Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(4):381-389. doi:10.7150/ijbs.23517 This issue Cite
Research Paper
Expanding the Oro-Dental and Mutational Spectra of Kabuki Syndrome and Expression of KMT2D and KDM6A in Human Tooth Germs
Thantrira Porntaveetus1 ![]() , Mushriq F Abid2, Thanakorn Theerapanon3, Chalurmpon Srichomthong4,5, Atsushi Ohazama6, Katsushige Kawasaki6, Maiko Kawasaki6, Kanya Suphapeetiporn4,5, Paul T. Sharpe2, Vorasuk Shotelersuk4,5
, Mushriq F Abid2, Thanakorn Theerapanon3, Chalurmpon Srichomthong4,5, Atsushi Ohazama6, Katsushige Kawasaki6, Maiko Kawasaki6, Kanya Suphapeetiporn4,5, Paul T. Sharpe2, Vorasuk Shotelersuk4,5
1. Craniofacial Genetics and Stem Cells Research Group, Department of Physiology, Faculty of Dentistry, Chulalongkorn University, Bangkok 10330, Thailand
2. Centre for Craniofacial and Regenerative Biology, Dental Institute, King's College London, London, SE1 9RT, UK
3. Excellence Center in Regenerative Dentistry, Faculty of Dentistry, Chulalongkorn University, Bangkok, 10330, Thailand
4. Center of Excellence for Medical Genetics, Department of Pediatrics, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand
5. Excellence Center for Medical Genetics, King Chulalongkorn Memorial Hospital, the Thai Red Cross Society, Bangkok 10330, Thailand
6. Division of Oral Anatomy, Niigata University, Niigata 951-8514, Japan
Received 2017-10-27; Accepted 2018-2-26; Published 2018-3-9
Abstract

Kabuki syndrome is a rare genetic disorder characterized by distinct dysmorphic facial features, intellectual disability, and multiple developmental abnormalities. Despite more than 350 documented cases, the oro-dental spectrum associated with kabuki syndrome and expression of KMT2D (histone-lysine N-methyltransferase 2D) or KDM6A (lysine-specific demethylase 6A) genes in tooth development have not been well defined. Here, we report seven unrelated Thai patients with Kabuki syndrome having congenital absence of teeth, malocclusion, high-arched palate, micrognathia, and deviated tooth shape and size. Exome sequencing successfully identified that six patients were heterozygous for mutations in KMT2D, and one in KDM6A. Six were novel mutations, of which five were in KMT2D and one in KDM6A. They were truncating mutations including four frameshift deletions and two nonsense mutations. The predicted non-functional KMT2D and KDM6A proteins are expected to cause disease by haploinsufficiency. Our study expands oro-dental, medical, and mutational spectra associated with Kabuki syndrome. We also demonstrate for the first time that KMT2D and KDM6A are expressed in the dental epithelium of human tooth germs.
Keywords: Craniofacial anomalies, genetics, tooth development, hypodontia
Introduction
Kabuki syndrome or kabuki make-up syndrome (KS, MIM #147920) is a rare congenital dysmorphic syndrome that was originally described in 1981 [1, 2]. KS was initially considered to be discretely found in Japan, but later it has been recognized in other parts of the world. Estimated birth prevalence ranges from 1:32,000-86,000 [3, 4]. KS is clinically diagnosed by a peculiar face characterized by long palpebral fissures with eversion of the lateral third of the lower eyelids, arched eyebrows, long eyelashes, depressed nasal tip, and prominent ears. Patients also have postnatal growth retardation, intellectual disability, dermatoglyphic abnormalities with persisting fingerpads, and skeletal and dental anomalies.
Type 1 Kabuki syndrome (KS1, MIM #147920) is inherited in an autosomal dominant manner accounted for 55-80% of patients. KS1 is caused by mutations in KMT2D (histone-lysine N-methyltransferase 2D), also known as MLL2 (myeloid/ lymphoid or mixed-lineage leukemia 2; NM_003482.3) gene on chromosome 12q13. KS type 2 (KS2, MIM #300867), roughly found in 5% of patients, has been linked to mutations in the KDM6A (lysine(K)-specific demethylase 6A, NM_021140.3) located on chromosome Xp11.3 [5]. The underlying genetic mutations remain unknown in approximately 25% of patients [6]. Although the etiologic roles of KMT2D and KDM6A have been proposed in the function of several organs, evidence that these genes are involved in tooth development is very limited.
Oro-dental abnormalities were randomly reported in patients with KS1 and KS2. Common features include hypodontia, supernumerary teeth, cleft palate, small dental arch, and malocclusion [7-9]. Inconsistent oro-dental findings included microdontia, enamel hypoplasia [7], tooth gemination and fusion [10], and prominent upper incisors [11] were found in KS.
