene copy = 1 | Gene copy = 2 | Gene copy = 3 to 5 | |
|---|---|---|---|
| A. castellanii | 58 | 8 | 4 |
| E. histolytica | 11 | 19 | 44 |
Majority of the genes in both amoebas are transcribed into mRNA at high expression levels (ranks 7 and 8). While there is no solid evidence for a specific duplication of ribosomal protein encoding genes in parasitic organisms, several studies describe the phenomena of acquiring new functions by ribosomal protein paralogs. Many works demonstrated functional differentiation of ribosomal protein copies in Arabidopsis thaliana (for example (49)). Similarly, in S. cerevisiae 75% of ribosomal proteins have gene multiple copies, and depletion of some of them in cultured yeasts resulted in different phenotypes (50). Komili et al. even offered the term “ribosome code”, referring to the multilevel complexity of translation regulation (50). The “ribosome code” includes post-translational modifications of ribosomal proteins, rRNA modifications and functional diversification of duplicated ribosomal proteins. Moreover, the recent study of the Eh virulence highlighted importance of changes in protein expression levels during transition of the amoeba from commensal life style in human colon to invasive parasitical behavior (43). Together with the previous research, our observation suggests that E. histolytica may have developed intricate system to manipulate its translational machinery to efficiently respond to the host immune resistance and nutrition conditions.
Eh possesses 74 genes coding for different types of ribosomal proteins, many of them have 1 or multiple paralogs. Ac has 70 different types of ribosomal protein genes, that demonstrate lesser duplication level than that in Eh.
Another curious observation is gene copy number of aminoacyl-tRNA synthetases (also called ligases), mainly expressed at ranks 6 and 7 in both species. These are proteins that ligate amino acids to the corresponding tRNA molecules. Eh genome contains one isoform for each enzyme, except ligases for phenylalanine, serine, valine and tryptophan that have two or three gene copies. At the same time Ac encodes for 2-4 copies for most of the enzymes, except for those aimed at arginine, lysine and glutamic acid. To note, only four aminoacyl-tRNA ligase genes for histidine, isoleucine, glutamate, asparagine are recognized as HGT-acquired in Ac (8) that points on the underlying gene duplication mechanism. Having multiple copies of aminoacyl-tRNA synthetase encoding genes is known phenomenon in living organisms, but yet not fully explained. In prokaryotes it can be the way to resist toxic compounds, including antibiotics (51). Different isoforms of these enzymes may perform distinct functions, like RNA trafficking, rRNA synthesis, proof-reading; in some bacteria these enzymes can form homo- and heterodimers to improve translation efficiency in zinc-deficient conditions (52-54). For eukaryotic cells it is usual to have at least two gene copies for each enzyme due to the mitochondrial needs (55). Thus, decreased repertoire of aminoacyl-tRNA synthetases in Eh can be explained by the loss of most of the mitochondrial genes. Moreover, it has been noticed that Eh has high copy number of tRNA genes (4500 copies) (56), and this can be a mechanism compensating poor gene representation of aminoacyl-tRNA synthetases. It is interesting fact that Ac and Eh chose different directions in developing their translation machineries: one in duplication of ribosomal proteins, another in expansion of aminoacyl-tRNA ligases.
Cellular processes
Pathways related to the functioning of phagosomes, lysosomes and cell cycle are equally enriched in both amoebas at the level of the top expression ranks (8 and 7), but the lower expression ranks in these groups are under-represented in proteins number in Eh as compared to Ac. Peroxisome pathway is poorly pronounced in Eh, but extensive in Ac; endocytosis pathway is the only pathway in this block that is enriched in proteins as good in Eh as in Ac, what proves the crucial role of extracellular metabolites uptake for the parasitic growth and propagation. There is evidence for such processes as autophagy, apoptosis and p53 protein-stress signaling pathway to be present. Although it is not known if these processes exist in amoeba, it is possible that at least their reduced or modified variants may function in these organisms. For example, Ac expresses cathepsin B and cyclin-dependent kinase, enzymes specific for the suggested pathways.
Characteristic differences between Ac and Eh are detected for cytoskeletal proteins and cytoskeletal regulation pathway. In general, both amoebas express the same set of protein at the top three expression ranks (ranks 8, 7, 6), these are: actin, ARP2/3, PIP5K, RAC1, KRAS GTPase, Rho GEF, profilin, cofilin, paxillin, cell division controlling protein 42; however, only few genes are expressed in Eh at the low expression ranks (Table S2, Supplementary). Thus, the total protein count in Eh almost three times lesser than in Ac. Cytoskeletal proteins specific to E. histolytica are grainins, the calcium-binding vesicular proteins. They contain EF-hand motif that is typical for calcium-regulated polypeptides. Two grainin genes are expressed at ranks 8 and 7 and are likely to partially compensate absence of Golgi apparatus and rough EPR (57).
