Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(3):358-368. doi:10.7150/ijbs.23325 This issue Cite
Research Paper
Bmi1 Deficient Mice Exhibit Male Infertility
Xiuliang Dai1,2*, Qian Zhang2*, Zhenzhen Yu2, Weiwei Sun2, Rong Wang2, Dengshun Miao2 ![]()
1. Department of Reproductive Medicine Center, Affiliated Changzhou Maternity and Child Health Care Hospital, Nanjing Medical University, Changzhou, Jiangsu, China
2. State Key Laboratory of Reproductive Medicine, The Research Center for Bone and Stem Cells, Department of Anatomy, Histology and Embryology, Nanjing Medical University, Nanjing, Jiangsu, China
* These authors contributed equally to this work.
Received 2017-10-14; Accepted 2018-1-21; Published 2018-3-9
Abstract

Previous studies have demonstrated that the polycomb repressor Bmi1 is universally expressed in all types of testicular cells and might regulate the spermatogonia proliferation, however, it is unclear whether Bmi1 plays a critical role in maintaining normal male fertility in vivo. To answer this question, we first confirmed that Bmi1 is universally expressed in all types of testicular cells and found that the gene relative expression levels of Bmi1 in testis were the highest relative to other organs. Then we investigated the role of Bmi1 in maintaining normal male fertility using Bmi1 knockout male mouse model. Our results demonstrated that Bmi1 deficiency resulted in totally male infertility with smaller testis, severe oligospermia and sperm malformation. Mechanistically, decreased serum testosterone levels with down-regulating 3βHSD and 17βHSD expression levels, reduced germ cell proliferation, increased germ cell apoptosis with up-regulating p16, p19, p53 and p21 expression levels, increased reactive oxygen species (ROS) and H2O2 levels with down-regulating gene expression levels of anti-oxidant enzymes, and increased 8-OHdG and γ.H2AX positive cells in testis were observed in Bmi1 deficient mice compared with wild-type mice. These results indicate that Bmi1 deficiency results in male infertility by reducing testosterone syntheses, increasing oxidative stress and DNA damage, activating p16 and p19 signaling pathway, inhibiting germ cell proliferation and inducing germ cell apoptosis and sperm malformation. Thus, Bmi1 may be a novel and potential target for the clinic treatment of male infertility.
Introduction
Human epidemiological studies showed that about 15% of all couples in the world who are in the age of child-bearing are suffering infertility [1], and male factor contributes half [2]. Clinically, the male sterility mainly refers to oligospermia, asthnospermia and sperm deformity [3]. Male fertility is maintained directly by continuous spermatogenesis which is a process from spermatogonia to final spermatozoa production [4]. Steroidogenesis conducted by testicular leydig cells is also required for normal spermatogenesis and male reproductive function [5]. Therefore, spermatogenesis is complexly regulated not only by spermatogenic cells but also by somatic cells, and a failure occurring in any part will result in male subfertility or infertility. Although great deals of researches have been done on male infertility, new genes that are crucial for spermatogenesis are still need to be found and investigated.
B lymphoma Mo-MLV insertion region 1 (Bmi1), a member of the polycomb family of transcriptional repressors, is first identified as an oncogene [6, 7]. However, later studies demonstrated that Bmi1 is also crucial for normal tissue function [8, 9]. Bmi1 knockout mice (Bmi1-/-) exhibit premature ageing phenotypes of brain and hematopoiesis [8, 9]. Our previous studies have demonstrated that Bmi1 protects mice from premature osteoporosis and kidney ageing [10,31]. Several studies have indicated a possible role of Bmi1 in male reproductive function. A previous study has demonstrated that Bmi1 was expressed in testis and the knockdown of Bmi1 decreased the proliferation of cultured spermaogonia [11]. Another study has demonstrated that Bmi1 is universally expressed in all types of testicular cells including spermatogonia, spermcytes, round sperms and sertoli cells [12]. However, it is unclear whether Bmi1 deficiency results in male infertility.
Studies focusing on molecular mechanisms of Bmi1 revealed that p16 and p19 proteins and reactive oxygen species (ROS) are the key mediators [13, 14]. Bmi1 can transcriptionally inhibit the expressions of p16 and p19 which are well-known cell cycle dependent kinase inhibitors (CDKIs), thus promoting cell cycle progression [15]. Additionally, p19 can stabilize p53 via competently binding with Mdm2 [16]. Bmi1 also plays a crucial role in balancing cellular oxidative stress. It has been reported that Bmi1 deficiency resulted in ROS accumulation and tissue oxidative damage including DNA damage. Several studies have indicated that the elevated expressions of p16 and p53 in testis resulted in decreased proliferation of spermatogonia and increased germ cell apoptosis respectively, thereby impairing spermatogenesis [17-19]. Moreover, oxidative stress is thought as a common cause for male infertility [20, 21]. However, it is unclear whether Bmi1 deficiency induced-defective spermatogenesis is associated with up-regulated expressions of p16, p53 or enhanced oxidative stress.
In this study, we investigated the role of Bmi1 in maintaining normal male fertility by using Bmi1 knockout male mouse model. Reproductive phenotypes were analyzed and compared between wild type (WT) and Bmi1-/- male mice using histopathological, cellular and molecular techniques, including alterations of testis size and weight, tissue structures of testes and epididymis, sperm number and morphology, proliferation and apoptosis of spermatogenic cells, oxidative stress and DNA damage in testis and the expression of cell cycle and apoptosis regulating proteins. This study will elucidate mechanisms of role of Bmi1 in maintaining male reproductive function, thus will provide a novel and potential target for the clinic treatment of male infertility.
Materials and Methods
Breeding, genotyping and treating mice
All the required animals in this study were bred and maintained in an SPF laboratory animal center in Nanjing Medical University. Bmi1 heterozygous null (Bmi1+/-) mice were kindly provided by professor Anton Berns. Adult Bmi1+/- null male mice were crossed with adult Bmi1+/- null female mice to obtain Bmi1 homozygous (Bmi1-/-) mice. The mice genotyping results were presented in Figure S1. All the animal experiments were strictly in compliance with, and approved by the Institutional Animal Care and Use committee of Nanjing Medical University.