We previously reported clinical features but not detailed dental features with no molecular studies of Thai patients with KS [12]. Here, we characterize oro-dental manifestations and physical features of patients with novel KMT2D and KDM6A mutations. The mutations were identified by Whole Exome Sequencing (WES) and confirmed by Sanger sequencing. In addition, the expression of KMT2D and KDM6A during tooth development was demonstrated by in situ hybridization using human embryo sections.
Materials and Methods
Enrollment of Human Subjects
Participants in this study were from seven unrelated Thai families. Written informed consents were obtained from all participants. The study protocol was approved by the Human Ethical Review Board at the Faculty of Dentistry, Chulalongkorn University, Thailand. Physical and dental examinations were performed. Oro-dental characteristics were verified by panoramic radiographs.
DNA Isolation and Exome Sequencing
Genomic DNA was extracted from peripheral blood leukocytes and sent to Macrogen, Inc. (Seoul, Korea). Mutation analysis was performed as described previously [13]. Briefly, the DNA sample was prepared as an Illumina sequencing library enriched by SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA) and sequenced onto Hiseq 4000 (Illumina, San Diago, CA). The Burrows-Wheeler Aligner (BWA) was used to map raw data to the human reference genome version 19. GATK and HaplotypeCaller were used for variant calling. SNVs and Indels were annotated using SnpEff, dbpSNP 142, ClinVar, 1000 Genome, and ESP. The variants were filtered out using our in-house database of 700 unrelated Thai exomes. The novel variants were screened in the Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/index.php) and the Exome Aggregation Consortium database (exac.broadinstitute.org).
Polymerase Chain Reaction (PCR) and Sequencing
The most likely pathogenic variants were confirmed by PCR and Sanger sequencing. DNA of the parents was also Sanger sequenced to determine the presence of the mutations. Primers were designed for exons 7, 11, 22, 29, 39, and 51 of the KMT2D gene and exon 3 of the KDM6A gene (Table S1).
Human Embryos
Embryos were collected at different stages of gestation (approximately 8-12 weeks post-fertilization) from the Human Developmental Biology Resource Birth Defects Research Centre at the Institute of Child Health, University College London. Embryos were stored in phosphate buffered saline and delivered immediately following retrieval by courier. The ethical approval is held by Institute of Child Health (UCL) and King's College London has a subscription to obtain samples from this centre. Embryos were fixed in 4% paraformaldehyde (PFA) at 4°C and 10% EDTA (pH 7.4) was used to decalcify for 7.5-12 weeks (depending on the stage) at 4°C. They were embedded in paraffin wax after being dehydrated in a graded ethanol series, sectioned at 8 μm and mounted on slides.
In Situ Hybridization
Digoxigenin (DIG) - labeled in situ hybridization was performed as previously described [14]. The tooth sections were derived from two different samples at each developmental stage. Human cDNA clones for KDM6A (IMAGE ID, 1032414), KMT2D (IMAGE ID, 111472) were ordered from Source Bioscience. Images of the sections were photographed using a Zeiss microscope.
Results
Clinical Manifestations of Seven Thai Patients with Kabuki Syndrome
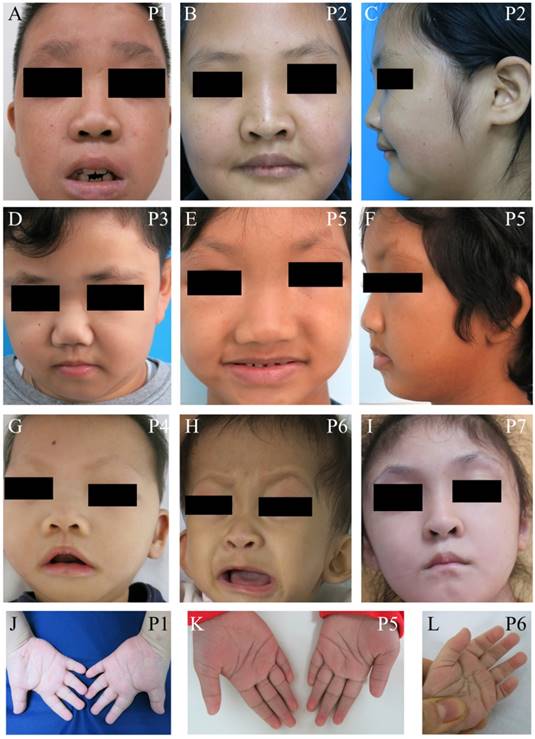
We identified seven unrelated Thai patients with a clinical diagnosis of KS. Five were female and two were male. The age of patients ranged from 2-15 years. They presented with typical facial dysmorphism, including long palpebral fissures with eversion of the lower lateral lids, arched eyebrows with the lateral one-third dispersed or sparse, depressed nasal tips, and prominent ears. Post-natal proportionate short stature, mild to moderate degree of intellectual disability, prominent finger fat pads, and fifth finger clinodactyly were common findings in our patients (Figure 1 and Table 1). Muscular hypotonia was common and appeared to be the most prominent in the neonatal period and to improve overtime. Most patients manifested feeding difficulties, delayed speech, and psychomotor development.