Environmental information processing
Both, Ac and Eh, conduct complex interactions with extracellular environment. This is the most enriched in proteins block for Eh, and some pathways are even better represented in Eh than in Ac, for example, Wnt pathway (cell specification and cell division), NF-kappa B pathway (immunity, cell survival), HIF-1 pathway (gene transcription in low-oxygen conditions). These processes have not been identified in amoebas and are common for multicellular organisms. But as only half of the detected genes in Ac and Eh have assigned functions, it is possible that more participants from these full or modified pathways yet are to be discovered. Thus, Ac (but no Eh) transcribes mRNA for frizzled protein, the key component in Wnt pathway, and casein kinase that participates in DNA repair and Wnt pathway. InParanoid analysis revealed orthology between casein kinase I in Ac and Eh, the former having one isoform in rank 4, the latter - four isoforms, expression ranks 2, 4, 6, 7. Casein kinase II was not identified as ortholog to casein kinase I and shows abundance in E. histolytica (three genes in Ac and eight in Eh). Overall in our data Ac expresses 11 tyrosine kinases, 132 serine/threonine kinases, 44 histidine kinases, and Eh expresses 50 tyrosine kinases, 20 serine/threonine kinases, and no histidine kinases, which in eukaryotes are responsible for stress resistance.
The following reference pathways are equally represented in Eh and Ac in respect to protein number and expression levels: Calcium signaling pathway, cGMP-PKG pathway (sensing NO levels), Hippo pathway (sensitive to cells density), VEGF pathway (cell growth and migration, so far known mostly in epithelial cells). Pathways that exhibit similar protein enrichment for both species only at expression ranks 8 and 7 are MAPK pathway, phosphatidylinositol, sphingolipid and phospholipase D pathways, PI3K-Akt and mTOR signaling pathways. Ras and Rap1 pathways, cAMP and AMPK signaling, as well as mTOR and FoxO cascades are prevalent in Ac as compared with Eh. All these processes are responsible for basic living functions, including energy metabolism, cell growth, biosynthesis. Again, not all of these processes are proved to be present in amoebas so far, and further biochemical experiments are required to clarify whether some of these biochemical reactions are, indeed, present or these GO annotations are non-specific. Altogether, these observations demonstrate high protein number and expression level for the Environmental information processing as in Ac as in Eh.
Metabolism
Pathways of carbohydrate metabolism are the most developed in Eh within this block and important for energy transformation and biosynthesis. It is also related to the already mentioned above lysis of mucus during tissue invasion that is based on degradation of long-chain carbohydrates by beta-amylase. Proteins supporting fructose/mannose and pentose phosphate pathways, as well, inositol phosphate metabolism, oxidative phosphorylation and glycolysis, are present in both species, although the protein number in Eh is half of that in Ac.
Majority of lipid and amino acid metabolic pathways are absent in Eh. Exceptions are fatty acid elongation and degradation, sphingolipid metabolism, glycerolipid and glycerophospholipid metabolisms. The last two pathways are the most represented groups in both amoebas, as they are closely related to basic life processes, such as glycolysis and fatty acid degradation. Two enzymes, previously reported in Eh as pathogenic factors (18) are outstanding in our RNAseq data in terms of gene copy number and expression levels: lecithin:cholesterol acyltransferase has seven paralogs in Eh with the expression up to rank 7, while Ac has only one ortholog expressed at rank 3; and long-chain-fatty-acid-CoA ligase with thirteen genes (expression ranks 2 to 7) in Eh; Ac has three isoforms expressed at ranks 2, 6, 6. Not all acyl-CoA ligases in E. histolytica were identified as homologous to that in A. castellanii, that signifies the diversity of these enzymes in the parasite. The only amino acid metabolisms detected in E. histolytica are that of glycine-serine-threonine and of cysteine-methionine. The importance of these pathways can be explained by the fact that cysteine is an important antioxidant agent in the absence of glutathione synthesis in Eh (13), and serine is a substrate for phosphatidylserine synthesis. Indeed, PS-synthase is detected in Eh (XP_657102.1), as well as in Ac (XP_004368267.1), both orthologs are expressed at rank 6. Methionine and threonine degradation can be an efficient way to produce ATP in anaerobic conditions (16). In support to this hypothesis Eh expresses threonine dehydratase (ranks 2, 3, 7) and methionine-gamma-lyase (ranks 1, 7, 7) - enzymes that are required for these amino acids degradation. Interesting observation concerns nucleotides metabolism: majority of the proteins involved are expressed at ranks 4-7 in both species, but the ratio of proteins participating in purine (adenine, guanine) and pyrimidine (thymine, cytosine) metabolism is different. It is close to one for Eh and is around 1.5 for Ac, with prevalence for purines. The explanation may lie in the understanding of the fact that purines (cAMP, ATP and GTP) are essential molecules for regulation of cell growth and multiple physiological processes that in Eh are greatly dependent on the host metabolites.