Computer assisted sperm analysis (CASA), H&E and Mitotracker staining
The caudal of mouse epididymis was cut into several pieces and immersed in PBS at 37℃ for 15 min. For CASA, 10μL supernatant was used to detect sperm motility by CASA system. Then the PBS containing caudal tissue was filtered by using 200 meshes gauze to purify the sperms. After centrifugation (2000r/min, 5 min), the sperms were re-suspended in 200μL PBS. 10μL sperm was then smeared on slides. After dry and fixation in 4% formaldehyde solution in PBS, sperm slides were respectively stained with haematoxylin and eosin solution and Mitotracker Green (Invitrogen, USA)counterstaining with DAPI..
Electron microscopy analysis
The epididymis caudal of mice were removed and fixed in glutaraldehyde. After dehydration and embedding in epoxy resin (CEM Corporation, Japan), 70 nm sections were made and loaded on copper screen. Followed by staining with uranium acetate and lead citrate, the images were collected by Jeol 1200EX (Tokyo, Japan).
Serum testosterone assay
Mice were anaesthetized by 2% chloral hydrate at a dose of 20 μl/g and blood was harvested from heart. Followed by placing at room temperature (RT) for 2 hours, the blood was centrifuged at 3000r/min for 15 min and the supernatant namely serum was collected. Testosterone levels were determined by testosterone assay ELISA kit (H090, Nanjing Jiancheng Bioengineering Institute, China) according to the instructions of manufacture.
Histology
The testis and epididymis were removed from mice and fixed in 4% formaldehyde solution in PBS. After dehydration, permeabilization, wax-dip and embedding, 5μM sections were cut. After dewaxing and hydration, sections were stained with haematoxylin and eosin solution. The micrographs were taken.
Immunohistochemistry
Paraffin embedded testicular sections were dewaxed and re-hydrated. Sections were immersed in solution of methanol:hydrogen peroxide (1:10) to block endogenous peroxidase activity. After antigen retrieving with boiling PBS, the sections were blocked with 10% goat serum at RT for 1 hour. And then, the primary antibodies were added on the sections and stored at 4℃ overnight. The primary antibodies we used in this study were including Ki67 (Abcam, USA), p16 (Santa Cruz Biotechnology, USA), p-p53 (Cell Signaling Technology,USA), 8-OHdG (Novus, USA) and γ.H2AX (Cell Signaling Technology, USA). The slides were rinsed in PBS and incubated with the biotin-conjugated secondary antibodies including goat anti-rabbit or goat anti-mouse (KPL, USA) at RT for 1hour. After being washed with PBS, the sections were incubated with Elite ABC (Vector, USA) at RT for 1 hour. And then, the sections were visualized by 3,3-diaminobenzidine (DAB) (Vector, USA) and counter-stained with Hematoxylin. The micrographs were taken.
TUNEL assay
Testicular sections were dewaxed and rehydrated and TUNEL assay kit (Roche, Switzerland) was used to detect cell apoptosis. We performed this experiment strictly following the instructions of manufacture.
Western blot
The fresh or frozen testis was homogenized in Radio-Immunoprecipitation Assay (RIPA) lysis buffer containing 10% phenylmethanesulfonyl fluoride (PMSF). After 1 hour for ice-bath, the extracts were centrifuged at 13000r/min for 15 min. The supernatant was acquired and mixed with loading buffer. And then, proteins was put in boiling water for 5 min and stored at -80℃ for late use. The protein samples were subjected to electrophoresis. After this, the separated proteins were blotted in PVDF membrane by transmembrane. After being washed in PBS, the membranes were soaked in 5% milk/PBST to block none specific proteins. And then the membranes were incubated with corresponding primary antibodies including p16 (Santa Cruz Biotechnology, USA), p19 (Santa Cruz Biotechnology, USA), p53 (Abcam,USA), p21 (Santa Cruz Biotechnology, USA), 3βHSD (Santa Cruz Biotechnology, USA) ,17βHSD (Santa Cruz Biotechnology, USA) and β-actin (Abcam, USA) at 4℃ overnight. Followed by being washed in PBST, membranes were incubated with HRP-conjugated goat-anti rabbit, goat anti-mouse or rabbit anti-goat antibodies (KPL, USA) at RT for 1hour. Finally the target proteins bands were visualized by ECL solution (Nanjing Jiancheng bioengineering Institute, China) and quantitated by Scion Image Beta 4.02 (Scion Corporation, NIH).
Real-time RT-PCR
The fresh testes were homogenized in Trlzol solution (Sigma, Japan), mRNA was extracted by trichloromethane and isopropanol. For reverse transcription, mRNA was used as template for single strand DNA synthesis. The commercial RT-kit (Vazyme, China) was used here. For real-time PCR, the single strand DNA we synthesized above was used as template with specific primers for different genes that we detected in this study. Primers sequences were list in Table S1. Commertial kit (Vazyme, China) was used for detecting mRNA expression.
Biochemistry assay
Fresh testes were homogenized in cold salt and centrifuged at 4000r/min for 10min. Supernatant was obtained for biochemical assays. The H2O2 level and total antioxidant capacity were determined by using the commercial kits (Nanjing Jiancheng Bioengineering Institute, China).
Flow cytometry analysis
Fresh testes were collected and placed in 4℃PBS containing 10% BSA. Followed by removing the tunica albuginea, testis was squeezed gently through a 200 meshes gauze to acquire the single cell suspension. For ROS detection, cells were incubated with 5mM diacetyldichlorofluorescein (DCFDA) (Sigma, Japan) at 37℃ for 30min. Cells were then centrifuged at 2000r/min for 5min and re-suspended in PBS. This was repeated 3 times to remove the DCFDA. Finally, samples were analyzed by FACScalibur flow cytometer (Becton Dickinson, Heidelberg, Germany).