Clinical photographs of the patients. (A-I) Craniofacial photographs show distinct facial dysmorphism, long palpebral fissures with eversion of the lower lateral lids, arched eyebrows with the lateral one-third dispersed or sparse, depressed nasal tips, and prominent ears. (J-L) Hand photographs showed prominent finger fat pads. P, patient.
Clinical, radiographic, and laboratory manifestations observed in the study
| Clinical manifestation | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | |
|---|---|---|---|---|---|---|---|---|
| General information | Gender | M | F | M | F | F | F | F |
| Birth weight (g) | 3030 | 2675 | 3630 | 2190 | 2730 | 3200 | 3790 | |
| Duration of pregnancy | Full term | 30 weeks | Full term | 37 weeks | Full term | 38 weeks | 38 weeks | |
| Age at description (y) | 15 | 15 | 9 | 2 | 9 | 2 | 7 | |
| Recent height (cm, centile) | 138.4, <3rd | 139, <3rd | 126.4, 3rd-10th | 81, 3rd-10th | 161, 50th | 70, <3rd | 119, 25-50th | |
| Recent weight (kg, centile) | 60.7, >97th | 46.9, 50-75th | 44.9, >95th | 9.4, <3rd | 32.2, 50-75th | 6.6, <3rd | 28, 75-90th | |
| Head (cm, age, centile) | 51, 5y, 50th | 46, 4y, <3rd | 48.5, 5y, 3rd | 40, 18m, <3rd | 46, 3y, <3rd | 42, 1y, 3rd-10th | 47.5, 3y, 25th | |
Craniofacial features | High/sparse eyebrows | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Long palpebral fissures | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Eversion of lateral portion of lower eyelid | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Depressed nasal tip | Yes | Yes | Yes | Yes | Yes | Yes | No | |
| Ophthalmological anomalies | No | Epiphora, nodule on lowerlids, hypertelorism | No | ONH, CNLDO | Bilateral hyperopia | No | No | |
| Large protuberant ears | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Other anomalies | No | No | No | Under-developed superior crura | Rt preauricular sinus | No | Malformed pinnas | |
Oro-dental features | Micrognathia | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Dentition | Permanent | Permanent | Permanent | Primary | Mixed | Primary | Mixed | |
| Number of tooth agenesis | 4/28 | 8/28 | 2/28 | NA | 11/28 | NA | Multiple | |
| High-arched palate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Other palatal anomalies | Bifid uvula | Cleft soft palate and short uvula | No | Inverted v-shaped CP | No | No | No | |
| Dental caries | Yes | Yes | Yes | No | Yes | No | Yes | |
| Malocclusion | Yes | Yes | Yes | NA | Yes | NA | Yes | |
| Other oro-dental features | Diminutive incisors | Pits, nodules on the lower lip, delayed TD | Abnormal tongue movement | Nodules on the upper lip and gingiva | No | No | Hypoplasitc alveolar ridge | |
Medical findings | Hypotonia in infancy | No | NA | Yes | Yes | No | No | Yes |
| Feeding difficulties | No | Yes | Yes | Yes | No | No | Yes | |
| Learning disability | Yes IQ = 62 | Yes IQ = 44 | Yes | Yes | Yes IQ = 47 | Yes | Yes | |
| Motor delay | No | No | Yes | Yes | No | Yes | Yes | |
| Speech delay | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Seizures | No | No | Yes | Yes | No | No | No | |
| Hearing loss | No, serous otitis media | No | No | No | No | No | No | |
| Other known neurological features | HHD | No | No | Rt coronal CS, HIE, PVL | No | No | Cerebral nodules at Rt PFL and basal ganglion | |
Medical findings | Cardiac anomalies | No | No | No | No | No | CoA, PMVSD, TV aneurysm, isolated PFO | No |
| Neontal/ chronic hypoglycemia | Yes | No | No | No | No | No | Yes | |
| Renal anomalies | No | NA | NA | Lt hydronephrosis | No | No | No | |
| Other anomalies | DI, GHD, micropenis | Subclinical hypothyroid | Abnormal segmentation T11-T12, Fusion of T4-6 | Anovestibular fistula | Early puberty | Hepatomegaly, Splenomegaly | NA | |
Extremities | Brachydactyly | Yes | Yes | Yes | NA | Yes | Yes | No |
| Fifth finger clinodactyly | Yes | Yes | NA | NA | Yes | Yes | No | |
| Prominent finger fat pads | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Other anomalies | Enlarged 5th toes | Simian crease, single crease on the 5th fingers | No | No | No | Hypoplastic middle phalanges of 5th fingers | No | |
g, gram; y, year; cm, centimeter; NA, not applicable; Rt, right; Lt, left; ONH, optic nerve hypoplasia; CNLDO, congenital nasolacrimal duct obstruction; CP, cleft palate; TD, tooth development; HHD, hypothalamo-hypophyseal dysfunction; CS, craniosynostosis; HIE, hypoxic-ischemic encephalopathy; PVL, periventricular leukomalacia; PFL, parietofrontal lobe; CoA, coarctation of aorta; PMVSD, perimembranous ventricular septal defect; TV, tricuspid valve; PFO, patent foramen ovale; DI, diabetes insipidus; GHD, growth hormone deficiency.