Glycans related reactions are rare examples of intense metabolic activity in parasitic E. histolytica. Glycans play important role in substrate recognition by the cell and equally important for free-living Ac and for Eh during tissue intrusion. Thus, GPI (Glycosylphosphatidylinositol) anchor is a part of intermediate subunit of Gal/GalNAc lectins, an important pathogenic factor in the parasitic amoeba (19,58). Moreover, the intermediate domain of Gal/GalNAc lectins has been identified only in Eh and Entamoeba dispar (11). GPI lipids are considered as one of the main toxic immune suppressor factors in protozoa parasites (59), and lipophopshoglycans are important agents to resist the host protection mechanisms (58). Unlike E. histolytica, A. castellanii exhibits metabolism of co-factors and vitamins, as well terpenoids and polyketides metabolism. Xenobiotics degradation is very important for Ac as this amoeba constantly encounters multiple aggressive agents during its life cycle. Eh expresses only few proteins in this category, most likely mis-assigned. As expected, antibiotics biosynthesis is stronger represented in Ac, but also one of the most developed in Eh. The Entamoeba evolution in response to medical drugs is poorly studied subject, though is definitely intriguing question from the perspective of medicine.
Amoebiasis
Genes maintaining pathogenicity are highly expressed in Eh even when it grows in axenic environment, as in our experiment. Amoebapores A, B and C are unique for parasitic Eh and are among the most abundant transcripts (three genes expressed at the rank 8). They are orthologous to Ac saposins, lysosomal proteins that assist membrane lysing enzymes (60). It is not known yet how Ac destroys cell membranes when it parasites on humans, and saposins are possible candidates for the lysis molecules (61). Only one out of four Ac saposin encoding genes shows expression in our RNAseq data (rank 5). Leishmanolysin (or cell surface protease gp63) was originally identified in the parasite Leishmania major as a crucial pathogenic factor. This enzyme from Eh is most related to L. major enzyme among all discovered eukaryotic leishmanolysin-like proteins (62). Two cell surface proteases gp63 are expressed in Eh at ranks 6 and 7, and one is expressed in Ac at rank 2. The role of the protein in L. major is to resist to the host immune system and to prevent parasite recognition by the host phagocytes (62). Although functions of this protein in Eh is not fully understood, it is considered to influence amoeba's motility and participate in destruction of the host cells. Moreover, antibodies against surface protease gp63 prevent amoebiasis development in animal model (63,64). Gal/GalNac lectins have been already mentioned throughout this work as participants of mucus digestion. Eh has 11 genes coding for light and heavy lectin subunits, eight of them are expressed at ranks 6, 7, 8. Ac genome encodes for only one galactose-binding-lectin-domain containing protein with no orthology relation to Eh actins, expression rank 7. Phagosome-associated TMK96 kinase (PATMK) is expressed at the low level (rank 2) in Eh and is important phagocytosis element which is claimed to contribute to the host erythrocytes digestion (65). No Ac analog is found. The high level of expression and the high number of orthologs for proteases, mucus lysis enzymes and membrane channeling proteins are clearly observed for Eh and point out the extreme importance of these proteins for the parasite.
Genes specific to E. histolytica
During manual scanning of our data we identified several highly expressed proteins, possibly involved in pathogenicity of E. histolytica (Table 4). Two of them are cysteine protease inhibitors type 1 and 2 (chagasin family peptidase inhibitor I42 domain), expressed at ranks 8 and 7, respectively. Their function is not yet clear but it is suggested that inhibitor 1 participates in regulation of proteolitic activity within the amoeba cell, while inhibitor type 2 protects against host proteases (66).