For 3βHSD positive cells detection, testicular cells were fixed in 2% formaldehyde for 15min., Cells were then mixed with 0.2% Triton X-100 for 30min to penetrate membranes. Then, primary antibody 3βHSD (Santa Cruz Biotechnology, USA) was added in cells at RT for 2 hours. Followed by rinsing the cells, the secondary antibody Alexa 594-conjugated donkey-anti goat antibodies (Invitrogen, USA) were added into cells at RT for 1 hour in dark. Then, cells were rinsed by PBS and analyzed by FACS calibur flow cytometer (Becton Dickinson, Heidelberg, Germany).
Statistical analysis
All data were analyzed by using SPSS software (Version 16.0, SPSS Inc., Chicago, IL, USA) and presented as mean±SEM. For comparison between two groups, Student's t test was used to analyze the difference. P<0.05 was considered statistically significant.
Results
The expression and localization of Bmi1 in testis
To determine whether Bmi1 exhibits a relative high expression in testis, the Bmi1 mRNA levels were examined by real-time RT-PCR in multiple organs from WT mice, including heart, liver, spleen, kidney, parotid gland, skin, brain, testis and epididymis. Results showed that the expression levels of Bmi1 in testis were the highest when compared with other organs (Fig.1A). In addition, the mRNA levels of Bmi1 in epididymis were also abundant.
Bmi1 expression and localization in testes. (A) Real-time RT-PCR analysis of multiple organs extracts from 7-week old male mice for Bmi1 expression. Messenger RNA expression assessed by real-time RT-PCR is calculated as a ratio relative to GAPDH, and expressed relative to liver. Representative micrograph of testicular sections from 7-week old WT mice for (B) Bmi1 and PLZF double immunofluoresent staining and (C) Bmi1 immunohistochemical staining. Magnification is ×400 in B and C. Stars: spermatogonia. Arrows: sertoli cells.
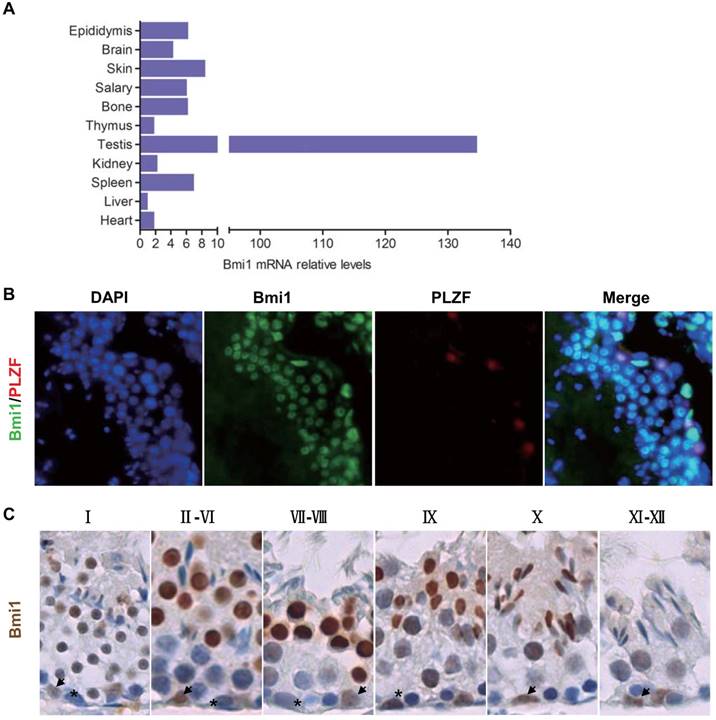
To confirm whether Bmi1 was localized in spermatogonial stem cells (SSC), we performed immunofluorescence on testicular sections of WT male mice by using Bmi1 antibody and Plzf antibody (a maker of SSC). Results showed that Bmi1 was not only expressed in SSC but also expressed in near all types of testicular cells, including spermatocytes, sperms and sertoli cells (Fig.1B). Immunohistochemistry for Bmi1 not only verified that Bmi1 was localized universally in all types of testicular cells, but also showed that Bmi1 was higher expressed in II-X stage unmatured spermatids (Fig.1C).
Bmi1 deficiency induces male infertility with severe oligospermia in mice
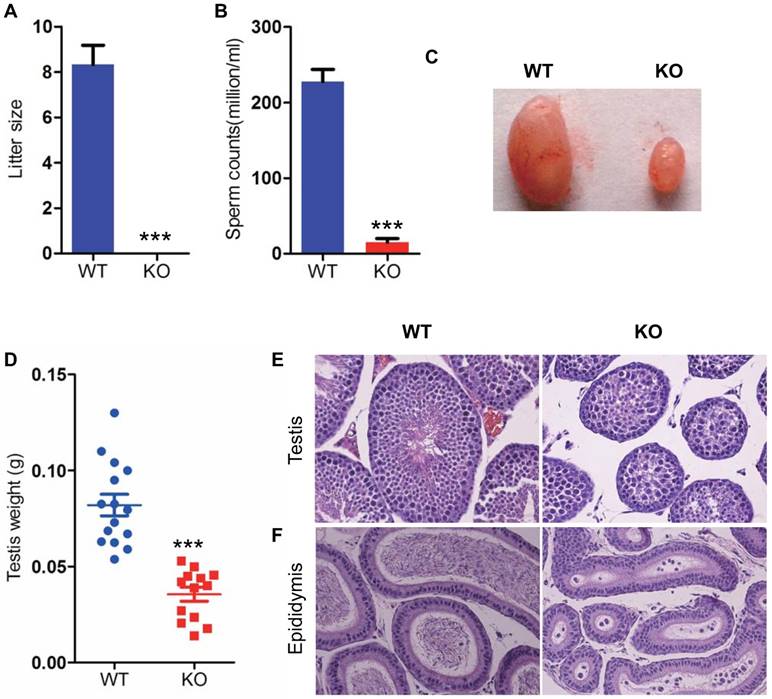
To determine whether Bmi1 was required for maintaining male fertility, we crossed WT or Bmi1-/- male mice with adult WT female mice for 1 month. The average litter size of WT male mice is 8, however, Bmi1-/- male mice were totally infertile (Fig. 2 A). To assess whether male infertility induced by Bmi1 deficiency was associated with the dysfunction of spermatogenesis, we compared testis size, testis weight, sperm counts and seminaferous epithelium between WT and Bmi1-/- male mice. Results showed that testis size, weight and sperm production were reduced significantly in Bmi1-/- mice compared with WT mice (Fig. 2B-D). More importantly, Bmi1 deficiency resulted in a highly disorganization of seminaferous epithelium including reduced germ cell counts, tubule diameters and sperms (Fig. 2E). In addition, round immature germ cell like cells were observed in epididymis of Bmi1-/- male mice (Fig. 2F).