A variety of physical phenotypes were observed in KS involving brain, heart, and endocrine system. Two patients were complicated by seizures. Multiple brain abnormalities including coronal synostosis, hypoxic-ischemic encephalopathy, and periventricular leukomalacia were observed in Patient 4. Patient 7 had cerebral nodules at the right parietofrontal lobe and basal ganglion. Cardiac malformations including aortic coarctation, perimembranous ventricular septal defect with tricuspid valve aneurysm, and isolated patent foramen ovale were detected in Patient 6. Abnormal skeletal segmentation of T11 - T12 and fusion of T4 - 6 were observed in Patient 3. Patient 4 suffered hydronephrosis.
Patient 1 possessed multiple abnormalities comprising short stature, micropenis, delayed development, and intellectual disability. Brain MRI illustrated a markedly thickened pituitary stalk, an absent posterior pituitary bright spot, and a slightly decreased height of anterior pituitary gland suggesting hypothalamo-hypophyseal dysfunction. Serum levels of testosterone (<0.087; 24.2 nmole/L) and IGF-1 (26.6; 99-633 ng/ml) were below normal range. The levels of cortisol, FT4, TSH, FSH, and LH were within normal range. The patient was diagnosed with diabetes insipidus and growth hormone deficiency. Endocrinological anomalies were also observed in Patient 2 with hypothyroidism and Patient 5 with precocious puberty.
Craniofacial and Oro-Dental Features
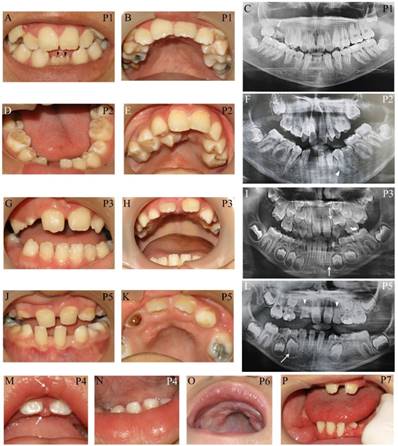
Apart from typical facial features, the common craniofacial phenotype of our KS patients included tooth absence, malocclusion, micrognathia, and high-arched palate/cleft palate. Abnormal tooth size and shape, deviated tooth roots, lip pits and nodules, and dental caries were frequently found. Patient 1 at 13 years presented pitted primary mandibular incisors, malocclusion, high-arched palate, bifid uvula, retained roots of maxillary right first molars with a periapical infection, and absence of four permanent mandibular incisors and maxillary right third molar (Figure 2A-C). Patient 2 at 15 years showed cleft soft palate, short uvula, pits and nodules on the lower lip, severe tooth crowding, root dilaceration, delayed tooth development, and absence of multiple permanent teeth including maxillary four lateral incisors, one mandibular central incisor, three second premolars, and two second molars (Figure 2D-F). Patient 3 at 7 years had high-arched palate and absence of permanent maxillary left lateral incisor and mandibular left canine (Figure 2G-I). Patient 5 at 9 years demonstrated spaced dentition, high arched palate, short truncated roots of maxillary lateral incisors, malalignment of unerupted mandibular right first premolar, and absence of multiple permanent teeth including four lateral incisors, four canines, maxillary first premolars, and mandibular left second premolar. Third molars were not present (Figure 2J-L). Patient 4 at 14 months showed the nodules on the upper lip and gingiva, cleft palate, and six primary teeth including two maxillary central incisors, and four mandibular incisors (Figure 2M, N). High-arched palate was observed in Patient 6 at 2 years (Figure 2 O). Patient 7 at 7 years had only 6 permanent teeth and severe hypoplasia of alveolar ridge (Figure 2P). The number of tooth agenesis ranged from two to eleven permanent teeth excluding the third molars. The most common missing teeth were lateral incisors and second premolars. The clinical, radiographic, and laboratory manifestations including physical features, medical conditions, craniofacial and oro-dental characteristics were summarized in Table 1 and Figure 2.