Genes specific to E. histolytica with the role in the pathogenicity
| Protein name | Gene ID | Expression rank |
|---|---|---|
| Cysteine protease inhibitor type 1 | EHI_040460 | 7 |
| Cysteine protease inhibitor type 2 | EHI_159600 | 8 |
| Rubrerythrin | EHI_134810 | 7 |
| Flavoprotein | EHI_067720 | 1 |
| EHI_135180 | 1 | |
| EHI_152650 | 1 | |
| EHI_022600 | 1 | |
| EHI_096710 | 1 | |
| EHI_129890 | 1 | |
| EHI_103260 | 1 | |
| EHI_181710 | 2 | |
| EHI_189480 | 2 | |
| EHI_025710 | 3 | |
| EHI_138480 | 5 | |
| EHI_064530 | 5 | |
| EHI_134740 | 7 | |
| EHI_159860 | 7 | |
| AIG1 | EHI_067730 | 0 |
| EHI_079610 | 1 | |
| EHI_176280 | 1 | |
| EHI_115170 | 1 | |
| EHI_157260 | 2 | |
| EHI_102600 | 2 | |
| EHI_025990 | 2 | |
| EHI_199470 | 2 | |
| EHI_195270 | 2 | |
| EHI_119040 | 3 | |
| EHI_129470 | 3 | |
| EHI_115150 | 3 | |
| EHI_022500 | 4 | |
| EHI_115160 | 4 | |
| EHI_195250 | 4 | |
| EHI_089670 | 5 | |
| EHI_072850 | 5 | |
| EHI_195260 | 5 | |
| EHI_176590 | 5 | |
| EHI_136940 | 5 | |
| EHI_109120 | 6 | |
| EHI_176580 | 6 | |
| EHI_126550 | 6 | |
| EHI_157360 | 6 | |
| EHI_136950 | 6 | |
| EHI_180390 | 7 | |
| EHI_126560 | 7 | |
| EHI_144270 | 7 | |
| EHI_176700 | 7 |
Antioxidant protection is required in Eh during tissue invasion, when the parasite confronts oxygen in blood and ROS generated by macrophages. There are two antioxidant protectors in Eh that resemble bacterial proteins. In particular, rubrerythrin (one gene, expression rank 7) neutralizes peroxide and is found, too, in anaerobic bacteria. Within Amoebozoa it is encountered only in E. histolytica, Entamoeba invadens and Entamoeba dispar. Flavoproteins, proteins of DNA repair and antioxidative protection systems, are numerous in Eh (14 genes) and are widely expressed (ranks 1-7); Ac possesses three flavoproteins, but not orthologous to those from Eh. Acanthamoeba's flavoproteins are members of electron transfer chain in mitochondria and include two characteristic domains: electron transfer domain (AANH like superfamily) and FAD-binding domain (ETF-alpha superfamily). Entamoeba's genes encode for iron-sulfur flavoproteins that belong to NADPH-dependent FMN reductase superfamily and perform anti-oxidative protection typical for bacteria and anaerobic protozoan parasites (13,15,67). Interestingly, flavoproteins activate metranidozole and chloramphenicol drugs that are used for amoebiasis treatment (68).
AIG proteins are not firmly assigned with any function in amoebas; in plants they are known antibacterial protectors (17) and developed diverse functions in animals. Nevertheless, it is obvious that these proteins are important to both species, especially to E. histolytica: Ac expresses one AIG1 (rank 1) and five AIG2 (ranks 4-6); whereas Eh has 28 AIG1 gene copies being not orthologous to those in Ac and highly expressed (14 transcripts in ranks 5, 6 and 7). Several studies speculated about the role of AIG1 proteins for host tissues invasion (18,19,43). Within Amoebozoa AIG2 is found only in Ac and contains Gamma-glutamyl cyclotransferase (GGCT) like domain, it is likely to participate in gamma-glutamyl cycle. Macrophage migration inhibitory factor-like protein (XP_655608) is represented in Eh by only one gene copy and is highly expressed (rank 7). This protein has no homologs in other Amoebozoa, except E. invadens, and shares sequence similarity with bacterial proteins from 4-Oxalocrotonate tautomerase superfamily. In bacteria this tautomerase usually participates in isomerization of unsaturated alpha-beta-ketones, the downstream product can vary from cysteine proteases inhibitor (69) to antibiotics (70). It is a potential drug target, as its single gene copy is highly expressed (therefore its depletion can not be balanced by the cell) and has low similarity to the analogical protein in the host.
Considering the overall reduction in gene number in E. histolytica it is exciting to identify proteins that are absent in Ac but highly expressed in Eh. Many of such proteins, as mentioned above, are related to the parasite's pathogenicity and protection against host resistance, therefore their investigation may help to suppress the progression of amoebiasis.
Conclusions
The two amoebas, A. castellanii and E. histolytica, demonstrate grand differences in the architecture of their genomes: Eh genome encodes about half of the protein coding genes comparing to Ac, with six times smaller number of introns per gene. In addition Eh is characterized by relatively high percentage of transposable and repetitive elements (46). Orthology analysis has showed that different protein families underwent expansion in Ac and in Eh, with the tendency in Ac towards proteins mediating response to the external signals; and in Eh - towards proteins providing it with nutrients from the host, either by phagocytosis or by lysis of host tissues. Eh reveals multiple cases of gene duplication which occurs in the greater extent than in Ac; the latter in its turn benefits by assimilating many genes involved in its metabolic pathways via horizontal gene transfer (8). Obviously, the two species had diverged due to their needs for adjusting to different environmental conditions, though why and how these changes appeared is mostly not clear. Many highly expressed and widely represented genes have yet not been studied in amoebas and functions of their products are poorly understood. For example, TolA like proteins and serpins in Ac, biotin protein ligases in Eh, and AIG proteins. Here we also highlighted importance of investigating three poorly studied E. histolytica cytoskeletal proteins (XP_655533.1, XP_652727.1 and XP_652978.1). Two of them (XP_655533.1 and XP_652727.1) contain FtsY-like domain and are especially interesting as they might be the E. histolytica's invention to substitute for the absence of endoplasmic reticulum. Another three under-investigated proteins are possibly related to host immune system resistance (macrophage migration inhibitory factor XP_655608.2; cysteine protease inhibitor-2 XP_649363.1 and cysteine protease inhibitor-1 XP_653255.1) and are interesting targets for the future experimental research. We believe that their functions specifically contribute to the parasite's thriving in the human colon environment.