Bmi1 deficiency induces male infertility with severe oligospermia in mice. (A) Litter size. (B) CASA analysis of sperm counts in caudal of epididymis from 7-week old male mice. (C) Testis size and (D) weight from 7-week-old mice. Representative micrographs of H&E staining for (E) testicular sections, (F) epididymis sections from 7-week-old mice. Magnification is ×400 in E and F. Each value is the mean ± SEM of determinations in 13-15 mice of each group. * * *: P < 0.001 compared with WT mice.
Effect of Bmi1 deficiency on leydig cells and testosterone synthesis
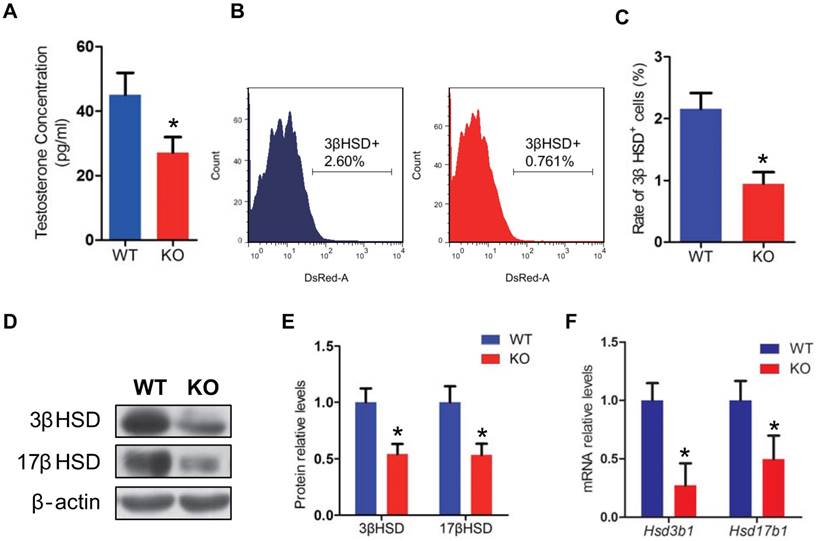
To investigate whether the male infertility caused by Bmi1 deficiency was associated with abnormal testosterone production, we assessed the testosterone levels in serum from WT and Bmi1-/- male mice. Testosterone levels were reduced significantly in Bmi1-/- mice compared with WT mice (Fig. 3A). In consistent with this, leydig cells population (3βHSD positive cells) was significantly lower in testis of Bmi1-/- male mice than WT male mice (Fig. 3B, C). Furthermore, both mRNA and protein levels of 3βHSD and 17βHSD were down-regulated significantly in Bmi1-/- male mice (Fig. 3D-F).
Effect of Bmi1 deficiency on leydig cells and testosterone synthesis. (A) Serum testosterone levels from 7-week-old mice. (B) Representative graphs of flow cytometry analysis of testicular germ cells stained for 3βHSD. (C) Quantitative percentage of 3βHSD positive cells relative to total germ cells from 7-week-old mice. (D) Western blot and (F) Real-time RT-PCR analysis of testis extract from 7-week-old mice for expressions of Hsd3b1 and Hsd17b1. Messenger RNA expression determined by real-time RT-PCR is calculated as a ratio relative to GAPDH, and expressed relative to WT mice. (E) 3βHSD and 17βHSD protein levels were assessed by densitometry analysis calculated as a ratio relative to β-actin protein levels and expressed relative to levels of WT mice. Each value is the mean ± SEM of determinations in 5 mice of each group. *: P < 0.05compared with WT mice.
Effect of Bmi1 deficiency on germ cell proliferation, apoptosis and sperm morphology
To determine whether the severe oligospermia caused by Bmi1 deficiency was associated with alterations of the germ cell proliferation and apoptosis, we performed Ki67 immunostaining and TUNEL assay in testicular sections from WT and Bmi1-/- mice. Results showed that Ki67 positive cells were reduced dramatically, whereas TUNEL positive cells were increased obviously in Bmi1-/- mice compared with WT mice (Fig. 4A-D).
Effect of Bmi1 deficiency on germ cell proliferation, apoptosis and sperm morphology. Representative micrographs of testicular sections from 7-week-old mice stained (A) immunohistochemically for Ki67, (C) for TUNEL. The numbers of (B) Ki67 and (D) TUNEL positive cells per tube. (E) Representative micrograph of sperms from 7-week-old Bmi1-/- mice stained for H&E (a: normal head b: irregular head, c & d: big head) (F) Evaluated abnormality rate of sperm from 7-week-old mice. (G) Representative micrographs of ultrastructure of sperms photoed by Electron microscopy. (H) Real-time RT-PCR analysis of testis extracts from 7-week-old mice for Tnp1, Tnp2, Prm1, Prm2, H1fnt and Spem1 expressions. Messenger RNA expression determined by real-time RT-PCR is calculated as a ratio relative to GAPDH, and expressed relative to WT mice. (I) Representative immunofluorescent micrographs of sperms stained with Mito-tracker. Magnification is ×400 in A, C, E and I, and is ×15000 in G. Each value is the mean ± SEM of determinations in 5 mice of each group. *: P < 0.05; **: P < 0.01; * * *: P < 0.001 compared with WT mice.