Oro-dental photographs and panoramic radiographs of the patients. (A-C) Patient 1 at 13 years presents pitted primary mandibular incisors (arrowheads), malocclusion, high-arched palate, absence of four permanent mandibular incisors and maxillary right third molar, retained roots of maxillary right first molars with a periapical infection (arrow). (D-F) Patient 2 at 15 years shows cleft soft palate, severe tooth crowding, root dilaceration (arrowhead), delayed tooth development, and absence of multiple permanent teeth including maxillary four lateral incisors, one mandibular central incisor, three second premolars, and two second molars. (G-I) Patient 3 at 7 years has high-arched palate and absence of permanent maxillary left lateral incisor and mandibular left canine (arrows). (J-L) Patient 5 at 9 years demonstrates spaced dentition and high arched palate. Radiograph reveals the truncated roots of maxillary lateral incisors (arrowheads) and malalignment of unerupted mandibular right first premolar (arrow), and absence of multiple permanent teeth including four lateral incisors, four canines, maxillary first premolars, mandibular left second premolar. Third molars were not present. (M, N) Patient 4 at 14 months shows the nodules on the upper lip and gingiva (arrows), cleft palate, and six primary teeth including two maxillary central incisors and four mandibular incisors. (O) High-arched palate is observed in Patient 6. (P) Patient 7 at 7 years has only 6 permanent teeth and severe hypoplasia of alveolar ridge. P, patient.
Mutation Analysis
Exome sequencing successfully identified mutations in all seven patients. Six (Patients 1-6) were heterozygous for mutations in KMT2D (NM_003482.3) while Patient 7 was heterozygous for a mutation in KDM6A (NM_021140.3). Of the six KMT2D mutations, five were novel. These comprised three frameshift deletions, c.855_856delCT, p.S286X, in exon 7 (Patient 2); c.3699delG, p.G1235VfsX95, in exon 11 (Patient 4), and c.6115delT, p.S2039QfsX8, in exon 29 (Patient 5), and two nonsense mutations, c.5317C>T, p.Q1773X, in exon 22 (Patient 3) and c.11824C>T, p.Q3942X, in exon 39 (Patient 6) of the KMT2D gene. Patient 1 harbored a known missense mutation c.16294C>T, p.R5432W, in exon 51 of KMT2D (Figure 3A, B). Prediction software predicted the variant to be deleterious (PROVEAN score -7.39: Deleterious, SIFT score 0.00: Damaging, REVEL score 0.907: HIGH). The p.286X was located in PHD finger, p.S2039QfsX8 in HMG box, p.Q3942X in polyglotamine tract, and p.R5432W in SET domain of the KMT2D protein (Figure 3C). Patient 7 possessed a novel frameshift deletion c.327_333delTCCAAAA, p.Y109X, in exon 3 of the KDM6A gene. The p.Y109X was located in tetratricopeptide repeat of the KDM6A protein (Figure 3D,E). Exome findings were validated by Sanger sequencing. All mutations were de novo except those of Patient 2 and 5 which are inconclusive due to unavailable parent DNA. Variants analysis was summarized in Figure 3 and Table 2.
Genetic analysis of KS patients and overview of the identified KMT2D and KDM6A mutations relative to a schematic representation of their genes and protein structures. (A) Chromatograms of the patients, parents, and controls. The mutations are indicated above the chromatograms. (B) The mutation, c.855_856delCT is located in exon 7, c.3699delG in exon 11, c.5317C>T in exon 22, c.6115delT in exon 29, c.11824C>T in exon 39, and c.16294C>T in exon 51 of the KMT2D gene. (C) The variant p.286X is located in PHD finger, p.S2039QfsX8 in HMG box, p.Q3942X in polyglutamine tract, and p.R5432W in SET domain of the KMT2D protein. (D) The mutation, c.327_333delTCCAAA, is localized in exon 3 of the KDM6A gene. (E) The p.Y109X is located in tetratricopeptide repeat of the KDM6A protein.