Pathway analysis allowed us to make an interesting observation about differences in the amoebas' expression patterns (Figures 2 and 3). Ac reveals distinct patterns for different biological processes, proving its expression plasticity and ability to quickly rearrange its behavior according to the current needs. Eh genes are relatively equally distributed throughout eight expression ranks for all pathway blocks, with the slight tendency towards the top expressed ranks, so that ranks 5-7 usually show maximum in protein enrichment. This feature can compensate overall low number of genes in the parasite's genome, while in general Eh expression pattern reflects its great dependency on the host resources. The strategy of genome reduction and at the same time increasing expression of the present genes in general is more advantageous and economical for endo-parasitism than maintaining complex genome structure. In agreement with that, the processes responsible for transcription, translation and post-translational protein processing did not undergo reduction in Eh as most of its biological pathways. Moreover, we have predicted that E. histolytica orchestrates specific regulation of its translational machinery: it expresses high number of ribosomal protein gene copies and atypically low for eukaryotes gene number for amino-tRNA ligases. The situation is reverse in Ac. Other features of different environmental adaptation between the two amoebas can be reflected by anti-oxidant protection (rubrerythrin and flavoproteins in Eh and superoxide dismutase in Ac); usage of the different types of cysteine proteases and protein phosphatases, preference for the different amylases (alpha-amylase in Ac and beta-amylase in Eh); preference for AIG1 protein in Eh and AIG2 in Ac; as well, the metabolisms of purines and pyrimidines, as Ac has enhanced activity for pathways involving pyrimidines (pyrimidines are precusors of ATP and GTP and the main energetic and signaling factors in the living cell). Similarly, expression of different actin isoforms signifies differences in cytoskeleton arrangement in Ac and Eh and suggests that the requirement of particular actin genes varies throughout life cycle of the amoebas. However, more research is required for full understanding of cytoskeleton diversity and regulation in Amoebozoa.
In this work, we aimed to study commensal life stage of the parasite rather than aggressive parasitic behavior in E. histolytica that allowed us to analyze genetic determinants of parasitism, i.e. those genes that are important for Eh thriving and are expressed even at the latent, asymptomatic stage of amoebiasis. Thus, we highlighted genes of beta-amylases, cell surface protease gp63, amoebapores, Gal/GalNac lectins and cysteine proteinases that are expressed at top ranks in our data and are crucial for E. histolytica. It is considered that evolution of amoeba species is relatively fast process (71) but we do not know what allows these organisms such impressive adaptive abilities. Therefore, further studies of horizontal gene transfer, gene duplication and expression regulation mechanisms are important for understanding ecology and genetics of Amoebozoa.
Supplementary Material
Table S1.
Table S2.
Table S3.
Table S4.
Figures S1-S2.
Acknowledgements
We thank M. Jakalski (University of Gdansk, Gdansk, Poland) for his valuable advice in data analysis and N. Grundmann (Institute of Bioinformatics, Münster, Germany) for his assistance in informatics part of the data processing. We also thank Malgorzata Wojtkowska (Laboratory of Bioenergetics, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University) for the help in RNA extraction.
Funding
The current study was funded by Institute of Bioinformatics, Muenster, Germany.
We acknowledge support by Open Access Publication Fund of University of Muenster.
Availability of data and materials
The RNAseq data has been submitted to DNA Data Bank of Japan (DDBJ), accession number: DRA006231.