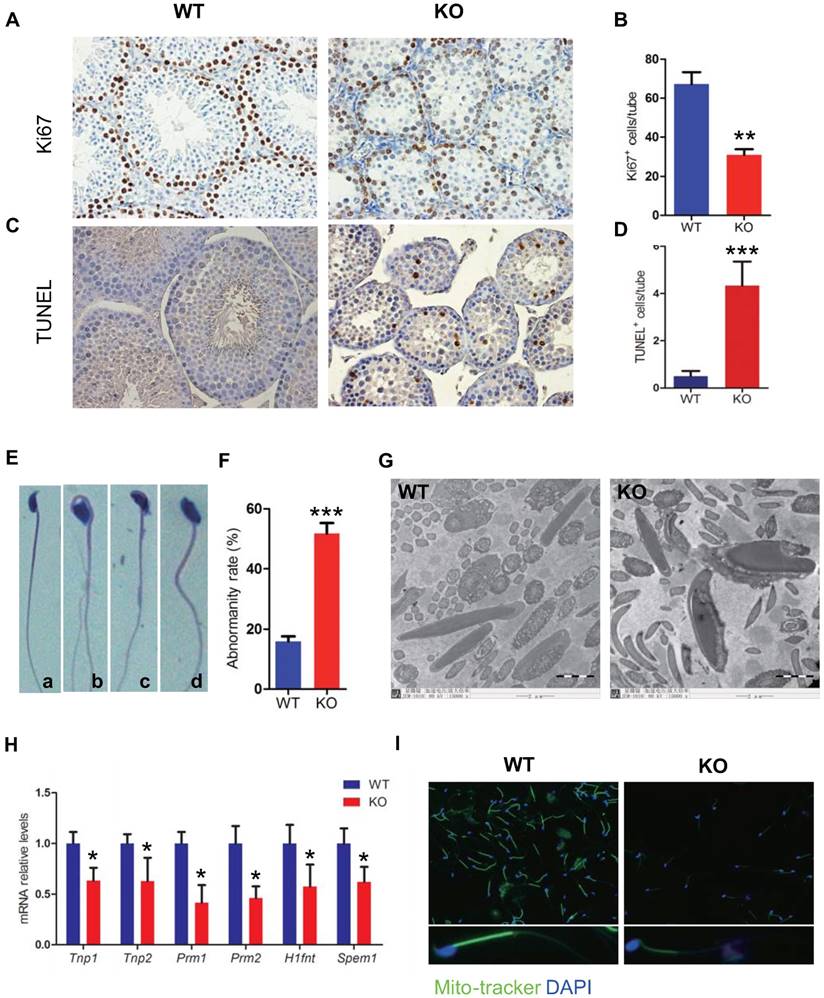
To assess effect of Bmi1 deficiency on sperm transition, the sperm morphology was examined. Numerous sperm head defects were observed in Bmi1-/- mice, including irregular head, and big head (Fig. 4E). The abnormality rate of sperms with defective sperm nuclear condensation and acrosome attachment was more than 50% in Bmi1-/- mice (Fig. 4F, G). A panel of genes including Tnp1, Tnp2, Prm1, Prm2, Hifnt and Spem1, which were reported necessarily for sperm shaping, were significantly down regulated in Bmi1-/- mice (Fig. 4H). In addition, mitochondrial contents in mitochondrial sheath were reduced significantly in Bmi1 null sperms compared with WT sperms, demonstrated by mitotracker staining (a kind of mitochondria dye) (Fig. 4I).
Effect of Bmi1 deficiency on p16 /p19 signaling pathway in testis
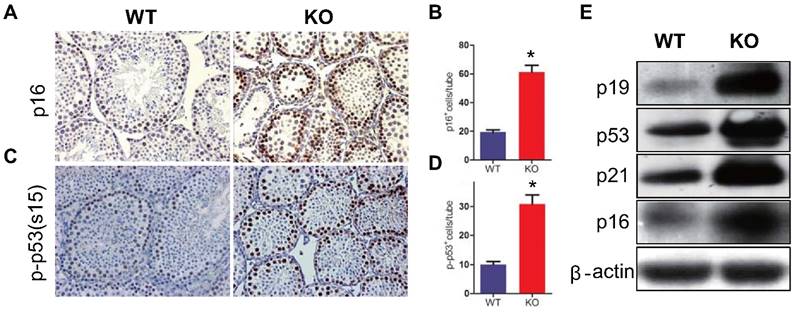
To determine whether the reduction of germ cell proliferation and induction of germ cell apoptosis caused by Bmi1 deficiency were associated with activation of p16 and p19 signaling pathways in testis, the expression levels of p16 or p19 pathway associated molecules were examined. Both p16 and p53 immunopositive cells in testis were increased significantly and the protein expression levels of p16, p19, p53 and p21were up-regulated dramatically in testis extracts from Bmi1-/- mice (Fig. 5A-I).
Effect of Bmi1 deficiency on p16 /p19 signaling pathway in testes. (A) Representative micrographs of testicular sections from 7-week-old mice stained immunohistochemically for (A) p16 and (C) p-p53. Magnification is ×400 in A and C. Quantitative numbers of (B) p16 and (D) p-p53 positive cells per tube. (E) Western blot analysis of testis extracts from 7-week-old mice for expressions of p19, p53, p21 and p16. Each value is the mean ± SEM of determinations in 5 mice of each group. *: P < 0.05compared with WT mice.
Effect of Bmi1 deficiency on oxidative stress status and DNA damage in testis
To determine whether Bmi1 deficiency resulted in oxidative stress in testis, the levels of ROS and H2O2 were examined in testis. ROS and H2O2 levels in testes were increased significantly in Bmi1-/- mice compared with WT mice (Fig. 6A-C). To further investigate whether Bmi1 deficiency induced-oxidative stress in testes is associated with declined antioxidant capacity, the total antioxidant capacity and a panel of genes that encode proteins against oxidative stress were examined in testis tissues. Results showed that the total antioxidant capacity and the expression levels of antioxidant enzyme genes including Gpx1, Gsr, Cat, Txnrd1 were decreased in Bmi1-/- mice (Fig. 6D, E).