Summary of mutations in the KMT2D and KDM6A genes identified in the study.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Gene | KMT2D | KMT2D | KMT2D | KMT2D | KMT2D | KMT2D | KDM6A |
| Exon | 51 | 7 | 22 | 11 | 29 | 39 | 3 |
| Inheritance | AD | AD | AD | AD | AD | AD | X-link |
| Chromosomal location | chr12: 49416417 | chr12: 49447088-9 | chr12: 49437653 | chr12: 49443672 | chr12: 49435767 | chr12: 49426664 | chrX: 44820628-35 |
| Mutation | c.16294C>Ta | c.855_856delCT | c.5317C>T | c.3699delG | c.6115delT | c.11824C>T | c.327_333delTCCAAAA |
| Amino acid changes | p.R5432W | p.S286X | p.Q1773X | p.G1235VfsX95 | p.S2039QfsX8 | p.Q3942X | p.Y109X |
| Type of mutation | Missense | Frameshift deletion | Stop gain | Frameshift deletion | Frameshift deletion | Stop gain | Frameshift deletion |
| Novel | No | Yes | Yes | Yes | Yes | Yes | Yes |
| Paternal age (Y) | 46 | 29 | 38 | 35 | 34 | 24 | 31 |
| Maternal age (Y) | 29 | 34 | 34 | 32 | 20 | 23 | 29 |
| De novo | Yes | NA | Yes | Yes | NA | Yes | Yes |
Expression of the KMT2D and KDM6A Genes
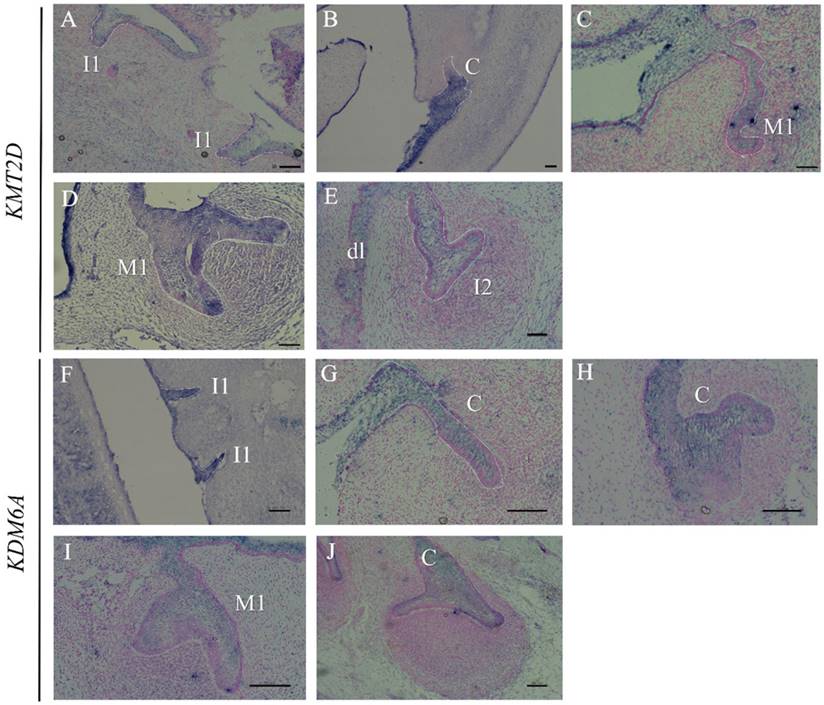
Since multiple dental anomalies have been found in patients with KMT2D and KDM6A mutations, the localization of these genes during tooth development was investigated. We performed in situ hybridization of KMT2D and KDM6A in human tooth germs at 7.5 - 12 weeks of gestation. The expression of KMT2D and KDM6A was observed in the dental epithelium of primary incisors, canine, and molars at the bud and cap stages of human tooth development (Figure 4).
Expression of KMT2D and KDM6A using in situ hybridization on frontal sections of human embryos during tooth development. Expression of KMT2D in dental epithelium (A) the primary lower incisors at 8 weeks (bud stage), (B) the primary lower canine at 8 weeks, (C) the primary upper first molar at 9 weeks, (D) the primary upper first molar at 10 weeks, (E) the dental lamina and the primary upper lateral incisor at 11 weeks. Expression of KDM6A in the epithelium (F) the primary incisors at 8 weeks (bud stage) (G) the canine at 8 weeks, (H) the upper primary canine at 9 weeks (early cap stage), (I) the upper primary first molar at 10 weeks, (J) the lower primary canine at 12 weeks. [I1, primary central incisor; I2, primary lateral incisor; C, primary canine; M1, primary first molar; dl, dental lamina (Scale bar: 100μm).
Discussion
We are the first study revealing comprehensive data of oro-dental anomalies incorporated with medical features and genetic mutations in Thai patients with KS, and the expression of KS-causing genes, KMT2D and KDM6A, in human tooth germs. Congenitally missing teeth, micrognathia, malocclusion, and high-arched palate were consistent oro-dental phenotypes found in our KS patients. The number of tooth agenesis ranged from two to eleven permanent teeth. The most commonly missing teeth, excluding the third molars, were lateral incisors and second premolars. The absence of canine teeth which is the least common missing teeth was observed in our KS patient.