Authors' contribution
VS performed the analysis of RNAseq raw data, GO analysis, data interpretation and drafted the manuscript; all authors contributed to the writing of the final version of the manuscript; TK assisted in bioinformatics data processing; YS performed RNAseq experiment; HK and WM designed the project. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
References
1. Sluse FE, Jarmuszkiewicz W. Uncoupling proteins outside the animal and plant kingdoms: functional and evolutionary aspects. FEBS Lett. 2002Jan16;510(3):117-20
2. Anderson IJ, Watkins RF, Samuelson J, Spencer DF, Majoros WH, Gray MW. et al. Gene Discovery in the Acanthamoeba castellanii Genome. Protist. 2005;156(2):203-14
3. Tekle YI, Anderson OR, Katz LA, Maurer-Alcalá XX, Romero MAC, Molestina R. Phylogenomics of “Discosea”: A new molecular phylogenetic perspective on Amoebozoa with flat body forms. Mol Phylogenet Evol. 2016Jun;99:144-54
4. Amaral Zettler LA, Nerad TA, O'Kelly CJ, Peglar MT, Peglar MT, Gillevet PM. et al. A Molecular Reassessment of the Leptomyxid Amoebae. Protist. 2000Oct;151(3):275-82
5. Cavalier-Smith T, Chao EE-Y, Oates B. Molecular phylogeny of Amoebozoa and the evolutionary significance of the unikont Phalansterium. Eur J Protistol. 2004;40:21-48
6. Lahr DJG, Grant J, Nguyen T, Lin JH, Katz LA. Comprehensive Phylogenetic Reconstruction of Amoebozoa Based on Concatenated Analyses of SSU-rDNA and Actin Genes. Ouzounis CA, editor. PLoS One. 2011Jul28;6(7):e22780
7. Cavalier-Smith T, Fiore-Donno AM, Chao E, Kudryavtsev A, Berney C, Snell EA. et al. Multigene phylogeny resolves deep branching of Amoebozoa. Mol Phylogenet Evol. 2015;83:293-304
8. Clarke M, Lohan AJ, Liu B, Lagkouvardos I, Roy S, Zafar N. et al. Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol. 2013;14(2):R11
9. Alsam S, Jeong SR, Sissons J, Dudley R, Kim KS, Khan NA. Escherichia coli interactions with Acanthamoeba: a symbiosis with environmental and clinical implications. J Med Microbiol. 2006Jun1;55(6):689-94
10. Matin A, Jung S-Y. Interaction of Escherichia coli K1 and K5 with Acanthamoeba castellanii Trophozoites and Cysts. Korean J Parasitol. 2011Dec;49(4):349
11. Weedall GD, Sherrington J, Paterson S, Hall N, Su X. Evidence of Gene Conversion in Genes Encoding the Gal/GalNac Lectin Complex of Entamoeba. Lohia A, editor. PLoS Negl Trop Dis. 2011Jun28;5(6):e1209
12. Chavez-Tapia NC, Hernandez-Calleros J, Tellez-Avila FI, Torre A, Uribe M. Image-guided percutaneous procedure plus metronidazole versus metronidazole alone for uncomplicated amoebic liver abscess. In: (ed.) Chavez-Tapia NC. Cochrane Database of Systematic Reviews. Chichester, UK: John Wiley & Sons, Ltd. 2009
13. Loftus B, Anderson I, Davies R, Alsmark UCM, Samuelson J, Amedeo P. et al. The genome of the protist parasite Entamoeba histolytica. Nature. 2005Feb24;433(7028):865-8
14. Jeelani G, Nozaki T. Metabolomic analysis of Entamoeba: applications and implications. Curr Opin Microbiol. 2014Aug;20:118-24
15. Vicente JB, Ehrenkaufer GM, Saraiva LM, Teixeira M, Singh U. Entamoeba histolytica modulates a complex repertoire of novel genes in response to oxidative and nitrosative stresses: implications for amebic pathogenesis. Cell Microbiol. 2009Jan;11(1):51-69
16. Anderson IJ, Loftus BJ. Entamoeba histolytica: Observations on metabolism based on the genome sequence. Exp Parasitol. 2005;110(3):173-7
17. Gilchrist CA, Houpt E, Trapaidze N, Fei Z, Crasta O, Asgharpour A. et al. Impact of intestinal colonization and invasion on the Entamoeba histolytica transcriptome. Mol Biochem Parasitol. 2006Jun;147(2):163-76
18. Thibeaux R, Weber C, Hon C-C, Dillies M-A, Avé P, Coppée J-Y. et al. Identification of the virulence landscape essential for Entamoeba histolytica invasion of the human colon. PLoS Pathog. 2013;9(12):e1003824
19. Davis PH, Schulze J, Stanley SL. Transcriptomic comparison of two Entamoeba histolytica strains with defined virulence phenotypes identifies new virulence factor candidates and key differences in the expression patterns of cysteine proteases, lectin light chains, and calmodulin. Mol Biochem Parasitol. 2007Jan;151(1):118-28