Effect of Bmi1 deficiency on oxidative stress status and DNA damage in testes. (A) Representative graphs of flow cytometry analysis of testicular germ cells from 7-week-old mice stained for DCFDA. (B) ROS levels were assessed by mean fluorescence intensity analysis and presented relative to levels of WT mice. Biochemistry analysis of testis extracts from 7-week-old mice for (C) H2O2 levels and (D) total antioxidant capacity. (E) Real-time RT-PCR analysis of testis extracts from 7-week-old mice for expressions of Gpx1, Gpx4, Gsr, Cat and Txnrd1. Messenger RNA expression determined by real-time RT-PCR is calculated as a ratio relative to GAPDH, and expressed relative to WT mice. (F) Representative micrographs of testicular sections from 7-week-old mice stained immunohistochemically for (F) 8-OHdG and (H) γ.H2AX. Magnification is ×400 in F and H. Quantitative numbers of (G) p16 and (I) γ.H2AX positive cells per tube. Each value is the mean ± SEM of determinations in 5 mice of each group. *: P < 0.05; * * *: P < 0.001 compared with WT mice.
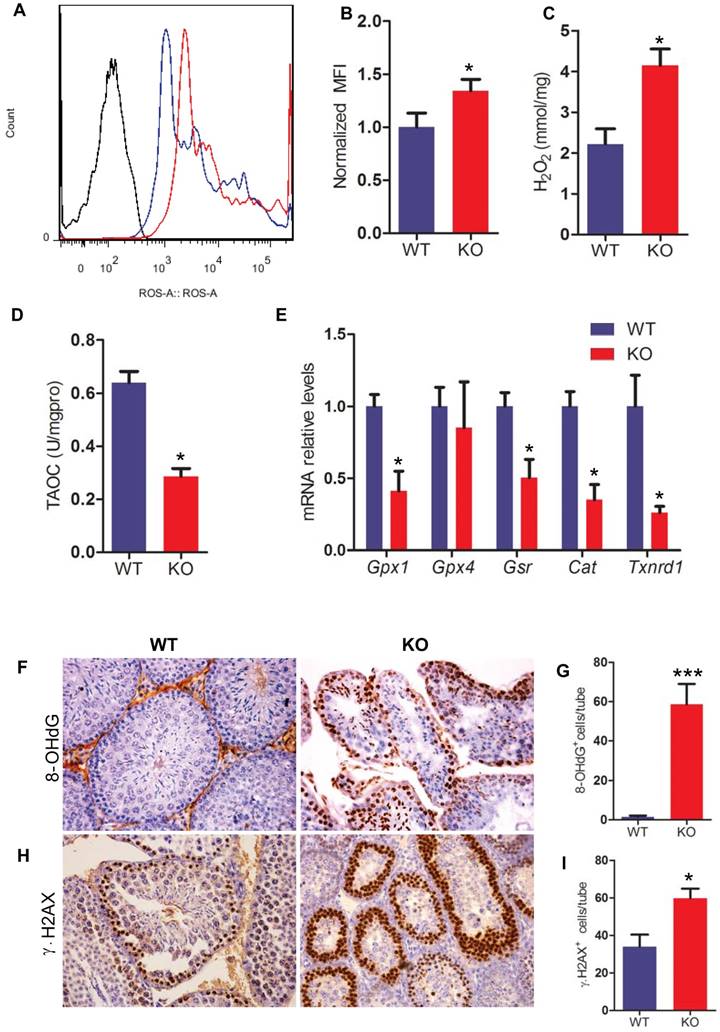
To evaluate whether increased oxidative stress in testes resulted in germ cell DNA damage, we performed immunohistochemistry on testicular sections by using 8-OHdG (a maker of DNA oxidation) and γ.H2AX (a maker of DNA damage response) antibodies. We found that the numbers of 8-OHdG positive cells and γ.H2AX positive cells in testis were increased dramatically in Bmi1-/- mice compared with WT mice (Fig. 6F-I).
Discussion
In this study, we first examined gene expression levels of Bmi1 in testis relative to other organs and its localization in testicular cells. Bmi1 localization in testicular cells reported from different laboratories was not consistent. One has reported that Bmi1 mainly localized in SSC [11], other demonstrated that Bmi1 has a universal expression in testis, such as sertoli cells, spermatocytes and sperms [12]. Our results confirmed that Bmi1 was universally expressed in all types of testicular cells including SSC, spermatocytes, sperms and sertoli cells. In addition, we found that the gene relative expression levels of Bmi1 in testis were the highest relative to other organs. Therefore, we speculated that Bmi1 might play a critical role in maintaining normal male fertility. Although a previous study has demonstrated that the knockdown of Bmi1 decreased the proliferation of cultured spermaogonia [11], however, it is unclear whether Bmi1 deficiency results in male infertility.
Next, we investigated the role of Bmi1 in maintaining normal male fertility using Bmi1 knockout male mouse model. Our results not only demonstrated that Bmi1 deficient male mice were absolute infertility with smaller testis, severe oligospermia and sperm malformation, but also revealed that Bmi1 deficiency results in male infertility by reducing testosterone syntheses, increasing oxidative stress and DNA damage, activating p16 and p19 signaling pathway, inhibiting germ cell proliferation and inducing germ cell apoptosis.
In view of the fact, common causes for male infertility include testosterone deficiency (hypogonadism) and sperm disorders, such as impaired sperm production or delivery, abnormal sperm development or shape, we examined serum testosterone levels and sperm number and shape. We found that Bmi1 deficiency resulted in hypogonadism and severe oligospermia and sperm malformation. Androgens are needed for puberty, male fertility and male sexual function. High levels of intratesticular testosterone, secreted by the leydig cells, are necessary for spermatogenesis [22].
Previous studies showed that the deletion of leydig cells in testis resulted in a failure of spermatogenesis [5, 23]. In current study, we found that Bmi1 deficiency not only dramatically reduced the number of leydig cells in testis, but also decreased testosterone production with down-regulated the expression levels of testosterone-synthesis enzymes 3βHSD and 17βHSD in testes. Androgens exert their action through androgen receptors and its signaling in the testis is essential for spermatogenesis. Androgen receptors signaling in spermatogenesis has been investigated in rodent models where testosterone levels are chemically suppressed or models with transgenic disruption of androgen receptors. These models have demonstrated the essential nature of androgens in the promotion of male fertility. Therefore, hypogonadism induced by Bmi1 deficiency partially contributed to male infertility.