With WES, we successfully identified etiologic mutations in all KS patients including six KMT2D and one KDM6A heterozygous mutations. The high detection rate of mutations (100%) was expected due to our strict criteria for clinical diagnosis of KS. Five out of six mutations in KMT2D were novel. Three were heterozygous frameshift deletions and two were nonsense mutations in the KMT2D gene. One was a known missense mutation in the KMT2D gene located within a SET highly conserved functional domain of the KMT2D (Figure 3B and C) [15, 16]. This mutation was recently shown to disrupt the Kabuki interaction interface (KIS) required for a formation of a second active site within SET1 family core complexes [16, 17]. KMT2D mutations observed in our group of patients showed no significant gathering in any specific exon or a mutational hot spot. One patient was heterozygous for a novel de novo frameshift deletion in the KDM6A gene (Figure 3D) located in tetratricopeptide repeat which was required as an interaction module and multiprotein complex mediator (Figure 3E) [18]. The mutation represented an X-linked dominant inheritance. Since all six novel mutations, four frameshift deletions and two nonsense mutations, were truncating mutations, they are likely to be subject to nonsense mediated mRNA decay (NMD) and expected to be loss-of-function mutations. Taken together with the fact that previous loss-of-function mutations causing KS were leading to haploinsufficiency [19-23], we speculate that the six novel mutations found in our patients cause KS by haploinsufficiency.
KMT2D and KDM6A have been shown to directly interact and function as parts of the activating signal cointegrator-2-containing complex involving in epigenetic control of transcriptionally active chromatin [24]. Interaction between both proteins is involved in the transcription of a wide range of downstream genes including retinoic acid receptor, p53, WNT/β-catenin, and HOX genes which are important for craniofacial development [25-27]. Human dentition has the characteristics of a complex adaptive system showing phenotypic and genetic heterogeneity influenced by epigenetic regulation [28]. One of the consistent dental feature of KS patients is congenital absence of teeth. We detected the expression of KMT2D and KDM6A in dental epithelium at early stages of human tooth development suggesting their roles in odontogenesis. The mutations of genes exclusively expressed in embryonic dental epithelium have been shown to cause tooth agenesis [29-31]. The majority of our mutations observed were truncating mutations [19-23]. These suggest that the deviated expression of KMT2D and KDM6A could disturb their roles, genes and signaling in dental epithelium, and/or epigenetic influence, causing a disruption of human tooth formation.
In summary, our identification of six novel truncating mutations in the KMT2D and KDM6A genes expands mutation spectra of the KMT2D and KDM6A genes associated with KS. Our study shows that WES is a promising method to identify both KMT2D and KDM6A mutations in KS patients. Congenital absence of teeth, high-arched palate, and micrognathia could be considered as one of the diagnostic features of KS. Abnormal tooth shape and size such as pitted incisors and truncated tooth roots add the range of dental features to the KS clinical phenotype. These incorporated with medical conditions would enable health care practitioners to provide an improved health care for KS patients. Our study also reveals for the first time that KMT2D and KDM6A are expressed in the dental epithelium of human tooth germs confirming their roles in tooth development. These pave a crucial way and highlight a requirement for further investigation of KMT2D and KDM6A in the development oro-dental structures.
Supplementary Material
Supplementary table S1.
Acknowledgements
This study was supported by the Thailand Research Fund (TRF) and Office of Higher Education Commission (OHEC) Thailand (MRG6080001), Faculty of Dentistry, Chulalongkorn University (DRF61004), the Chulalongkorn Academic Advancement Into Its 2nd Century Project, the Ratchadapisek Sompoch Endowment Fund.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99:570-3
2. Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99:565-9
3. Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H. et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31:565-89
4. Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet. 2005;67:209-19
5. Miyake N, Koshimizu E, Okamoto N, Mizuno S, Ogata T, Nagai T. et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med Genet A. 2013;161A:2234-43