20. Acanthamoeba castellanii (ID 278) - Genome - NCBI.
21. Entamoeba histolytica (ID 27) - Genome - NCBI.
22. Eichinger L, Noegel AA. Comparative genomics of Dictyostelium discoideum and Entamoeba histolytica. Curr Opin Microbiol. 2005Oct;8(5):606-11
23. Song J, Xu Q, Olsen R, Loomis WF, Shaulsky G, Kuspa A. et al. Comparing the Dictyostelium and Entamoeba Genomes Reveals an Ancient Split in the Conosa Lineage. PLoS Comput Biol. 2005;1(7):e71
24. Watkins RF, Gray MW. Sampling Gene Diversity Across the Supergroup Amoebozoa: Large EST Data Sets from Acanthamoeba castellanii, Hartmannella vermiformis, Physarum polycephalum, Hyperamoeba dachnaya and Hyperamoeba sp. Protist. 2008Apr;159(2):269-81
25. Jarmuszkiewicz W, Wagner AM, Wagner MJ, Hryniewiecka L. Immunological identification of the alternative oxidase of Acanthamoeba castellanii mitochondria. FEBS Lett. 1997;411(1):110-4
26. Diamond LS, Harlow DR, Cunnick CC. A new medium for the axenic cultivation of Entamoeba histolytica and other Entamoeba. Trans R Soc Trop Med Hyg. 1978;72(4):431-2
27. Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data.
28. Aho A V, Aho A V, Kernighan BW, Weinberger PJ. Awk - A Pattern Scanning and Processing Language (Second Edition). 1978.
29. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013Apr25;14(4):R36
30. Home - Genome - NCBI.
31. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010May2;28(5):511-5
32. Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22
33. Remm M, Storm CEV, Sonnhammer ELL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314(5):1041-52
34. Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. Vol. 428, Journal of Molecular Biology. 2016
35. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997Sep1;25(17):3389-402
36. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002Jul15;30(14):3059-66
37. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014May1;30(9):1312-3
38. Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation.
39. Draw Freely | Inkscape.
40. Levengood-Freyermuth SK, Click EM, Webster RE. Role of the carboxyl-terminal domain of TolA in protein import and integrity of the outer membrane. J Bacteriol. 1993Jan;175(1):222-8
41. Tang BL. Rab32/38 and the xenophagic restriction of intracellular bacteria replication. Microbes Infect. 2016Oct;18(10):595-603
42. Ruiz-Perez F, Nataro JP. Bacterial serine proteases secreted by the autotransporter pathway: classification, specificity, and role in virulence. Cell Mol Life Sci. 2014Mar;71(5):745-70
43. Weber C, Koutero M, Dillies M-A, Varet H, Lopez-Camarillo C, Coppée JY. et al. Extensive transcriptome analysis correlates the plasticity of Entamoeba histolytica pathogenesis to rapid phenotype changes depending on the environment. Sci Rep. 2016Oct21;6:35852
44. Sun J, Jiang H, Flores R, Wen J. Gene duplication in the genome of parasitic Giardia lamblia. BMC Evol Biol. 2010Feb17;10:49
45. DeBarry JD, Kissinger JC, Kissinger J, Rocchi M, Eichler E. A Survey of Innovation through Duplication in the Reduced Genomes of Twelve Parasites. Brayton KA, editor. PLoS One. 2014Jun11;9(6):e99213
46. Lorenzi HA, Puiu D, Miller JR, Brinkac LM, Amedeo P, Hall N. et al. New Assembly, Reannotation and Analysis of the Entamoeba histolytica Genome Reveal New Genomic Features and Protein Content Information. Carlton JM, editor. PLoS Negl Trop Dis. 2010Jun15;4(6):e716
47. Lahr DJG, Nguyen TB, Barbero E, Katz LA. Evolution of the Actin Gene Family in Testate Lobose Amoebae (Arcellinida) is Characterized by Two Distinct Clades of Paralogs and Recent Independent Expansions. Mol Biol Evol. 2011Jan1;28(1):223-36
48. Angelini S, Deitermann S, Koch H-G. FtsY, the bacterial signal-recognition particle receptor, interacts functionally and physically with the SecYEG translocon. EMBO Rep. 2005May;6(5):476-81
49. Carroll AJ, Heazlewood JL, Ito J, Millar AH. Analysis of the Arabidopsis Cytosolic Ribosome Proteome Provides Detailed Insights into Its Components and Their Post-translational Modification. Mol Cell Proteomics. 2007Oct13;7(2):347-69
50. Komili S, Farny NG, Roth FP, Silver PA. Functional specificity among ribosomal proteins regulates gene expression. Cell. 2007Nov2;131(3):557-71