The available evidence suggests that oxidative stress is central to the etiology of male infertility. High levels of oxidative stress result in damage to sperm DNA, RNA transcripts, and telomeres and, therefore might provide a common underlying etiology of male infertility [24]. Previous a study has demonstrated that cells derived from Bmi1 deficient mice had impaired mitochondrial function, a marked increase in the intracellular levels of reactive oxygen species and subsequent engagement of the DNA damage response pathway [14]. Thus, we examined alterations of parameters related oxidative stress and DNA damage in Bmi1 deficient testes and sperms. Our results demonstrated that ROS and H2O2 levels were significantly increased with down-regulating gene expression levels of anti-oxidant enzymes, and DNA damage and its response parameters, 8-OHdG and γ.H2AX positive cells in testes were dramatically increased in Bmi1 deficient mice. We also found that mitochondrial contents in mitochondrial sheath were reduced significantly in Bmi1 deficient sperms. Oxidative stress can damage the sperm nuclear and mitochondrial DNA, which is associated with shorter telomere length, formation of the oxidative base adduct 8-hydroxy-deoxyguanine (8-OHdG) and fragmentation of mitochondrial DNA or damage the sperm plasma membrane and thus affecting sperm motility [25]. The effects of 8-OHdG adducts in sperm DNA are more likely to lead to DNA damage owing to the limited capacity of spermatozoa to undergo DNA repair, leading to genome hypermutability, genetic instability and infertility. High levels of sperm DNA damage are found in infertile men and have a negative impact on male fertility potential [26]. Bmi1 not only plays a crucial role in balancing cellular oxidative stress, but also contributes to DNA damage repair. Loss of Bmi1 leads to impaired repair of DNA double-strand breaks by homologous recombination [27]. Thus, increased oxidative stress and DNA damage induced by Bmi1 deficiency play an important role in male infertility.
One critical target of Bmi1 is Ink4a/Arf, which encodes the cell cycle inhibitors p16ink4a and p19Arf [28]. Bmi1 regulates cell proliferation and apoptosis through the Ink4a/Arf locus, respectively [28, 29]. In this study, we examined alterations of p16/p19 signaling molecules and the proliferation and apoptosis of testicular cells. Our results demonstrated that expression levels of p16, p19, p53 and p21 in testes were up-regulated dramatically, the proliferation of testicular cells was reduced significantly and their apoptosis was increased markedly in Bmi1 deficient mice. Previous studies have reported a number of defects including severe neurological abnormalities, alterations in various haematopoietic cell lineages, a generalized failure-to-thrive and a markedly shortened lifespan in Bmi1 deficient mice [8, 15, 28, 30]. The importance of the transcriptional repression of the Ink4a/Arf locus is underscored by the observation that mice deficient in Bmi1 and also lacking p16, p19 or both of these gene products develop less severe neurological and hematological abnormalities than Bmi1 deficient mice [8, 28, 30]. Our previous studies have demonstrated that Bmi1 deficiency results in osteoporosis by up-regulating expression levels of p16 and p19, inhibiting the proliferation of bone marrow mesenchymal stem cells and inducing their apoptosis [10]. Recently, we also found that Bmi1 plays a critical role in protection from renal tubulointerstitial injury by maintaining redox balance, while p16 deletion ameliorated renal tubulointerstitial injury caused by Bmi1 Deficiency [31, 32]. Although there is no any report about effect of Bmi1 on male fertility, previous study has shown that the up-regulation of p16 is main responsible for germ cell cycle arresting in response to heat stimulation [33], whereas a high level of p53 resulted in germ cell apoptosis [17-19]. Results from current study indicate that Bmi1 deficiency can inhibit spermatogenic cell proliferation and induce their apoptosis by activating p16/p19 signaling pathways.
Spermiogenesis is a process of sperm transformation from round to long. Many events are involved in it, including acrosome formation, nucleus condensation and flagella formation [34]. Our results showed that high expression levels of Bmi1 were observed in immature sperms, while Bmi1 deficiency not only resulted in severe oligospermia, but also displayed sperm malformation, including big head, sharp head, irregular head. We also found that sperm nucleus in Bmi1 deficient mice were not well condensed and detached from acrosome. Sperm nucleus condensation is a very complicated process and regulated by many genes. Gene knockout model have identified a panel of necessary genes for nucleus condensation, including Tnp1, Tnp2, Prm1, Prm2, H1fnt, Spem1 [35-40]. In consistent with the nucleus condensation defects, the expression levels of nucleus condensation related genes in testes were all down-regulated in Bmi1deficient mice. These results indicate that Bmi1 also played an important role in spermatogenesis, although detail mechanism remains to be investigated.