6. Priolo M, Micale L, Augello B, Fusco C, Zucchetti F, Prontera P. et al. Absence of deletion and duplication of MLL2 and KDM6A genes in a large cohort of patients with Kabuki syndrome. Mol Genet and Metab. 2012;107:627-9
7. Matsune K, Shimizu T, Tohma T, Asada Y, Ohashi H, Maeda T. Craniofacial and dental characteristics of Kabuki syndrome. Am J Med Genet. 2001;98:185-90
8. Tuna EB, Marsan G, Gencay K, Seymen F. Craniofacial and dental characteristics of Kabuki syndrome: nine years cephalometric follow-up. J Clin Pediatr Dent. 2012;36:393-400
9. Teixeira CS, Silva CR, Honjo RS, Bertola DR, Albano LM, Kim CA. Dental evaluation of Kabuki syndrome patients. Cleft Palate Craniofac J. 2009;46:668-73
10. dos Santos BM, Ribeiro RR, Stuani AS, de Paula e Silva FW, de Queiroz AM. Kabuki make-up (Niikawa-Kuroki) syndrome: dental and craniofacial findings in a Brazilian child. Braz Dent J. 2006;17:249-54
11. Bogershausen N, Gatinois V, Riehmer V, Kayserili H, Becker J, Thoenes M. et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum Mutat. 2016;37:847-64
12. Shotelersuk V, Punyashthiti R, Srivuthana S, Wacharasindhu S. Kabuki syndrome: report of six Thai children and further phenotypic and genetic delineation. Am J Med Genet. 2002;110:384-90
13. Porntaveetus T, Srichomthong C, Ohazama A, Suphapeetiporn K, Shotelersuk V. A novel GJA1 mutation in oculodentodigital dysplasia with extensive loss of enamel. Oral Diseases. 2017;23:795-800
14. Oommen S, Otsuka-Tanaka Y, Imam N, Kawasaki M, Kawasaki K, Jalani-Ghazani F. et al. Distinct roles of MicroRNAs in epithelium and mesenchyme during tooth development. Developmental Dynamics. 2012;241:1465-72
15. Lindsley AW, Saal HM, Burrow TA, Hopkin RJ, Shchelochkov O, Khandelwal P. et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J Allergy Clin Immunol. 2016;137:179-87 e10
16. Schott DA, Blok MJ, Gerver WJ, Devriendt K, Zimmermann LJ, Stumpel CT. Growth pattern in Kabuki syndrome with a KMT2D mutation. Am J Med Genet A. 2016;170:3172-9
17. Shinsky SA, Hu M, Vought VE, Ng SB, Bamshad MJ, Shendure J. et al. A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J Mol Biol. 2014;426:2283-99
18. Zeytuni N, Zarivach R. Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure. 2012;20:397-405
19. Micale L, Augello B, Maffeo C, Selicorni A, Zucchetti F, Fusco C. et al. Molecular analysis, pathogenic mechanisms, and readthrough therapy on a large cohort of Kabuki syndrome patients. Hum Mutat. 2014;35:841-50
20. Lindgren AM, Hoyos T, Talkowski ME, Hanscom C, Blumenthal I, Chiang C. et al. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures and cleft palate. Hum Genet. 2013;132:537-52
21. Hannibal MC, Buckingham KJ, Ng SB, Ming JE, Beck AE, McMillin MJ. et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A. 2011;155a:1511-6
22. Bjornsson HT, Benjamin JS, Zhang L, Weissman J, Gerber EE, Chen YC. et al. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med. 2014;6:256ra135
23. Van Laarhoven PM, Neitzel LR, Quintana AM, Geiger EA, Zackai EH, Clouthier DE. et al. Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum Mol Genet. 2015;24:4443-53
24. Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359-83
25. Tucker A, Sharpe P. The cutting-edge of mammalian development; how the embryo makes teeth. Nat Rev Genet. 2004;5:499-508
26. Liu F, Millar SE. Wnt/β-catenin Signaling in Oral Tissue Development and Disease. J of Dent Res. 2010;89:318-30
27. Van Laarhoven PM, Neitzel LR, Quintana AM, Geiger EA, Zackai EH, Clouthier DE. et al. Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum Mol Genet. 2015;24:4443-53
28. Townsend GC, Richards L, Hughes T, Pinkerton S, Schwerdt W. Epigenetic influences may explain dental differences in monozygotic twin pairs. Aust Dent J. 2005;50:95-100
29. Ohazama A, Johnson EB, Ota MS, Choi HJ, Porntaveetus T, Oommen S. et al. Lrp4 modulates extracellular integration of cell signaling pathways in development. PLOS ONE. 2009;3:e4092
30. Han D, Gong Y, Wu H, Zhang X, Yan M, Wang X. et al. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur J Med Genet. 2008;51:536-46
31. Intarak N, Theerapanon T, Ittiwut C, Suphapeetiporn K, Porntaveetus T, Shotelersuk V. A novel PITX2 mutation in non-syndromic oro-dental anomalies. Oral Dis. 2017 doi:10.1111/odi.12804
Author contact
![]() Corresponding author: Dr Thantrira Porntaveetus, Craniofacial Genetics and Stem Cells Research Group, Department of Physiology, Faculty of Dentistry, Chulalongkorn University, Bangkok 10330, Thailand. Email: Thantrira.pac.th
Corresponding author: Dr Thantrira Porntaveetus, Craniofacial Genetics and Stem Cells Research Group, Department of Physiology, Faculty of Dentistry, Chulalongkorn University, Bangkok 10330, Thailand. Email: Thantrira.pac.th