51. Chaliotis A, Vlastaridis P, Mossialos D, Ibba M, Becker HD, Stathopoulos C. et al. The complex evolutionary history of aminoacyl-tRNA synthetases. Nucleic Acids Res. 2017Feb17;45(3):1059-68
52. Ribas de Pouplana L, Schimmel P. Aminoacyl-tRNA synthetases: potential markers of genetic code development. Trends Biochem Sci. 2001Oct;26(10):591-6
53. Rubio MÁ, Napolitano M, Ochoa de Alda JAG, Santamaría-Gómez J, Patterson CJ, Foster AW. et al. Trans-oligomerization of duplicated aminoacyl-tRNA synthetases maintains genetic code fidelity under stress. Nucleic Acids Res. 2015Nov16;43(20):9905-17
54. Lasonder E, Ishihama Y, Andersen JS, Vermunt AMW, Pain A, Sauerwein RW. et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002Oct3;419(6906):537-42
55. Hernández G, Jagus R. Evolution of the protein synthesis machinery and its regulation. 2016th ed. Springer International Publishing. 2016:564 p
56. Das K, Ganguly S. Evolutionary genomics and population structure of Entamoeba histolytica. Comput Struct Biotechnol J. 2014;12(20-21):26-33
57. Nickel R, Jacobs T, Urban B, Scholze H, Bruhn H, Leippe M. Two novel calcium-binding proteins from cytoplasmic granules of the protozoan parasite Entamoeba histolytica. FEBS Lett. 2000;486(2):112-6
58. Frederick JR, Petri WA. Roles for the galactose-/N-acetylgalactosamine-binding lectin of Entamoeba in parasite virulence and differentiation. Glycobiology. 2005Dec;15(12):53R-59R
59. Ramakrishnan S, Serricchio M, Striepen B, Bütikofer P. Lipid synthesis in protozoan parasites: A comparison between kinetoplastids and apicomplexans. Prog Lipid Res. 2013Oct;52(4):488-512
60. Munford RS, Sheppard PO, O'Hara PJ. Saposin-like proteins (SAPLIP) carry out diverse functions on a common backbone structure. J Lipid Res. 1995Aug;36(8):1653-63
61. Michalek M, Sönnichsen FD, Wechselberger R, Dingley AJ, Hung C-W, Kopp A. et al. Structure and function of a unique pore-forming protein from a pathogenic acanthamoeba. Nat Chem Biol. 2013Jan11;9(1):37-42
62. Tillack M, Biller L, Irmer H, Freitas M, Gomes MA, Tannich E. et al. The Entamoeba histolytica genome: primary structure and expression of proteolytic enzymes. BMC Genomics. 2007Jun14;8:170
63. Teixeira JE, Sateriale A, Bessoff KE, Huston CD. Control of Entamoeba histolytica adherence involves metallosurface protease 1, an M8 family surface metalloprotease with homology to leishmanolysin. Infect Immun. 2012Jun;80(6):2165-76
64. Roncolato EC, Teixeira JE, Barbosa JE, Zambelli Ramalho LN, Huston CD. Immunization with the Entamoeba histolytica Surface Metalloprotease EhMSP-1 Protects Hamsters from Amebic Liver Abscess. Appleton JA, editor. Infect Immun. 2015Feb;83(2):713-20
65. Boettner DR, Huston CD, Linford AS, Buss SN, Houpt E, Sherman NE. et al. Entamoeba histolytica Phagocytosis of Human Erythrocytes Involves PATMK, a Member of the Transmembrane Kinase Family. PLoS Pathog. 2008Jan;4(1):e8
66. Šarić M, Vahrmann A, Bruchhaus I, Bakker-Grunwald T, Scholze H. The second cysteine protease inhibitor, EhICP2, has a different localization in trophozoites of Entamoeba histolytica than EhICP1. Parasitol Res. 2006Dec27;100(1):171-4
67. Das A, Coulter ED, Kurtz DM, Ljungdahl LG. Five-gene cluster in Clostridium thermoaceticum consisting of two divergent operons encoding rubredoxin oxidoreductase- rubredoxin and rubrerythrin-type A flavoprotein- high-molecular-weight rubredoxin. J Bacteriol. 2001Mar;183(5):1560-7
68. Smutná T, Pilarová K, Tarábek J, Tachezy J, Hrdý I. Novel functions of an iron-sulfur flavoprotein from Trichomonas vaginalis hydrogenosomes. Antimicrob Agents Chemother. 2014Jun;58(6):3224-32
69. Vicik R, Busemann M, Baumann K, Schirmeister T. Inhibitors of cysteine proteases. Curr Top Med Chem. 2006;6(4):331-53
70. Huddleston JP, Burks EA, Whitman CP. Identification and characterization of new family members in the tautomerase superfamily: analysis and implications. Arch Biochem Biophys. 2014Dec15;564:189-96
71. Tekle YI, Grant J, Anderson OR, Nerad TA, Cole JC, Patterson DJ. et al. Phylogenetic placement of diverse amoebae inferred from multigene analyses and assessment of clade stability within “Amoebozoa” upon removal of varying rate classes of SSU-rDNA. Mol Phylogenet Evol. 2008Apr;47(1):339-52
Author contact
![]() Corresponding author: wojmakde
Corresponding author: wojmakde