Taken all together, we can draw a conclusion that Bmi1 deficiency results in male infertility by increasing oxidative stress and DNA damage, activating p16 and p19 signaling pathway and reducing testosterone syntheses, inhibiting germ cell proliferation and inducing germ cell apoptosis and sperm malformation. Thus, Bmi1 may be a novel and potential target for the clinic treatment of male infertility.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81471501 and 81230009) to D.M.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sciarra J. Infertility: an international health problem. Int J Gynaecol Obstet. 1994;46:155-63
2. Kumar N, Singh AK. Trends of male factor infertility, an important cause of infertility: A review of literature. J Hum Reprod Sci. 2015;8:191-6
3. Harris ID, Fronczak C, Roth L, Meacham RB. Fertility and the aging male. Rev Urol. 2011;13:e184-90
4. Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol. 2013;29:163-87
5. O'Hara L, Smith LB. Androgen receptor roles in spermatogenesis and infertility. Best Pract Res Clin Endocrinol Metab. 2015;29:595-605
6. Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753-63
7. van Lohuizen M, Frasch M, Wientjens E, Berns A. Sequence similarity between the mammalian bmi-1 proto-oncogene and the Drosophila regulatory genes Psc and Su(z)2. Nature. 1991;353:353-5
8. Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962-7
9. Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL. et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302-5
10. Zhang HW, Ding J, Jin JL, Guo J, Liu JN, Karaplis A. et al. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res. 2010;25:640-52
11. Zhang S, Li D, Li E, Li L, Wang J, Wang C. et al. Expression localization of Bmi1 in mice testis. Mol Cell Endocrinol. 2008;287:47-56
12. Gjerstorff MF, Relster MM, Greve KB, Moeller JB, Elias D, Lindgreen JN. et al. SSX2 is a novel DNA-binding protein that antagonizes polycomb group body formation and gene repression. Nucleic Acids Res. 2014;42:11433-46
13. Biehs B, Hu JK, Strauli NB, Sangiorgi E, Jung H, Heber RP. et al. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat Cell Biol. 2013;15:846-52
14. Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C. et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387-92
15. Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C. et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525-30
16. Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731-7
17. Wang H, Zhao R, Guo C, Jiang S, Yang J, Xu Y. et al. Knockout of BRD7 results in impaired spermatogenesis and male infertility. Sci Rep. 2016;6:21776
18. Fouchecourt S, Livera G, Messiaen S, Fumel B, Parent AS, Marine JC. et al. Apoptosis of Sertoli cells after conditional ablation of murine double minute 2 (Mdm2) gene is p53-dependent and results in male sterility. Cell Death Differ. 2016;23:521-30
19. Liang M, Wen J, Dong Q, Zhao LG, Shi BK. Testicular hypofunction caused by activating p53 expression induced by reactive oxygen species in varicocele rats. Andrologia. 2015;47:1175-82
20. Lavranos G, Balla M, Tzortzopoulou A, Syriou V, Angelopoulou R. Investigating ROS sources in male infertility: a common end for numerous pathways. Reprod Toxicol. 2012;34:298-307
21. Yu B, Lin H, Yang L, Chen K, Luo H, Liu J. et al. Genetic variation in the Nrf2 promoter associates with defective spermatogenesis in humans. J Mol Med (Berl). 2012;90:1333-42
22. Dohle GR, Smit M, Weber RF. Androgens and male fertility. World journal of urology. 2003;21:341-5
23. Beck-Peccoz P, Persani L, Lania A. Thyrotropin-Secreting Pituitary Adenomas. In: (ed.) De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM. et al. Endotext. South Dartmouth (MA). 2000
24. Bisht S, Faiq M, Tolahunase M, Dada R. Oxidative stress and male infertility. Nature reviews Urology. 2017;14:470-85
25. Bisht S, Dada R. Oxidative stress: Major executioner in disease pathology, role in sperm DNA damage and preventive strategies. Frontiers in bioscience. 2017;9:420-47
26. Schulte RT, Ohl DA, Sigman M, Smith GD. Sperm DNA damage in male infertility: etiologies, assays, and outcomes. Journal of assisted reproduction and genetics. 2010;27:3-12
27. Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M. et al. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Molecular and cellular biology. 2011;31:1972-82
28. Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164-8
29. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. The Journal of clinical investigation. 2004;113:175-9
30. Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E. et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes & development. 2005;19:1438-43
31. Jin J, Lv X, Chen L, Zhang W, Li J, Wang Q. et al. Bmi-1 plays a critical role in protection from renal tubulointerstitial injury by maintaining redox balance. Aging Cell. 2014;13:797-809
32. Jin J, Tao J, Gu X, Yu Z, Wang R, Zuo G. et al. P16 INK4a Deletion Ameliorated Renal Tubulointerstitial Injury in a Stress-induced Premature Senescence Model of Bmi-1 Deficiency. Scientific reports. 2017;7:7502
33. Xin-Chang Z, Peng W, Zhao-Yuan H, Xiao-Bin H, Ru-Jin Z, Yi-Xun L. Expression of P16(INK4a) in testis of rhesus monkey during heat stress and testosterone undecanoate induced azoospermia or oligozoospermia. Contraception. 2002;65:251-5
34. O'Donnell L. Mechanisms of spermiogenesis and spermiation and how they are disturbed. Spermatogenesis. 2014;4:e979623
35. Shirley CR, Hayashi S, Mounsey S, Yanagimachi R, Meistrich ML. Abnormalities and reduced reproductive potential of sperm from Tnp1- and Tnp2-null double mutant mice. Biol Reprod. 2004;71:1220-9
36. Fajardo MA, Haugen HS, Clegg CH, Braun RE. Separate elements in the 3' untranslated region of the mouse protamine 1 mRNA regulate translational repression and activation during murine spermatogenesis. Dev Biol. 1997;191:42-52
37. Yelick PC, Balhorn R, Johnson PA, Corzett M, Mazrimas JA, Kleene KC. et al. Mouse protamine 2 is synthesized as a precursor whereas mouse protamine 1 is not. Mol Cell Biol. 1987;7:2173-9
38. Lee K, Haugen HS, Clegg CH, Braun RE. Premature translation of protamine 1 mRNA causes precocious nuclear condensation and arrests spermatid differentiation in mice. Proc Natl Acad Sci U S A. 1995;92:12451-5
39. Martianov I, Brancorsini S, Catena R, Gansmuller A, Kotaja N, Parvinen M. et al. Polar nuclear localization of H1T2, a histone H1 variant, required for spermatid elongation and DNA condensation during spermiogenesis. Proc Natl Acad Sci U S A. 2005;102:2808-13
40. Zheng H, Stratton CJ, Morozumi K, Jin J, Yanagimachi R, Yan W. Lack of Spem1 causes aberrant cytoplasm removal, sperm deformation, and male infertility. Proc Natl Acad Sci U S A. 2007;104:6852-7
Author contact
![]() Corresponding author: Dr. Dengshun Miao, The State Key Laboratory of Reproductive Medicine, The Research Center for Bone and Stem Cells, Department of Anatomy, Histology and Embryology, Nanjing Medical University, Nanjing, Jiangsu, 210029, The People's Republic of China. Tel & FAX: 011-86-25-8686-9377; E-mail: dsmiaoedu.cn
Corresponding author: Dr. Dengshun Miao, The State Key Laboratory of Reproductive Medicine, The Research Center for Bone and Stem Cells, Department of Anatomy, Histology and Embryology, Nanjing Medical University, Nanjing, Jiangsu, 210029, The People's Republic of China. Tel & FAX: 011-86-25-8686-9377; E-mail: dsmiaoedu.cn